

Abstract
Nitrogenase catalyzes the sequential addition of six electrons and six protons to a N2 that is bound to the active site metal cluster FeMo-cofactor, yielding two ammonia molecules. The nature of the intermediates bound to FeMo-cofactor along this reduction pathway remains unknown, although it has been suggested that there are intermediates at the level of reduction of diazene (HN=NH, also called diimide) and hydrazine (H2N-NH2). Through in situ generation of diazene during nitrogenase turnover, we show that diazene is a substrate for the wild-type nitrogenase and is reduced to NH3. Diazene reduction, like N2 reduction, is inhibited by H2. This contrasts with the lack of H2 inhibition when nitrogenase reduces hydrazine. These results support the existence of an intermediate early in the N2 reduction pathway at the level of reduction of diazene. Freeze-quenching a MoFe protein variant with α-195His substituted by Gln and α-70Val substituted by Ala during steady-state turnover with diazene resulted in conversion of the S = 3/2 resting state FeMo-cofactor to a novel S = 1/2 state with g1 = 2.09, g2 = 2.01, and g3 ~ 1.98. 15N- and 1H-ENDOR establish that this state consists of a diazene-derived [-NHx] moiety bound to FeMo-cofactor. This moiety is indistinguishable from the hydrazine-derived [-NHx] moiety bound to FeMo-cofactor when the same MoFe protein is trapped during turnover with hydrazine. These observations suggest that diazene joins the normal N2-reduction pathway, and that the diazene- and hydrazine-trapped turnover states represent the same intermediate in the normal reduction of N2 by nitrogenase. The implications of these findings for the mechanism of N2 reduction by nitrogenase are discussed.
Keywords: FeMo-cofactor, EPR, ENDOR, Active Site, Substrate
Nitrogenase is the enzyme responsible for catalyzing biological reduction of N2 to two NH3, an essential reaction in the global biogeochemical nitrogen cycle (1–3). The minimum stoichiometry for the nitrogenase catalyzed reduction of N2 involves delivery of 8e− and 8H+ (eqn 1).
| (eqn 1) |
The Mo-based nitrogenase is comprised of two component proteins called the Fe protein and the MoFe protein. The Fe protein contains a single [4Fe-4S] cluster plus two MgATP binding sites (4). It functions to deliver one electron at a time to the MoFe protein in a reaction coupled to the hydrolysis of the two MgATP molecules (5, 6). The Fe protein dissociates from the MoFe protein after each electron transfer and hydrolysis of two MgATP (7), necessitating eight rounds of Fe protein binding to, and dissociation from, the MoFe protein for each N2 reduced. The MoFe protein contains an [8Fe-7S] cluster called the P-cluster (8) that is proposed to broker electrons between the Fe protein and the N2-binding site, a [7Fe-9S-Mo-X-homocitrate] cluster called the FeMo-cofactor (Figure 1).
Figure 1. FeMo-cofactor.
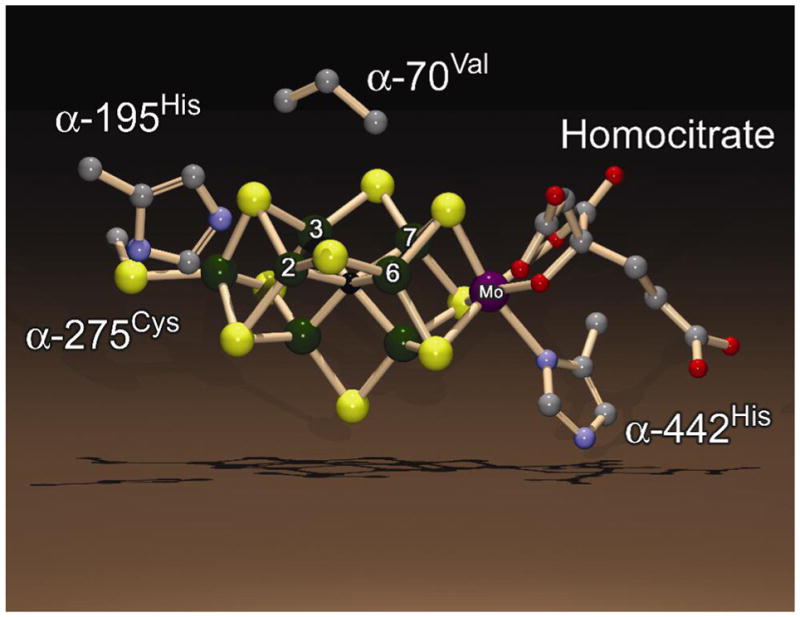
Shown is the structure of the FeMo-cofactor along with the side chains for a few amino acids from the MoFe protein. Fe atoms 2, 3, 6, and 7 are labeled. The color scheme is Fe in green, Mo in purple, C in grey, N in blue, O in red, and S in yellow. The atom at the center of FeMo-cofactor (X) is shown in black. The structure is based on the PDB coordinate file 1M1N and was generated using the programs DS ViewerPro and POV-ray.
