

Abstract
The cation-pi interaction between positively charged and aromatic groups is a common feature of many proteins and protein complexes. The structure of the complex between cytochrome c2 (cyt c2) and photosynthetic reaction center (RC) from Rhodobacter sphaeroides exhibits a cation-pi complex formed between Arg-C32 on cyt c2 and Tyr-M295 on the RC (Axelrod et. al (2002) J. Mol. Biol. 319, 501–515). The importance of the cation-pi interaction for binding and electron transfer was studied by mutating Tyr-M295 and Arg-C32. The first and second order rates for electron transfer were not affected by mutating Tyr-M295 to Ala indicating that the cation-pi complex does not greatly affect the association process or structure of the state active in electron transfer. The dissociation constant KD showed a greater increase when Try-M295 was replaced by non-aromatic Ala (3-fold) than by aromatic Phe (1.2-fold) characteristic of a cation-pi interaction. Replacement of Arg-C32 by Ala increased KD (80-fold) largely due to removal of electrostatic interactions with negatively charged residues on the RC. Replacement by Lys, increased KD (6-fold) indicating that Lys does not form a cation-pi complex. This specificity for Arg may be due to a solvation effect. Double mutant analysis indicates interaction energy between Tyr-M295 and Arg-C32 of about −24 meV (−0.6 kcal/mole). This energy is surprisingly small considering the widespread occurrence of cation-pi complexes and may be due to the trade-off between the favorable cation-pi binding energy and the unfavorable desolvation energy needed to bury Arg-C32 in the short-range contact region between the two proteins.
Keywords: electron transfer, protein association, binding free energy, site-directed mutations, bacterial photosynthesis
Non-covalent molecular interactions play an important role in molecular association processes in biological systems. (1–3) Among these interactions are hydrogen bonds, hydrophobic interactions, and ion-paired salt bridges. (2,3). In addition to these, the cation-pi interaction, the short range electrostatic interaction between a positively charged cation and pi electrons in an aromatic group, (4) has been found to play a role in biological systems. The cation-pi interaction is important in ligand binding (5), in protein structures (6) and in protein-protein complexes (7). This work concerns a cation-pi interaction found in the interface between the cytochrome c2 (cyt c2) and reaction center (RC) of Rhodobacter sphaeroides that is formed between Arg-C32 on the cyt c2 and Tyr-M295 on the RC (8). The importance of this cation –pi interaction on the binding and electron transfer reactions between the two proteins was studied by site directed mutation of both the Arg-C32 on the cyt c2 and Tyr-M295 on the RC.
The RC (9,10) is a membrane bound pigment-protein complex in photosynthetic bacteria that performs the initial light-induced electron transfer reactions to convert sunlight into chemical energy (11). Light absorbed by the RC induces electron transfer from a special bacteriochlorophyll dimer, D, the primary donor through series of bound electron acceptors to a bound ubiquinone (QB). The photo-oxidized donor (D+) is reduced by electron transfer from a water soluble cyt c2 allowing flow of electrons through a membrane associated electron transfer chain in a cycle that is coupled to proton pumping that drives ATP synthesis. Operation of the photocycle relies on efficient reactions between the mobile cyt c2 and the membrane-bound RC. For efficient operation, cyt c2 must associate with the RC, transfer electrons and dissociate within the time scale of electron turnover in the cycle (~ 10−3 s) (12).
The binding and electron transfer rates of isolated cyt c2 and RC have been extensively studied using laser pulse kinetic measurements (13–18). The reduction of the oxidized donor D+ by reduced cyt c2 shows two kinetic phases following a single laser flash – a fast (μs) first order phase (independent of cyt c2 concentration) due to electron transfer from bound cyt c2 to the photo-oxidized donor of the RC and a slower (ms) second order phase (dependent on cyt c2 concentration) due to the binding and subsequent electron transfer of free cyt c2. The observed biphasic kinetics can be explained by the following scheme (17).
| (1) |
where KD is the dissociation constant, kon is the association rate constant, koff is the dissociation rate constant and ke is the electron transfer rate constant in the bound state. The equilibrium between bound and free cyt c2 is achieved in the dark. Following a laser flash the re-reduction of D+ by cyt c22+ is biphasic. RCs with a bound cyt c2 undergo rapid electron transfer with a rate constant ke ( 106 s−1) (14). RCs without a bound cyt c2 undergo slower diffusion limited electron transfer with an observed second order rate constant k2 (~109 s−1M−1) (18). Since ke ≫ koff (19), the observed second order rate constant is the association rate, k2 ~ kon. (20) The fraction of RCs with a bound cyt c2 can be determined by the ratio of the fast and slow phases. The issociation constant KD can be determined from a plot of the fraction of RCs with bound cyt c2 versus the free cyt c2 concentration. The importance of electrostatic interactions on binding and electron transfers between cyt c2 and the RC has been established by the ionic strength dependence of k2 (13), the effect of site directed mutation of charged residues (17,21), chemical cross-linking (22–24) and by electrostatic modeling (15,25,26). Hydrophobic interactions, particularly those of Tyr L162 on the RC have been shown to be important for binding and electron transfer. (18, 27).