Relatively little is known at a molecular level about the nitrogenase N2-reduction mechanism beyond the fact that N2 binds to and is reduced at one or more of the metal atoms of FeMo-cofactor (Figure 1) (9, 10). It is generally accepted that the sequential addition of electrons and protons to the N2 bound on FeMo-cofactor results in a series of semi-reduced and semi-protonated intermediates (11–19), ultimately yielding two ammonia molecules. In contrast to this situation, much more is known about how N2 is activated and reduced by metal complexes (20). Chatt and coworkers (21–23) developed a cycle for the reduction of N2 at a mononuclear Mo complex, with each of the key proposed intermediate states having been isolated and characterized. More recently, Schrock and coworkers (24–26) have demonstrated the catalytic reduction of N2 to two NH3 by a different mononuclear Mo complex, and have been able to capture and characterize many of the intermediate states along the reaction pathway. Both the Chatt and Schrock cycles belong to one fundamental class of potential nitrogenase mechanismsin which the first three ‘H-atoms’ (e−/H+) are sequentially added to a single N atom, in those instances the distal N of an end-on bound N2, followed by cleavage of the N-N bond and release of the first NH3 (this class of mechanisms thus has been termed ‘distal’) (27). The resulting metal (M)-bound nitride (M≡N) is reduced by three additional H-atoms to yield the second NH3. In contrast to this mechanism are potential nitrogenase mechanisms in which the first five H-atoms are added alternately to the two N atoms of N2, followed by cleavage of the N-N bond and release of the first NH3 (27). Reduction of the remaining metal bound amido (-NH2) by a sixth H-atom results in release of the second NH3 (this class of mechanisms has been termed ‘alternating’). Mechanisms of the two classes begin in the same way with N2 binding followed by the addition of two H-atoms, resulting in bound isomers of diazene (M-NH=NH). The mechanisms then diverge, with distal mechanisms having nitride (M≡N) and imido (M=NH) intermediates, while the alternating mechanisms include hydrazido (M-NH=NH2) and hydrazine (M-NH2-NH2) intermediates. The two mechanism classes then converge to an amido (M-NH2) state.
A major goal of nitrogenase research is to devise experiments to characterize the reaction intermediates during N2 reduction and thereby to determine the mechanism and its molecular details. Within the framework of the alternating class of mechanisms, areasonable starting assumption is that the nitrogenase reaction mechanism for N2reduction involves intermediates in which the FeMo-cofactor (denoted as M) binds N2-derived species at the levels of reduction of diazene (N2H2) and hydrazine (N2H4) (eqn. 2).
| (eqn 2) |
This view was supported by the early demonstration that hydrazine (H2N=NH2) is a substrate for nitrogenase, and is reduced by two electrons and two protons to yield two ammonia molecules (28). The assumption is that hydrazine joins into a late step in the same reaction pathway utilized during N2 reduction (11).
Despite these observations, little is known at the molecular level about the nature of N2-reduction intermediates actually bound to nitrogenase. While a FeMo-cofactor bound diazene seems likely as an early intermediate in the pathway, assessment of the interactions of diazene with nitrogenase have proven challenging because of the short half-life of diazene in aqueous solutions. McKenna and coworkers (29, 30) were able to demonstrate that dimethyldiazene (H3C-N=N-CH3) and diazirine (cyclic methyldiazene) are substrates for nitrogenase. More recently it was shown that methyldiazene (HN=N-CH3) could be used as a substrate for nitrogenase (27). An intermediate was trapped during turnover of this substrate, and ENDOR spectroscopy was used to show that it contains a methyldiazene-derived [-NHx] species bound to FeMo-cofactor. While the results obtained so far have been important in understanding early steps in the nitrogenase mechanism, the fact that they are obtained with diazene analogs requiresextrapolation to the N2-reduction mechanism.
Here, we employ the in situ generation of diazene itself to demonstrate that it is a nitrogenase substrate that is reduced to ammonia. Moreover, it is shown that the reduction of both N2 and diazene are inhibited by H2, indicating that diazene enters at an early step in the same reaction pathway. By substitution of amino acids in the MoFe protein near FeMo-cofactor, it has been possible to trap an intermediate in which a diazene-derived species is bound to FeMo-cofactor, and to establish the properties of this bound state by X/Q-band EPR and Q-band 15N-ENDOR spectroscopies. The properties of this diazene-derived state are directly compared to those of a hydrazine-derived state trapped during turnover. The features of diazene reduction by nitrogenase along with the properties of the diazene-derived intermediate provide new insights into the nitrogenase N2 reduction mechanism.
Materials and Methods
Materials and proteins
All reagents were obtained from Sigma-Aldrich Chemicals (St. Louis, MO) and were used as supplied unless stated otherwise. 15N-labeled hydrazine was obtained from Cambridge Isotopes (Andover, MA). Azotobacter vinelandii strains DJ995 (wild-type MoFe protein), DJ1310 (α-70Ala MoFe protein), DJ997 (α-195Gln MoFe protein), and DJ1316 (α-195Gln/α-70Ala MoFe protein) were constructed and nitrogenase proteins were expressed and purified as described previously (31). All proteins used were greater than 95% pure as judged by SDS-PAGE analysis using Coomassie blue staining. Manipulation of proteins was done in septum-sealed serum vials under an argon atmosphere. All transfer of gases and liquids was done using gas-tight syringes.
Azodiformate synthesis
Azodiformate (5 in Figure 2) is the precursor for diazene production. It is prepared from azodicarbonamide (4 in Figure 2) using the procedure of Graham Palmer (32), with slight modifications. Preparation of azodicarbonamide (4) was accomplished after first synthesizing biurea (3 in Figure 2) using the procedure of Audrieth and Mohr (33). The synthesis began with 200 mg of 14 or 15N-labeled hydrazine sulfate (1 in Figure 2) dissolved in a 3 mL solution of 0.5 M acetic acid in water. To this solution was added drop-wise 275 mg of potassium cyanate (2 in Figure 2) dissolved in 0.93 mL of water. The mixture was stirred gently for 4 hours at room temperature and then transferred to a 25 mL centrifuge tube. The biurea (3) product was crystallized by the addition of 2 mL water, and isolated as a white precipitate collected by centrifugation at 5000 × g for 5 minutes. The precipitate was washed twice with absolute ethanol followed by diethyl ether (2 mL each wash) with a yield of 150 mg. After being dried under vacuum for several minutes, the biurea (3) was used in the synthesis of azodicarbonamide (4 in Figure 2). The biurea was oxidized to azodicarbonamide using cupric acetate and nitrate following the procedure of Kepler (34). Biurea (150 mg) was added to a 3.25 mL solution of glacial acetic acid containing 3 mg of cupric acetate. This was followed by the addition of 146 mg of ammonium nitrate (solid). The resulting solution was refluxed at 115°C for 10 minutes and then cooled to 45°C. The azodicarbonamide (4) was isolated as an orange precipitate collected by centrifugation and was washed several times with cold water, yielding 100 mg of product with a calculated yield of ~70%. The azodicarbonamide (4) was dried overnight under vacuum and stored at room temperature. The azodicarbonamide (4) was dissolved in 2 mL of half-saturated KOH solution in H2O (about 2 grams in 4 mL H2O), generating azodiformate (5), which was then precipitated by adding ethanol (~20 mL). This was washed with 50% ether in ethanol, and then dried overnight under vacuum, giving a yield of ~ 90%. Dried azodiformate (5) was stable for many weeks when stored under argon in the dark.
Figure 2. Diazene synthetic scheme.
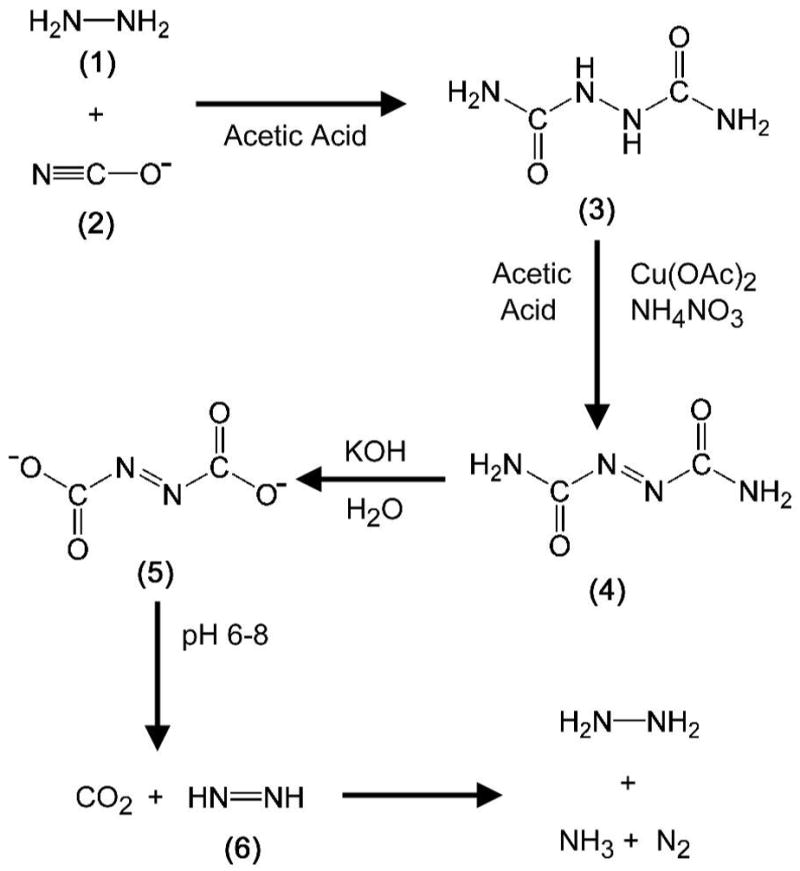
Biurea (3) was synthesized as a white precipitate following reaction of hydrazine (1, where the N atoms were either 14N or 15N) and cyanate (2). Biurea (3) was oxidized by copper acetate yielding the orange precipitate azodicarboamide (4). This was treated with base to form azodiformate (5) immediately prior to use. Neutralization of azodiformate (5) by addition to the buffered assay mixture rapidly yielded diazene (6) and CO2. The breakdown of diazene to hydrazine, N2, and ammonia is also shown. See Material and Methods for details.
Nitrogenase activity assays
Substrate reduction reactions for nitrogenase proteins were conducted at 30°C using a variation of a method described previously (35). The assay solution contained a MgATP regeneration system (5 mM ATP, 6 mM MgCl2, 30 mM phosphocreatine, and 0.2 mg/mL creatine phosphokinase) in MOPS buffer (200 mM, pH 7.2) with 1.2 mg/mL bovine serum albumin and 9 mM sodium dithionite. Solutions were degassed with oxygen-free argon and brought to a final pressure of 1 atm prior to the addition of dithionite. MoFe protein (200 μg) was added followed by Fe protein (500 μg) to initiate the reaction. Reactions proceeded with shaking at 30°C and were then quenched by the addition of 300 μL of 400 mM EDTA. Concentrations of azodiformate were established using the extinction coefficient of 33 M−1 cm−1 at 403 nm(32). A 500 mM azodiformate (5 in Figure 2) stock solution (made in a 150 mM KOH solution in water) was prepared just prior to starting the assay, and 5 – 50 μL of this solution was added to the assay solution immediately following initiation of the reaction by addition of the Fe protein. Upon addition of the azodiformate (5) solution to the assay solution, with the neutralization of the pH, an immediate reaction occurred, yielding the gases diazene and carbon dioxide. The quantity of diazene is presented as equal to the quantity of azodiformate added, although it is clear that the real concentration of diazene will always be lower than this value. Adding the same quantity of a 150 mM KOH solution without azodiformate to an assay had no noticeable affect on the proton reduction activity or pH of the solution.
Where indicated, the gases nitrogen, argon, or acetylene were added as an overpressure, and the vial was then vented to 1 atm final pressure. Hydrogen was quantified by gas chromatography. Ammonia was quantified using a fluorescence detection method (36) with slight modifications. A 25 μL aliquot of post-reaction solution containing NH3 was added to 1 mL of a solution containing 20 mM phthalic dicarboxyaldehyde, 3.5 mM 2-mercaptoethanol, and 5% (v/v) ethanol in a 200 mM potassium phosphate buffer, pH 7.3, and allowed to react in the dark for 30 min. The fluorescence (λexcitation/λemission 410 nm/472 nm) of the mixture was used to quantify ammonia by comparison to a standard made using NH4Cl, using a Shimadzu Model RF-5301 PC spectrofluorometer and the software provided with the instrument.