The structure of the cyt c2:RC complex was obtained by co-crystallizing the two proteins in a photochemically active complex and determining its x-ray crystal structure. (8) The structure of the complex shows the binding site to be between the cyt c2 and the periplasmic surface of the RC with a central short range contact region in which the exposed heme edge is in contact with Tyr-L162, directly above the BChl dimer (Figure 1). The close contact between the two proteins provides an efficient tunneling pathway for fast electron transfer in the bound state. In the central region the residues from the two proteins make contact through van der Waals, and hydrogen bonding interactions as well as the cation-pi interaction (Figure 1). The closest distance between the positively charged guanidinium group of Arg-C32 and the phenolic group of Tyr-M295 is less than 4 Å, characteristic of a cation-pi complex (6). Surrounding the central region of close contact is a solvent separated region having positively charged residues on the cyt c2 positioned opposite negatively charged residues on the RC interface between the two proteins. The charged residues do not form salt bridges but are separated by solvent.
Figure 1.
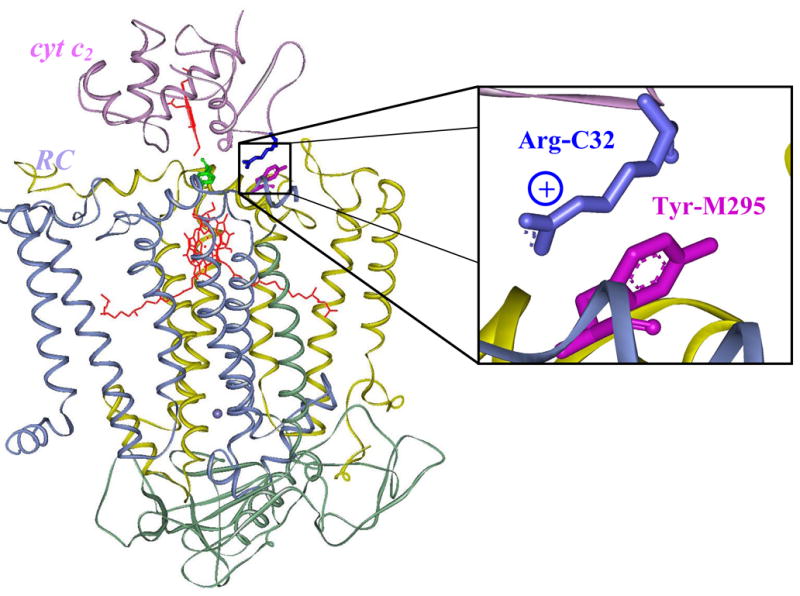
The cation-pi interaction in the RC: cyt c2 complex (PDB accession code 1L9B, ref. 8). The RC subunits L, M, and H are shown in yellow, blue, and green, respectively. The primary donor (bacteriochlorophyll dimer) of the RC is shown in red. The cyt c2 shown in lavender binds with its heme cofactor (red) just above the primary RC donor. Tyr L162, in contact with both cofactors is shown in green. Arg-C32 (blue) of cyt c2 and Tyr-M295 (purple) of the RC form a cation-π interaction in the docked complex. This interaction is enlarged in the box to the right; the view was slightly changed to better emphasize the interaction.
In this work, the functional role of the cation-pi interaction in the binding, association and electron transfer between cyt c22+ and the RC is addressed in order to answer the following questions. (1) What is the magnitude of this interaction? (2) Is this interaction important for the association of cyt c2 and the RC? (3) Is it important for inter-protein electron transfer? To answer these questions site directed mutants of the RC at Tyr-M295 and cyt c2 at Arg-C32 were constructed. Mutants were created that replaced the aromatic residue Tyr-M295 on the RC with either aromatic or non-aromatic residues, and replaced the cationic residue Arg-C32 on cyt c2 with either the cationic Lys residue or neutral residues. The effect of these mutations on binding and electron transfer rates were measured by transient absorption spectroscopy using flash photolysis and compared to native values.
MATERIALS AND METHODS
Site Directed Mutagenesis
The site directed mutations on the RC were constructed as previously described using the QuickChange Mutagenesis kit (Stratagene) and a Perkin Elmer PCR System (28). The site directed mutation on the cyt c2 gene were constructed as follows. First, cycA, the gene coding for the cyt c2 protein, was transferred on a 2.7 kb Pst I fragment from pC2P404.1 (18) to pBCSK(+) (Stratagene, La Jolla, CA) creating pBCcycA. Mutations were introduced into the plasmid via the QuickChange mutagenesis kit as discussed above. The mutation was confirmed by DNA sequence (UCSD Molecular Pathology Shared Resource in the UCSD Cancer Center). The Pst I fragment carrying cyc A was transferred into pRK404 creating pRKcycA*. pRKcycA* was transformed into S17-1 and conjugated into CYCA1, the Rb. sphaeroides host lacking cyc A (29), for transcription and translation of the modified gene sequence.
Protein Isolation and Purification
The bacteria harboring the modified RC or cyt c2 gene was grown in the dark as described (28). RCs from Rb. sphaeroides carotenoidless strain R26 (native) and mutant strains were isolated in 15mM Tris-HCl pH 8, 0.025% lauryl dimethylamine-N-oxide (LDAO), 0.1mM EDTA following published procedures (28).. The final ratio of absorbance, A280/A800, was ≤ 1.5. The RC samples were then dialyzed for two days against HM (10mM Hepes pH 7.5, 0.04% dodecyl-β-D-maltoside (Anatrace)).
Native and mutant cyt c2 proteins were isolated and purified as previously reported (30). Samples were purified to an absorbance ration A280/A412 ≤ 0.3. Mass spectroscopy was performed (UCSD Dept. of Chemistry, mass spectrometry facility) to verify the Arg-C32 →Gln and Arg-C32 →Lys substitutions in the mutant cyt c2’s.
Quinone/Quinol Preparations
Quinone (Q0) (2,3-dimethoxy-5-methylbenzoquinone) was obtained from Aldrich with ≥99% purity. Quinol (Q0H2) was synthesized by reducing quinone with hydrogen gas in the presence of platinum black also obtained from Aldrich.
Electron Transfer Measurements
Electron transfer kinetics between the RC and cyt c2 were measured by flash absorption spectroscopy as described (31). Absorbance changes were measured at 865 nm or 595 nm following a laser flash from a Nd:YAG laser with an tunable optical parametric oscillator (Opotek, λ=800nm, τ=10ns). All measurements were conducted at 23° C in a buffer of 10 mM Hepes and 0.04% β-maltoside at pH 7.5. Samples included 50 μM Q0 and Q0H2 to ensure that the cyt c2 was reduced prior to each laser flash.
The fraction of RC with and without a bound cyt c22+ were determined from the relative amplitudes of the fast and slow phase of the D+ reduction by the cyt c22+. The concentration of free cyt c22+ was obtained by subtracting the concentration of bound cyt c22+ from the total added. The value of the dissociation constant, KD was determined from the dependence of the fraction bound on the concentration of free cyt c22+. The change in free energy due to mutation of residue i was obtained from the relation as described in (18)
| (2) |
where, KD,i and KD,0 are the dissociation constants for mutant and native complexes respectively. The second order rate constant, k2, was determined from the concentration dependence of the measured rate constant for the slow phase of D+ re-reduction. When the concentration of cyt c2 is large compared to the RC, the reaction becomes pseudo-first order in cyt c2. To maximize the concentration range over which this condition holds, the RC concentration was kept low (0.2 μM). The first order electron transfer rate (ke) constants was measured at 595 nm with a higher RC concentration (5 μM RC, 30 μM cyt c2). With no cyt c2 present some mutant RCs produced a small transient signal at this wavelength, which was subtracted from the observed kinetic trace.
Determination of the cation-pi interaction energy by double mutant analysis
The specific cation-pi interaction energy can be estimated by double mutant analysis of changes due to mutation of Arg-C32 and Tyr-M295. The relation of the changes in free energy due to mutation and the cation-pi interaction energy is illustrated by the double mutant cycle. (31–33). The interaction energy between two residues can be estimated by the interaction energy ΔΔGint where
| (3) |
ΔΔGY, ΔΔGR, and are the changes in free energy obtained for the binding reaction of RCs modified at residue Tyr-M295 to native cyt c2 and cyt c2 modified at residue Arg-C32 with native RCs and ΔΔGYR is the change in free energy for the binding between the modified RC and modified cyt c2. KD,Y, KD,R and KD,YR are the corresponding changes in dissociation constant. KD,0 is the dissociation constant of native cyt c2 and RC.
The change in free energy due to mutation of Tyr-M295 is assumed to be due to contributions from the cation pi interaction of-Tyr-M295 with Arg-C32, ΔΔGcat-pi, and non-specific interactions with other residues ΔΔGYX.
| (4) |
The major contributions to ΔΔGcat-pi are the increase in free energy due to the loss of the cation-pi interaction and the decrease due to the loss of the solvation penalty; it will also include changes in other specific interactions between the two residues such as those due to van der Waals contacts. These cannot be separated in this analysis.
The change in binding free energy due to mutation of Arg-C32, ΔΔGR, is assumed to have contributions from ΔΔGcat-pi, between Arg-C32 and Tyr-M295 and interactions between Arg-C32 with other residues on the RC, ΔΔGRX. In addition we specifically include an additional term ΔΔGelect, which is due to electrostatic interaction of positively charged Arg-C32 with the negative potential of the RC surface that is lost due to mutation to a neutral residue X.
| (5) |
The change in free energy due to the mutation of the two interacting residues, ΔΔGRY can be expressed as
| (6) |
where it is assumed that the interactions changed by single mutations are the same in the double mutation, i.e. the structure of the complex containing single mutations is the same (apart from the local changes to the mutated residue) as that containing both mutations. From Eqns. 3–6 the interaction free energy between the mutated residues is the negative of the free energy increase due to loss of the cation-pi interaction.
| (7) |
Thus, the cation-pi interaction energy can be obtained from double mutant analysis.
RESULTS
Dissociation Constants, KD. KD and ΔΔG values were obtained from the dependence of the fraction of RC with a bound cyt c2, on the free concentration of cyt c2. The binding curves are presented in Figures 2 and 3 and the results are summarized in Table 1.
Figure 2.