Hydrazine was quantified using a previously reported colorimetric assay (11). Briefly, 5 μL of sample or standard (up to 5 mM) was added to a disposable 1 mL cuvette. Then 1 mL of the assay solution, containing 70 mM dimethylaminobenzaldehyde and 1 M HCl in 95% ethanol, was added to the cuvette, and allowed to sit for 15 minutes. The absorbance at 458 nm was then used to quantify amounts of hydrazine versus a blank and a standard curve prepared from hydrazine sulfate.
X-band EPR sample preparation and analysis
EPR samples were prepared in a solution containing a MgATP regeneration system (10 mM ATP, 15 mM MgCl2, 20 mM phosphocreatine, and 0.2 mg/mL phosphocreatine kinase) in 150 mM MOPS buffer, pH 7.3, with 50 mM dithionite. MoFe protein was added to a final concentration of ~75 μM. Azodiformate was added from a stock solution (described above). Turnover conditions were initiated by the addition of Fe protein to a final concentration of 50 μM. EPR samples under resting conditions were prepared as described above, except that Fe protein was not included. All X-band EPR samples were frozen in 4 mm calibrated quartz EPR tubes at 77 K. For the pH profile, the same solution as described above was used, except that the buffer was 50 mM MES, 50 mM TAPS and 50 mM MOPS and the pH was adjusted by the addition of HCl or NaOH. X-band EPR spectra were recorded using a Bruker ESP-300 E spectrometer with an ER 4116 dual-mode X-band cavity equipped with an Oxford Instruments ESR-900 helium flow cryostat. Spectra were obtained at a microwave frequency of ~ 9.65 GHz. Precise values of the frequency were recorded for each spectrum to determine proper g alignment. Initial spectra were obtained at a microwave power of 1.0 mW, with a modulation frequency of 1.26 mT, and a temperature of 8 K and were the sum of five scans. Subsequent data manipulation was done using IGOR Pro (WaveMetrics, Lake Osewego, OR). The temperature-dependence (4.8 to 14 K) of the EPR signal intensity was determined at 100 μW for the turnover and resting state signals. The microwave power dependence on EPR signal intensity was determined at 4.8 K with microwave powers ranging from 10 μW to 2 mW.
35 GHz EPR/ENDOR spectroscopy
Q-band samples were prepared as described above, except that the MoFe protein concentration was ~200 μM, and the reactions were initiated by addition of 100 μM of Fe protein. CW and Mims/ReMims pulsed 35 GHz ENDOR spectra were recorded at 2 K on spectrometers described previously (37). The ENDOR pattern for an I = ½ nucleus (1H, 15N) exhibits a ν(±) doublet that is split by the hyperfine coupling, A, and centered at the nuclear Larmor frequency. The Mims pulse sequence, [π/2-τ-π/2-T(rf)- π/2-detect], has the property that its ENDOR intensities follow the relationship, I(A) ~ 1 - cos(2πAτ) (38). As a result, the signals vanish (‘blind spots’) at Aτ= n, n = 0, 1, …, and show maximum intensities at Aτ= n+1/2. The reMims sequence [π/2-τ1-π/2-T(rf)- π/2-τ2-π-detect] is used to overcome spectrometer dead-time limitations (39), and to place all ‘blind spots’ with n > 0 at frequencies that are outside the ENDOR envelope. The full hyperfine tensor for an interacting nucleus is determined by analysis of a 2D field-frequency pattern comprised of numerous spectra collected across the EPR envelope, as described (40).
Results
Diazene is a substrate for nitrogenase
Despite the obvious importance of studying the reactions of diazene (6 in Figure 2) with nitrogenase, the short half-life of diazene in aqueous solutions (41) has made such studies difficult. Diazene undergoes both dismutation (eqn 3) and decomposition (eqn 4) reactions near neutral pH, with an estimated half-life at pH 8.0 of 5 s (32, 41).
| (eqn 3) |
| (eqn 4) |
Despite this short half-life, however, it was shown that diazene can be generated more rapidly than it decays, thus allowing a steady state concentration of diazene to accumulate in solution for short times (< 30 sec) (32). This is achieved by rapidly generating diazene by neutralization of azodiformate (5 in Figure 2). Azodiformate (5), which is synthesized from azodicarbonamide (4 in Figure 2) in KOH, is stable at room temperature for several hours when maintained at high pH. Upon neutralization of azodiformate (5) by adding a small quantity to an enzyme solution buffered near neutral pH, diazene and CO2 are rapidly liberated. This strategy was successfully employed to investigate the interaction of diazene with cytochrome c oxidase (32).
Here, we have similarly generated diazene in solution with nitrogenase under turnover conditions as a way to probe diazene as a possible substrate, inhibitor, and ligand. An important issue was to distinguish diazene interactions with nitrogenase from the possible interactions of diazene decay products (e.g., N2 and hydrazine). When nitrogenase is allowed to turn over (MoFe and Fe proteins, reductant, MgATP, and MgATP regeneration system) for five minutes in the presence of different concentrations of diazene (added as azodiformate1), total ammonia production is seen to increase with increasing diazene concentration, with saturation observed at the highest concentrations (Figure 3A, upper curve). It was important to determine how much of this ammonia production might be coming from nitrogenase reduction of diazene and how much was coming from other sources, such as the decomposition of diazene that is not catalyzed by nitrogenase. The latter contribution to the ammonia detected was established by repeating the turnover experiment except that the nitrogenase reaction quencher EDTA had been added to the assay at the beginning of the reaction before azodiformate was added. As can be seen in Figure 3A (lower curve), ammonia was detected under these conditions arising from non-enzymatic decay of diazene, but the quantity was much lower than that observed from the enzyme-catalyzed reaction.
Figure 3. Diazene is a substrate for nitrogenase.

(Panel A) Total ammonia detected following 5 minutes of reaction with 200 μg (800 nM) of wild-type MoFe protein and 500 μg (8 μM) of Fe protein in a total assay solution volume of 1 mL is shown as a function of the concentration of azodiformate added (△). Also shown is the total ammonia detected for a similar assay condition except that EDTA was added before initiation of the reaction (○), and for an assay where azodiformate was added 30 minutes prior to the initiation of the enzyme reaction by addition of the Fe protein (●). (Panel B) The total ammonia detected minus the ammonia assigned to non-nitrogenase catalyzed diazene breakdown and from non-diazene sources is plotted against the concentration of azodiformate added (▲). The points up to 15 mM azodiformate are fitted to the Michalis-Menten equation (line).