Binding data for the native cyt c2 with the native and mutant RCs. Shown is a plot of f, the fraction of RC with a bound cyt c2, versus the concentration of free cyt c22+. Binding of cyt c2 to RCs with an aromatic residue at M295 are shown as solid symbols (native ■, Tyr →Phe ●, Tyr →Trp ▲). Binding of cyt c2 to RCs without an aromatic residue are shown as open symbols (Tyr →Leu □, Tyr →Asn ○, Tyr →Ala △). The solid lines are the binding curves (Eqn. 3) using the averaged value of KD for the aromatic (0.34 μM) and non-aromatic (0.86 μM) mutant RCs. Note that KD for all of the aromatic residues at M295 are within error indistiguishable. Similarly, KD for all of the non-aromatic substitutions are within error indistinguishable, but differ by ~3-fold from the native. (Conditions: 10 mM Hepes pH 7.5, 0.04% β-maltoside, 50 μM Q0, 50 μM Q0H2).
Figure 3.

Binding data for mutant cyt c2 that lack the cationic Arg-C32 with the native and mutant RC lacking the pi system at M295. Diamonds represent mutations of Arg-C32 to Lys, circles represent mutation of Arg-C32 to Gln. Solid symbols indicate the binding mutant cyt c2 to the native RC. Open symbols indicate the binding data of the mutant cyt c2 to the Tyr-M295 →Leu mutant RC. The binding curve for the native system is shown for reference. Note that the binding of the mutant cyt c2 is essentially the same to either the native or mutant RC indicating that the mutations introduce little if any secondary changes that affect the binding. (Conditions as in Figure 2).
Table 1.
Binding and Electron Transfer Parameters for single and double mutant reactions of RCs modified at Tyr-M295 and cyt c2 modified at Arg-C32 (pH = 7.5)a
| RC Strain | cyt c2 | ke(× 106s−1) | k2 (× 108M−1s−1) | KD(μM) | ΔΔG (meV) |
|---|---|---|---|---|---|
| Native | Native | 1.0 | 13.1 | 0.30 | 0 |
| Tyr-M295 →Phe | Native | 1.1 | 13.4 | 0.38 | 6 |
| Tyr-M295 →Trp | Native | 0.9 | 12.5 | 0.44 | 10 |
| Tyr-M295 →Asn | Native | 1.0 | 13.2 | 0.78 | 24 |
| Tyr-M295 →Leu | Native | 0.9 | 13.1 | 0.89 | 28 |
| Tyr-M295 →Ala | Native | 1.0 | 12.1 | 0.91 | 28 |
| Native | Arg-C32→Lys | 0.9 | 4.6 | 1.7 | 44 |
| Native | Arg-C32→Gln | 0.6 | 7.8 | 15 | 100 |
| Native | Arg-C32→Ala | 0.8 | 6.1 | 24 | 112 |
| Tyr-M295 →Leu | Arg-C32→Lys | 0.9 | 3.4 | 1.8 | 46 |
| Tyr-M295 →Leu | Arg-C32→Gln | 0.6 | 13.1 | 17 | 103 |
| Tyr-M295 →Ala | Arg-C32→Ala | 0.7 | 6.7 | 31 | 118 |
ke is the rate constant for the fast, first order electron transfer. kon is the second order rate constant. KD is the dissociation constant for the cyt c2:RC complex. ΔΔG = RTln(KD,i/KD,0) is the difference in binding free energy between the mutant and native RCs. The experimental precision for the measured values of the rates and KD is ± 15%. The uncertainty in the free energies is ± 5meV
Reaction Center Mutations
Mutation of Tyr-M295 on the RC to aromatic residues Phe and Trp had small effects on KD, (increases by factors of 1.3 and 1.5, respectively) indicating that these aromatic residues can also participate in a cation-pi interaction with Arg-C32. Mutation of Tyr-M295 to Asn, Leu and Ala produced larger changes in KD (up to a factor of 3). The changes in free energy due to these mutations increased up to a maximum ΔΔG = 28 meV for mutations Tyr-M295 to Leu and Ala. The free energy changes can be attributed largely to the loss of the cation-pi interaction.
Cytochrome Mutations
Mutation of Arg-C32 to Lys, Gln and Ala produced increases in KD by factors of 5.6, 50 and 80. These changes were larger than those produced by mutation of Tyr- M295. The larger changes free energy (100 and 112 meV) due to the mutation of the cationic Arg to neutral Gln and Ala result from the additional loss of stabilizing electrostatic interaction between Arg and the negative potential due to acidic residues on the RC surface (17).
Second Order Association Rate Constants, k2
Second order rate constants were obtained from plots of the observed kinetic rate against the concentration of free cyt c2 (17,31). Mutant RCs had the same second order rate constant within experimental error (kon=12–13×108M−1s−1) as the native RC for the native cyt c2. The mutations to cyt c2 (Arg-C32 →Lys, Arg-C32 →Glu and Arg-C32 →Ala) resulted in smaller values of kon (5 ×108M−1s−1, 8×108M−1s−1 and 6×108M−1s−1, respectively) that accompanied the increase in dissociation constant KD.
First Order Electron Transfer Rate Constants, ke
The first order electron transfer rate constant for the native RC: cyt c2 system was found to be ke=1 × 106 s−1, as previously reported (14–18). All mutant RCs bound with native cyt c2 had the same first order rate constant (ke = 0.9–1.1 × 106 s−1). Similarly, ke = 0.9 × 106 s−1 for electron transfer between the bound Arg-C32 →Lys mutant cyt c2 and the native RC. Small changes were observed in ke for electron transfer between the native RC and the Arg-C32 →Gln cyt c2 (ke = 0.6 × 106 s−1) of the Arg-C32 →Ala cyt c2 (ke = 0.8 × 106 s−1).