The other potential source of ammonia in these assays would be nitrogenase reduction of diazene breakdown products (e.g., N2 and hydrazine). To establish the maximum contribution to ammonia production from nitrogenase reduction of diazene breakdown products, an assay was conducted where azodiformate was added 30 min prior to the initiation of the enzyme catalyzed reaction. In this case, the diazene would fully decompose prior to initiation of the assay (32, 41). Nitrogenase added after the 30 min pre-incubation was then allowed to react for 5 min prior to quenching with EDTA (Figure 3, middle curve). The ammonia produced under these conditions results from the breakdown of diazene directly to ammonia and from nitrogenase reduction of diazene breakdown products. This ammonia production represents the upper limit to the amount of ammonia produced during normal diazene turnover that does not come from nitrogenase reduction of diazene itself.
The difference between the ammonia production in the presence of diazene and that ascribed to non-diazene and non-nitrogenase reactions represents the ammonia produced from nitrogenase reduction of diazene. This corrected ammonia production from nitrogenase reduction of diazene is plotted as a function of diazene (azodiformate) concentration (Figure 3B). It is evident that nitrogenase reduces diazene to ammonia in a concentration-dependent reaction, with saturation at approximately 12 mM diazene (added azodiformate). It is noted that at still higher concentrations of diazene, inhibition of diazene reduction to ammonia is observed. This inhibition could reflect competition for the nitrogenase active site from N2 and hydrazine (breakdown products of diazene) or from substrate inhibition. For the lower concentrations of diazene, the data were fit to the Michaelis-Menten equation to obtain estimates of kinetic parameters for nitrogenase reduction of diazene. While it is not possible to establish an accurate Vmax and Km for diazene because the substrate concentration is changing during the course of the assay, it is possible to obtain limits. From the results in Figure 3B, a minimum Vmax for diazene reduction by nitrogenase is found to be 400 nmol NH3/min/mg MoFe protein. This value compares favorably with the Vmax for N2 reduction by nitrogenase determined in a parallel experiment of 600 nmol NH3/min/mg MoFe protein (35). Likewise, an upper limit on the Km for diazene is calculated to be 4.5 mM, compared to the Km for N2 of 60 μM (35).
Hydrogen inhibits diazene reduction by nitrogenase
An important mechanistic aspect of N2 reduction by nitrogenase is that this reaction is inhibited by H2 (1, 10, 42). Several proposals have been put forward to explain this inhibition, with all sharing the assumption that H2 and N2 interact with the same or similar sites and reduction states of FeMo-cofactor (1, 10, 43). Consistent with this proposal is the observation that H2 does not inhibit hydrazine reduction, which is presumed to occur later in the N2 reduction pathway. Likewise, H2 does not inhibit reduction of non-physiological substrates such as acetylene (results for acetylene and hydrazine are shown in Table 1).
Table 1.
Inhibition of Substrate Reduction by H2
| Additionsa | Specific Activity (nmol NH3 or C2H4/min/mg)b | Percent Inhibition of Maximal Activity |
|---|---|---|
| 0.11 atm N2c | 172 ± 15 | |
| 0.11 atm N2, 0.89 atm H2 | 63 ± 4 | 63 |
| 4.6 μazodiformated | 373 ± 36 | |
| 4.6 μmol azodiformate, 1 atm H2 | 189 ± 7 | 58d |
| 10 mM hydrazinee | 540 ± 10 | |
| 10 mM hydrazine, 1 atm H2c | 530 ± 5 | 2 |
| 0.01 atm acetylenec | 770 ± 16 | |
| 0.01 atm acetylene, 1 atm H2 | 755 ± 17 | 2 |
Gases are added as a partial pressure with argon added to achieve 1 atm total pressure.
Ammonia and ethylene were quantified as described in the Materials and Methods ection.
Nitrogen and acetylene reduction assays were performed at pH 7.0 for 10 minutes using 100 μg of MoFe protein.
Diazene reduction assays were performed at pH 7.0 for 5 minutes using 200 μg of MoFe protein.
Hydrazine reduction assays were performed at pH 7.2 for 10 minutes using 200 μg of the α-70Ala MoFe protein.
When H2 was included in a diazene reduction assay, significant inhibition of ammonia production (corrected to ammonia production from diazene reduction by nitrogenase) was observed (Table 1). The extent of H2 inhibition of diazene reduction was similar to that seen for H2 inhibition of N2 reduction under these conditions. Figure 4 shows the effects of increasing H2 partial pressure on nitrogenase-catalyzed ammonia production from either N2 or diazene. It can be seen that H2 inhibition of diazene reduction parallels the H2 inhibition of N2 reduction. Thus, N2 and diazene behave alike, and differently from hydrazine or acetylene, in that their reduction by nitrogenase is inhibited by H2.
Figure 4. Hydrogen inhibition of diazene and nitrogen reduction.