DISCUSSION
Role of the Cation-pi Interaction in Binding
The mutation of either the cationic or the aromatic partner in the cation pi complex resulted in changes in the binding of the cyt c2 to the RC. Replacement of Tyr-M295 with residues Ala, Asn or Leu lacking pi electrons had the effect of increasing KD (by up to 3-fold for Ala) while replacement of Tyr-M295 with the other aromatic residues Phe and Trp had relatively small effect. These results are characteristic of the formation of a cation-pi complex at the cyt c2:RC interface and indicate that an aromatic residue at position M295 assists the binding of the cyt c2 to the RC. The removal of the cationic Arg-C32 residue and replacement with neutral residues Ala and Gln resulted in a larger increase in KD (by up to 80-fold) corresponding to a change in binding free energy of ~110 meV. The larger effect upon mutation of the cationic residue can be attributed to the loss of the electrostatic interactions between the Arg-C32 and residues other than Tyr-M295. The largest interaction is likely to be the electrostatic interaction between Arg-C32 and Asp-L155 at a distance of 7Å away (6) The Coulomb interaction between the two charged residues would be ~100 meV, for a dielectric constant of 20.
The double mutant cycle provides a means to assess the magnitude of the cation-pi interaction energy between the two mutated residues. From the three double mutant cycles involving mutations in this study, (Arg-C32 → Lys/Tyr-M295 → Leu), (Arg-C32 → Gln/Tyr-M295 → Leu), and (Arg-C32 → Ala/Tyr-M295 → Ala), values for ΔΔGint = −26, −25 and −22 meV are obtained from Eqn. 3 giving an average of −24+2 meV. The values obtained for all three mutant cycles are in good agreement with each other. The cation pi interaction energy is close to the magnitude of the free energy change for mutation of Tyr-M295 to Ala indicating that the interactions of Tyr-M295 with residues besides Arg-C32 are small (ΔΔGYX ~ 4 meV, Eqn.. 4)
The specific site-site interaction between Arg-C32 and Tyr-M295 has a relatively small net contribution to the binding free energy of the complex, ΔΔGint = −24mev ( −0.6 kcal/mole ~ kBT). The cation-pi interaction between Arg-C32 and Tyr-M295 in the cyt c2:RC complex was identified by the CAPTURE program (6) (http://capture.caltech.edu/) that has been successfully used to identify cation pi interactions in many other protein structures. Gas phase estimates of the electrostatic and van der Waals contributions were computed to be ~ −4 kcal/mole (−170 meV) and ~ −2 kcal/mole (−85 meV), respectively (average of 3 structures in 2 crystal forms (pdb id.1L9B and 1L9J). Note that these values are much larger than the smaller observed net effect which includes the competing energy cost required to desolvate the cation. Since the guanidinium group is positively charged, it interacts with water dipoles and forms hydrogen bonds. In forming the cyt c2:RC complex Arg-C32 becomes largely inaccessible to solvent. A solvent accessibility for Arg-C32 side chain changes from 55% to 3% in forming the cyt c2:RC complex (determined using the CNS program (34).) Thus, a significant energy is required to desolvate the cationic guanidinium group to bring it into close contact with the Tyr.
Although Lys is capable of participating in a cation pi interaction, the change in the interaction energy between Lys-C32 and Tyr-M295 (ΔΔGint =−26 meV from the double mutant analysis) suggests that Lys-C32 does not form a cation-pi complex. The larger change in free energy ΔΔGR of 44 meV for mutation of the charged Arg-C32 to Lys than those found for the mutations the aromatic Tyr-M295 to either Leu or Ala could be due to changes in the electrostatic interaction due to the Arg → Lys mutation, i.e. ΔΔGelect in Eqn. 5 is not zero. This change could result either form a difference in the Coulomb interaction, due to a change in the position of the charge on the potential surface or due to a differences in the solvation of Lys and Arg.
The inability of Lys to form a cation-pi complex may be because the desolvation energy cost is larger due to the higher charge density for the ammonium cation than the guanidinium cation that has charge delocalized over 3 N atoms. The delocalizalized charge lowers the desolvation energy compared to Lys making the formation of the cation-pi complex with Arg more favorable (34). In addition, steric effects may also contribute. The aromatic ring of Tyr-M295 makes the closest contact with the ε-nitrogen atom of Arg-C32. The ζ-nitrogen atom of Lys-C32 is one bond length longer and may not be able to form a strong cation-pi bond. Crowley and Golovin (7) have recently examined a structural data base for protein structures having cation-pi interactions. They found that the most common cation-pi interaction in protein complexes was between Arg and Tyr with interactions between Lys and Tyr much less frequent. It is interesting to note that Arg-C32 is the only Arg residue in the interface region, compared to 5 Lys residues. This suggests that Arg-C32 is selected for its ability to form a cation-pi complex in close association with the RC surface.