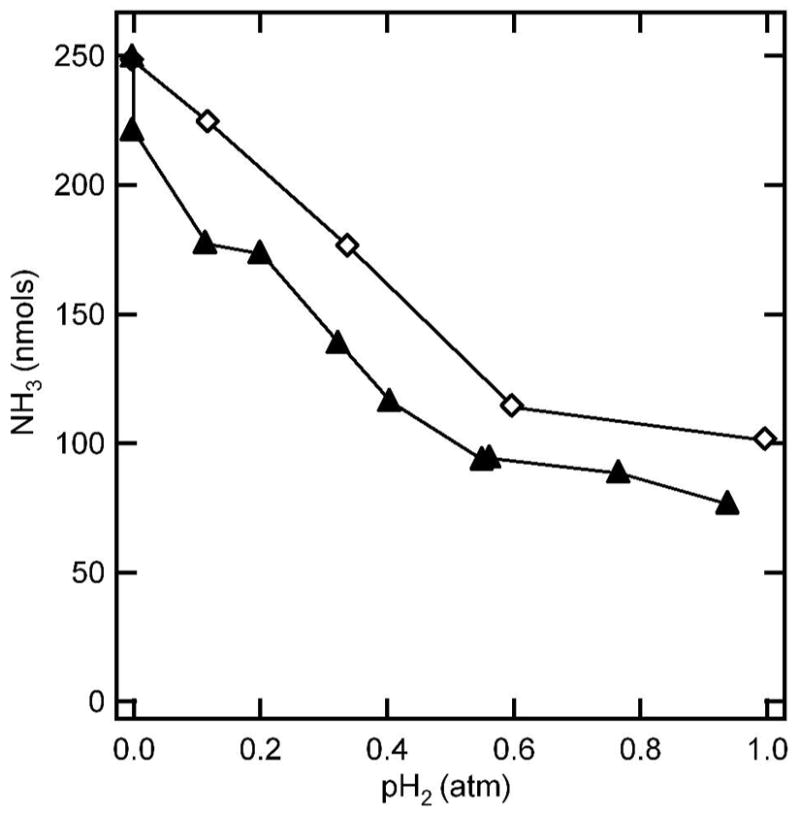
The quantity of amnmonia detected after 5 min of turnover under N2 (▲) or diazene (◇) is shown as a function of the partial pressure of H2 (pH2) with Ar as the supplementary gas. Assay conditions were as described in the legend to Figure 3.
Diazene inhibits proton reduction
Like any other substrate for nitrogenase, diazene should compete for electron flux through nitrogenase (1). Thus, it is expected that in the presence of diazene, the rate of H2 formation by nitrogenase should decrease as electrons are diverted to the competing substrate. Figure 5 shows the effects of increasing diazene concentration on the H2 evolution rate catalyzed by nitrogenase. The open symbols represent the H2 production rates observed when diazene is allowed to decompose prior to the initiation of the assay, and thus accounts for all non-diazene inhibitors and substrates. The lower (closed) data represent the H2 reduction rates when diazene is present. From the difference between these curves, it is apparent that diazene inhibits H2 evolution by nitrogenase, indicating competition for electron flux through nitrogenase.
Figure 5. Diazene inhibition of proton reduction.
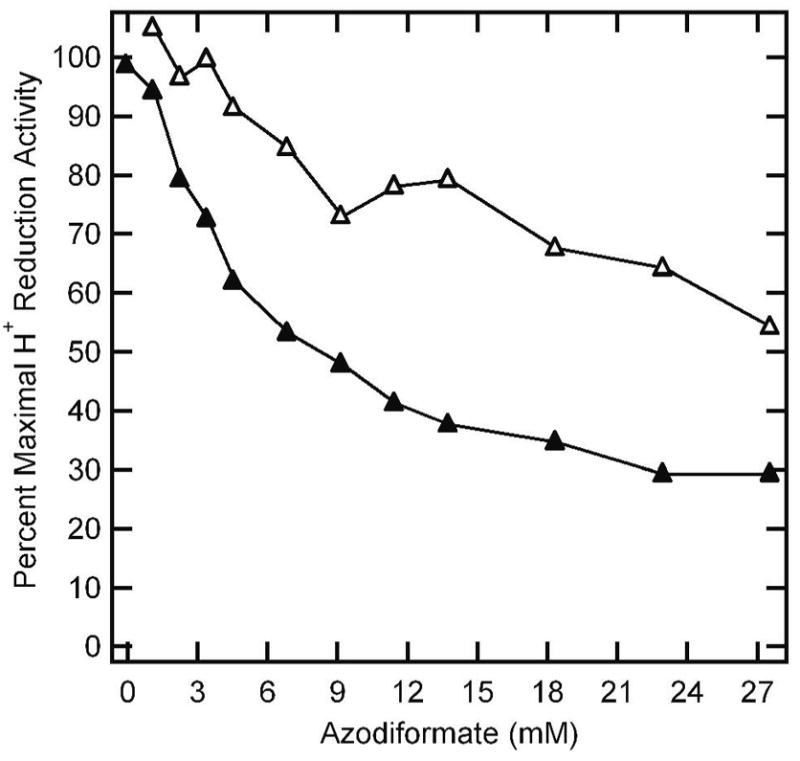
The percentage of the maximum proton reduction activity of wild-type MoFe protein is plotted against the concentration of azodiformate added for a reaction where azodiformate is added upon initiation of the reaction (▲) and when the reaction is initiated 30 minutes after addition of azodiformate (△). Assay conditions were as described in the legend to Figure 3.
Trapping a diazene-derived species bound to FeMo-cofactor
Given that diazene is a substrate for nitrogenase, we attempted to trap an intermediate with a diazene-derived species bound to FeMo-cofactor, as this might represent an early state along the reaction pathway. The trapping of substrate-derived species on FeMo-cofactor can be monitored by the change in the EPR spectrum of the FeMo-cofactor (27, 36, 44–49). In its resting state (called MN), FeMo-cofactor is in an S = 3/2 spin state with a characteristic EPR spectrum in the perpendicular mode with g values of 4.45, 3.56, and 2.00 (Figure 6). When nitrogenase is freeze-trapped under proton reduction conditions, this EPR signal is greatly diminish in intensity, which has been interpreted as the conversion of FeMo-cofactor to a more reduced, diamagnetic state (called MR) (50, 51). When the wild-type MoFe protein is freeze-trapped during turnover with diazene, the S = 3/2 resting-state FeMo-cofactor EPR signal partially converts to a new state with an S = 1/2 EPR signal (Figure 6). However, based on its g values, this appears to be from an N2-turnover state, probably formed with N2 generated as a breakdown product of diazene (49). In contrast, conversion to an intermediate with a distinct EPR signal is observed when the α-195Gln MoFe protein is trapped during turnover with diazene; the substitution of α-195His by Gln has been suggested to limit proton delivery for reduction of nitrogenous substrates, and thus to arrest the reduction of these substrates (36, 52, 53). The further substitution of α-70Val by Ala in the α-195Gln MoFe protein (double substituted MoFe protein) leads to a more complete conversion of the resting-state FeMo-cofactor EPR signal to the new S = ½ state. Substitution of α-70Val by Ala has been shown to open up the nitrogenase active site, allowing larger substrates to interact (54).