The Role of the Cation-pi interaction in electron transfer
The first order electron transfer rate constants ke for all mutated RCs in this study were very similar to that of native RCs (ke = 1.0 × 106 s−1) (Table 1). Since the first order rate is due to electron transfer from cyt c2 in its bound state on the RC surface, we conclude that the positioning of the cyt c2 on the RC surface is the same with or without the cation-pi interaction. Small changes in rate were observed for reaction of the mutant cyt c2 (ke = 0.6–0.9 × 106 s−1), which could be due to small changes in the redox potential of the mutant cyt c2, the reorganization energy for electron transfer or in the distance (<1Å) between the cofactors in the bound state (36–38).
These results can be rationalized with results from previous experiments using a model in which the electron transfer rate ke is determined by the close juxtaposition of the exposed heme edge with the short-range contact region on the RC centered around Tyr-L162 which is in close contact with the BChl2 (D) (8). Mutation of hydrophobic residues (particularly Tyr-L162) changed the electron transfer rate ke presumably due to changes in the distance between cofactors in the structure of the modified cyt c2:RC complex (18). In contrast, mutations of charged residues on the RC, located outside of the short-range contact region, did not change the electron transfer rate ke, although they modified the binding affinity (17). The modification of cation-pi contacts appears to have effects on ke similar to the charged mutations even though the cation-pi complex is part of the short range interaction region. This may be because the cation-pi complex is at the boundary of the short-range contact region. Thus, changes in the cation-pi bonding do not affect the structure in the central region around Tyr-L162 which largely determines the rate of electron transfer (39,40).
The Role of the Cation-pi interaction in cyt c2 association/dissociation
The measured value for the second order association rate constant was the same for all of the mutated RCs used in this study (k2 =kon = 12–13 × 108 M−1 s−1). Thus, the cation-pi interaction does not influence the rate of association. This is reasonable since the cation-pi interaction is a short-range interaction between a charged residue and the quadrupole moment of a neutral residue. Thus, the formation of the cation-pi complex is not expected to be important for the formation of the transition state for the association process which is located ~ 8–10 Å above the RC surface (17,25,26). This is consistent with the finding that mutations of hydrophobic residues on the RC do not appreciably affect kon (18).
Although the cation-pi interaction does not affect the association rate, it does affect the dissociation rate, koff. Since the loss of the cation– pi results in a 2– 3– fold increase in KD (Table 1) but no change in kon, the loss of the cation-pi interaction results in a 2– 3– fold increase in koff. (KD = koff/kon ). These changes reflect the slightly tighter binding of cyt c2 to the RC and an increase in the barrier for dissociation in the presence of the interaction. Thus, the cation-pi interaction increases the residence time by ~ 3– fold of the cyt c2 on the RC surface. If the binding were made substantially tighter, koff would be correspondingly smaller which would substantially reduce the turnover rate of photosynthetic electron transfer. For example, an increase of 60meV in the cation-pi interaction would result in a 10-fold decrease in koff and an ~10-fold decrease in the overall turnover rate of electron transfer (τ=25 ms). Thus, a strongly binding cation-pi interaction would have deleterious effect on the photosynthetic efficiency.
Mutations to the charged residue Arg-C32 resulted in decreased association rates (k2 = 5–8 × 108 M−1s−1). These changes can be attributed to changes in electrostatic interactions rather than cation-pi interactions. The changes in k2 are consistent with the changes of k2 due to mutation of charged residues in the interface (17,21) It is interesting to note that the Arg-C32 → Lys mutation had the largest change on k2 suggesting that the configuration of the two residues is significantly different.
Importance of the Arg-C32 - Tyr-M295 interaction
The magnitude for the cation-pi interaction on increasing the binding in the cyt c2:RC complex appears to be surprisingly small considering that such complexes have been found in many other systems (7) including the complex between cyt c and cyt bc1 from yeast (41). Thus, the cation-pi interaction in the cyt c2:RC complex is just one of many interactions (electrostatic, hydrophobic, hydrogen bonding) that optimize the protein-protein interface, rather than a dominant component of the binding energy. The widespread occurrence of cation-pi complexes may be due to the importance of electrostatic interactions (and thus positively charged residues) in the protein association process (7). The cationic Arg-C32 is one of several positively charged residues on the cyt c2 surface that serve to attract and orient the cyt c2 on the negatively charged binding surface of the RC form a loosely bound encounter complex. Further short range docking of the cyt c2 leads through a transition state to the bound state in which electron transfer occurs (26). In the bound state the Arg-C32 is buried in the central short range contact region of the interface where solvent is excluded. In this solvent excluded region the two interfaces are in close contact and optimized for electron transfer. In contrast, the other charged residues in the interface are separated by solvent. (8) The formation of a cation-pi complex provides a means to offset the cost of desolvating the charge (42), thereby providing a means to bury a positively charged group within the solvent excluded short-range interaction domain. Although the overall cation-pi interaction does not greatly increase the binding affinity, the binding is well optimized by other interactions so that a further increase in affinity is not necessary and might slow down the dissociation of the oxidized cyt c2 and thus reduce the overall turnover of the reaction.
Acknowledgments
We thank Ed Abresch, Roger Isaacson and Larry Gross for technical assistance and helpful discussions, Timothy Donohue for providing the cytochrome c2 deletion strain and George Feher for helpful discussions. DNA sequencing was performed by the DNA Sequencing Shared Resource, UCSD Cancer Center, which is funded in part by NCI Cancer Center Support Grant # 2 P30 CA23100-18.
Abbreviations
- cyt c2
cytochrome c2
- RC
reaction center
- bc1
cytochrome bc1
- Q10
ubiquinol-10
- D
primary electron donor (bacteriochlorophyll dimer)
- D+
photo-oxidized primary electron donor
- QB
bound acceptor quinone
- ke
first order electron transfer rate
- k2
observed second order rate constant
- KD
dissociation constant
- Q0
2,3-dimethoxy-5-methyl-1,4-benzoquinone
Footnotes
This work was supported by NIH Grant GM 41637
References
- 1.Kleanthous C. Protein-protein recognition. Oxford University Press; Oxford, UK: 2000. [Google Scholar]
- 2.Jones S, Thornton J. In: Protein-protein recognition. Kleanthous C, editor. Oxford University Press; New York: 2000. pp. 33–59. [Google Scholar]
- 3.Stites W. Protein-protein interactions: Interface structure, binding thermodynamics, and mutational analysis. Chem Rev. 1997;97:1233–1250. doi: 10.1021/cr960387h. [DOI] [PubMed] [Google Scholar]
- 4.Ma J, Dougherty D. The cation-pi interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 5.Zacharius N, Dougherty DA. Cation-pi interactions in ligand recognition and catalysis. Trends in Pharm Sci. 2002;23:281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
- 6.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowley P, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins: Struc Funct Bioinform. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 8.Axelrod HL, Abresch EC, Okamura MY, Yeh AP, Rees DC, Feher G. X-ray structure determination of the cytochrome c2: Reaction center electron transfer complex from Rhodobacter sphaeroides. J Mol Biol. 2002;319:501–515. doi: 10.1016/S0022-2836(02)00168-7. [DOI] [PubMed] [Google Scholar]
- 9.Gunner M. The reaction center protein from purple bacteria: Structure and function. Curr Top Bioenerg. 1991;16:319–367. [Google Scholar]
- 10.Feher G, Allen JP, Okamura MY, Rees DC. Structure and function of bacterial photosynthetic reaction centers. Nature(London) 1989;339:111–116. [Google Scholar]
- 11.Blankenship RE. Molecular mechanisms of photosynthesis. Blackwell Science Inc; London: 2002. [Google Scholar]
- 12.Crofts AR, Wraight CA. The electrochemical domain of photosynthesis. Biochim Biophys Acta. 1983;726:149–185. [Google Scholar]
- 13.Prince RC, Cogdell RJ, Crofts AR. The photo-oxidation of horse heart cytochrome c and native cytochrome c2 by reaction centres from Rhodopseudomonas spheroides R-26. Biochim Biophys Acta. 1974;347:1–13. doi: 10.1016/0005-2728(74)90194-7. [DOI] [PubMed] [Google Scholar]
- 14.Overfield RE, Wraight CA, Devault DC. Microsecond photooxidation kinetics of cytochrome c2 from Rhodopseudomonas sphaeroides: In vivo and solution studies. FEBS Lett. 1979;105:137–142. doi: 10.1016/0014-5793(79)80903-5. [DOI] [PubMed] [Google Scholar]
- 15.Tiede D, Vashishta A, Gunner M. Electron-transfer kinetics and electrostatic properties of the Rhodobacter sphaeroides reaction center and soluble c-cytochromes. Biochemistry. 1993;32:4515–4531. doi: 10.1021/bi00068a006. [DOI] [PubMed] [Google Scholar]
- 16.Moser C, Dutton PL. Cytochrome c and c2 binding dynamics and electron transfer with photosynthetic reaction center protein and other integral membrane redox proteins. Biochemistry. 1988;27:2450–2461. doi: 10.1021/bi00407a031. [DOI] [PubMed] [Google Scholar]
- 17.Tetreault M, Rongey SH, Feher G, Okamura M. Interaction between cytochrome c2 and the photosynthetic reaction center from Rhodobacter sphaeroides: Effects of charge-modifying mutations on binding and electron transfer. Biochemistry. 2001;40:8452–8462. doi: 10.1021/bi010222p. [DOI] [PubMed] [Google Scholar]
- 18.Gong X, Paddock ML, Okamura M. Interactions between cytochrome c2 and photosynthetic reaction center from Rhodobacter sphaeroides: Changes in binding affinity and electron transfer rate due to mutation of interfacial hydrophobic residues are strongly correlated. Biochemistry. 2003;42:14492–14500. doi: 10.1021/bi035603c. [DOI] [PubMed] [Google Scholar]
- 19.Gerencser L, Laczko G, Maroti P. Unbinding of oxidized cytochrome c from photosynthetic reaction center of Rhodobacter sphaeroides is the bottleneck of fast turnover. Biochemistry. 1999;38:16866–16875. doi: 10.1021/bi991563u. [DOI] [PubMed] [Google Scholar]
- 20.Bendall D. In: Protein electron transfer. Bendall D, editor. Bios Scientific Publishers Ltd; Oxford, UK: 1996. pp. 43–68. [Google Scholar]
- 21.Caffrey MS, Bartsch RG, Cusanovich MA. Study of the cytochrome c2-reaction center interaction by site-directed mutagenesis. J Biol Chem. 1992;267:6317–6321. [PubMed] [Google Scholar]