Figure 6. EPR spectra for various MoFe proteins trapped with diazene.
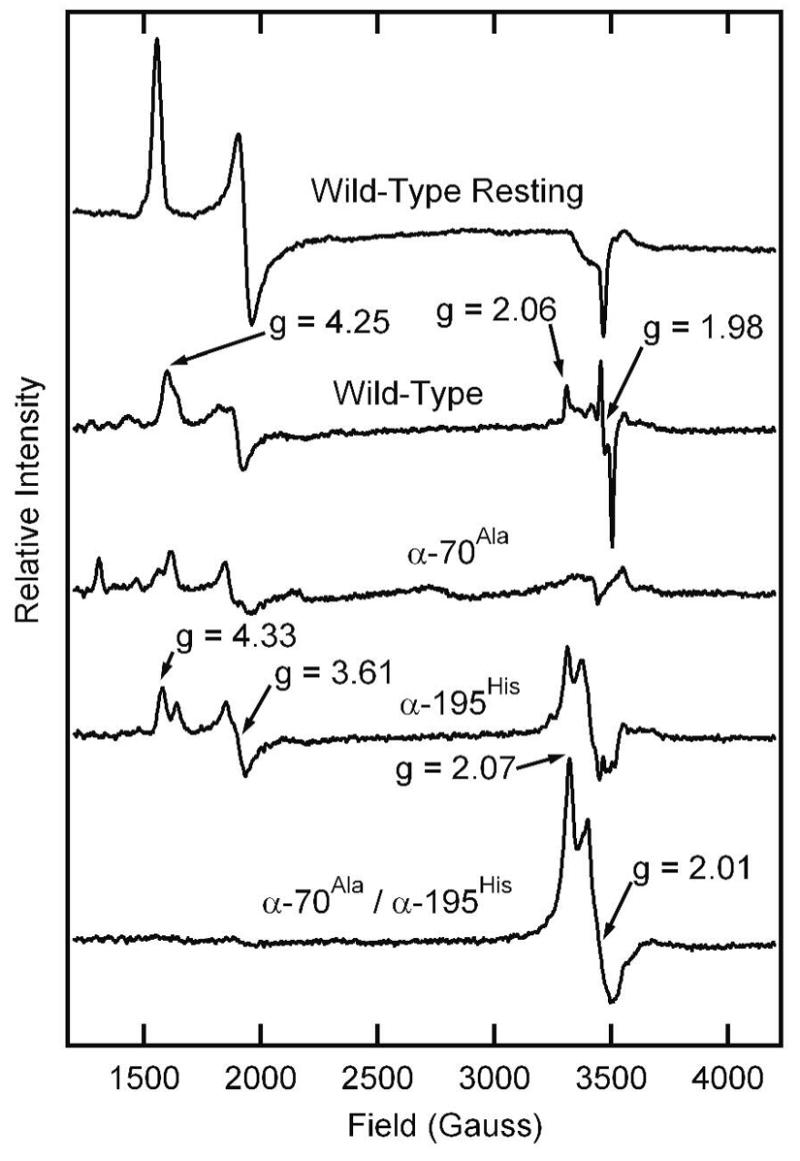
Shown are X-band EPR spectra for the wild-type MoFe protein in the resting state (top trace) and other MoFe proteins (15 μM) trapped by freezing during turnover in the presence of 5 μmol of azodiformate in a total liquid volume of 400 μL at pH 6.5. Turnover and EPR conditions are described in the Materials and Methods section.
It was important to establish that the trapped state observed by EPR is a result of diazene binding to or reacting with nitrogenase, and not one of the diazene breakdown products. In an experiment parallel to the a kinetic studies described above, diazene was allowed to decompose for 30 min in an EPR tube prior to the addition of the nitrogenase proteins. Following the addition of nitrogenase proteins, the reaction was allowed to react for 30 sec before being frozen in liquid nitrogen. The EPR spectrum of this sample was compared to the spectrum obtained when nitrogenase proteins were added to the reaction solution immediately after generating the diazene (Figure 7). It is evident that most of the new S = ½ EPR spectrum observed when nitrogenase is trapped during turnover with diazene results from nitrogenase interaction with diazene rather than with a diazene breakdown product.
Figure 7. EPR spectra for α-70Ala/α-195Gln MoFe protein trapped with diazene or diazene decomposition products.