- 22.Rosen D, Okamura MY, Abresch EC, Valkirs GE, Feher G. Interaction of cytochrome c with reaction centers of Rhodopseudomonas sphaeroides R-26: Localization of the binding site by chemical cross-linking and immunochemical studies. Biochemistry. 1983;22:335–341. doi: 10.1021/bi00271a016. [DOI] [PubMed] [Google Scholar]
- 23.Drepper F, Dorlet P, Mathis P. Cross-linked electron transfer complex between cytochrome c2 and the photosynthetic reaction center of Rhodobacter sphaeroides. Biochemistry. 1997;36:1418–1427. doi: 10.1021/bi961350u. [DOI] [PubMed] [Google Scholar]
- 24.Hall J, Zha X, Durham B, O’Brien P, Vieira B, Davis D, Okamura M, Millett F. Reaction of cytochromes c and c2 with the Rhodobacter sphaeroides reaction center involves the heme crevice domain. Biochemistry. 1987;26:4494–4500. doi: 10.1021/bi00388a048. [DOI] [PubMed] [Google Scholar]
- 25.Miyashita O, Onuchic JN, Okamura MY. Continuum electrostatic model for the binding of cytochrome c2 to the photosynthetic reaction center from Rhodobacter sphaeroides. Biochemistry. 2003;42:11651–11660. doi: 10.1021/bi0350250. [DOI] [PubMed] [Google Scholar]
- 26.Miyashita O, Onuchic J, Okamura M. Transition state and encounter complex for fast association of cytochrome c2 with bacterial reaction center. Proc Natl Acad Sci U S A. 2004;101:16174–16179. doi: 10.1073/pnas.0405745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wachtveitl J, Farchaus J, Mathis P, Oesterhelt D. Tyrosine 162 of the photosynthetic reaction center l-subunit plays a critical role in the cytochrome c2 mediated rereduction of the photooxidized bacteriochlorophyll dimer in Rhodobacter sphaeroides. 2. Quantitative kinetic analysis. Biochemistry. 1993;32:10894–904. doi: 10.1021/bi00091a045. [DOI] [PubMed] [Google Scholar]
- 28.Paddock ML, Adelroth P, Change C, Abresch EC, Feher G, Okamura MY. Identification of the proton pathway in bacterial reaction centers: cooperation between Asp-M17 and Asp-L210 facilitates proton transfer to the secondary quinone (QB) Biochemistry. 2001;40:6893–6902. doi: 10.1021/bi010280a. [DOI] [PubMed] [Google Scholar]
- 29.Donohue TJ, McEvan AG, Van Doren S, Crofts AR, Kaplan S. Phenotypic and genetic characterization of cytochrome c2 deficient mutants of Rhodobacter sphaeroides. Biochemistry. 1988;27:1918–1925. doi: 10.1021/bi00406a018. [DOI] [PubMed] [Google Scholar]
- 30.Bartsch R. In: The photosynthetic bacteria. Clayton R, Sistrom W, editors. Plenum Press; New York: 1978. pp. 249–279. [Google Scholar]
- 31.Tetreault M, Cusanovich M, Meyer T, Axelrod H, Okamura M. Double mutant studies identify electrostatic interactions that are important for docking cytochrome c2 onto the bacterial reaction center. Biochemistry. 2002;41:5807–5815. doi: 10.1021/bi012053e. [DOI] [PubMed] [Google Scholar]
- 32.Serrano L, Horovitz A, Avron B, Bycroft M, Fersht A. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry. 1990;29:9343–9352. doi: 10.1021/bi00492a006. [DOI] [PubMed] [Google Scholar]
- 33.Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- 34.Brünger AT, Adams PD, Clore GM, DeLanod WL, Gros P, Grosse-Kunstleve RW, Jiangf J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Mason P, Neilson G, Dempsey C, Barnes A, Cruickshank J. The hydration structure of guanidinium and thiocyanate ions: Implications for protein stability in aqueous solution. Proc Natl Acad Sci U S A. 2003;100:4557–4561. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcus RA, Sutin N. Electron transfer in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 37.Moser CC, Keske JM, Warnke K, Farid RS, Dutton PL. The nature of biological electron transfer. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 38.Beratan D, Betts J, Onuchic J. Protein electron transfer rates set by the bridging secondary and tertiary structure. Science. 1991;252:1285–1288. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 39.Aquino A, Beroza P, Beretan D, Onuchic J. Docking and electron transfer between cytochrome c2 and the photosynthetic reaction center. Chem Phys. 1995;197:277–288. [Google Scholar]
- 40.Miyashita O, Okamura MY, Onuchic JN. Theoretical understanding of the interprotein electron transfer between cytochrome c2 and the photosynthetic reaction center. J Phys Chem B. 2003;107:1230–1241. [Google Scholar]
- 41.Hunte C, Solmaz S, Lange C. Electron transfer between yeast cytochrome bc(1) complex and cytochrome c: A structural analysis. Biochim Biophys Acta. 2002;1555:21–28. doi: 10.1016/s0005-2728(02)00249-9. [DOI] [PubMed] [Google Scholar]
- 42.Gallivan J, Dougherty D. A computational study of cation-pi interactions versus salt bridges in aqueous media: Implications for protein engineering. J Am Chem Soc. 2000;122:870–874. [Google Scholar]