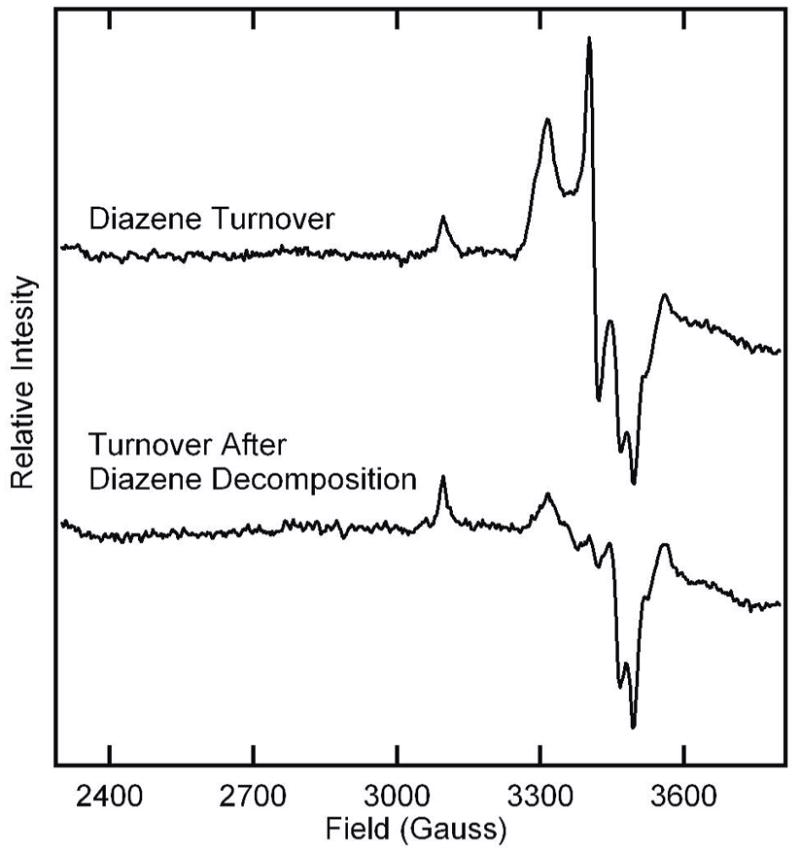
Shown are X-band EPR spectra for the α-70Ala/α-195Gln MoFe protein trapped during turnover with 4 mM azodiformate (upper trace) or trapped during turnover with 4 mM azodiformate that was allowed to decompose for 30 min prior to the initiation of the reaction (lower trace). Turnover and EPR condition are described in the Materials and Methods section.
The X-band EPR signal of the diazene-trapped state of α-195His/α-70Val MoFe protein appears axial. To increase the g-resolution for comparison between the diazene-and hydrazine-trapped states, EPR spectra were collected at Q-band. Figure 8 presents a Q-band absorption-display EPR spectrum of the diazene-trapped state and its derivative, each overlaid with the corresponding EPR spectra of the hydrazine-trapped state in the same α-70Ala/α-195Gln MoFe protein. Overall, the spectra of the inter mediates are highly similar, and again appear to be axial, with g⊥ = 2.01 and g|| ~ 2.1. However, there is interference in the low-g region from the Fe protein EPR signal at g = 1.93, and the collection of ENDOR spectrum over a range of fields (not shown) suggests that the spectra of both intermediates in fact have a rhombic splitting, with g2 = 2.01 and g3 ≤ 1.98. The shapes of the g1 ~ 2.1 features of the spectra further suggest that both intermediates are heterogeneous. Spectra taken over a range of incident microwave powers (see Supporting Information, Figure S1) confirm that the signal of each intermediate is a superposition of spectra with g|| = 2.11 and g|| = 2.085, presumably from two major substates with slightly different properties; the g|| = 2.085 conformation dominates in the diazene-derived intermediate whereas the two have comparable contributions in the hydrazine-derived intermediate. Quantification of the diazene-dependent EPR signal by comparison to a CuEDTA standard solution indicates that this signal represents ~ 60% spin conversion of FeMo-cofactor.
Figure 8. Q-band EPR spectra of diazene- and hydrazine-dependent intermediates.
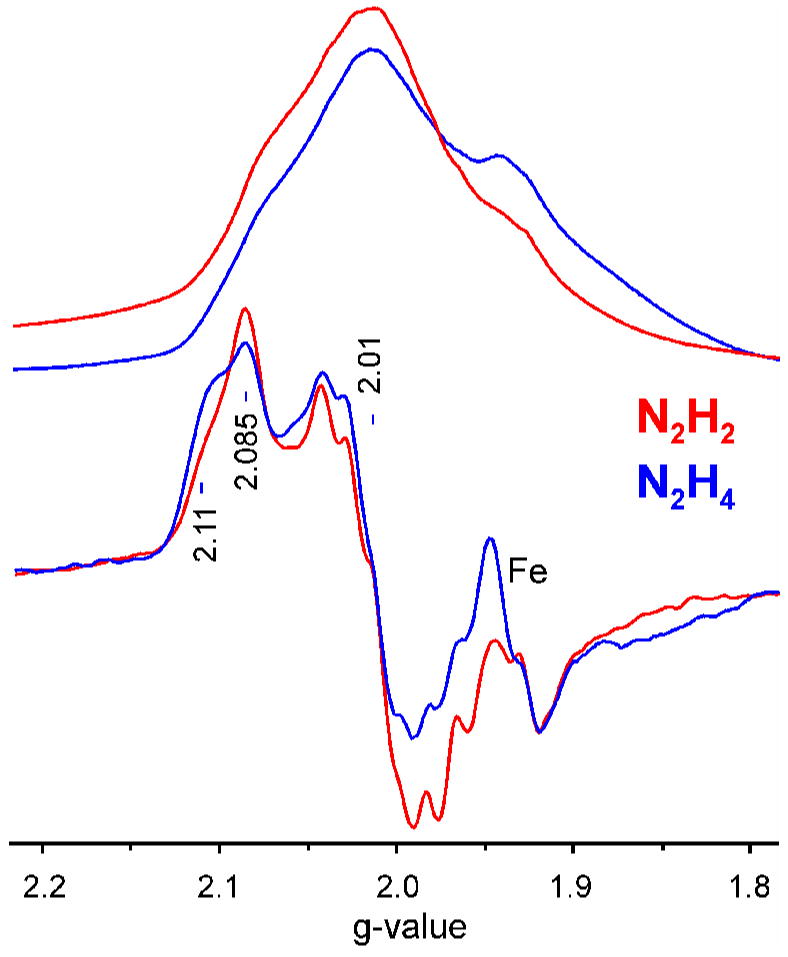
(Upper traces) Absorption-display rapid-passage spectra. (Lower traces) digital derivatives. These enhance the tiny signal from adventitious Mn2+. Conditions: Microwave frequency, 35.025 – 35.041 GHz; microwave power, ~ 1 mW; modulation amplitude, 1.3G; sweep rate, 33G/sec; time constant, 128 ms; temperature, 2K.
The pH dependence of the intensities of the EPR signals of the diazene- and hydrazine-derived intermediates were determined (see Supporting Information, Figure S2). The EPR signal of the diazene-trapped state was most intense at the lowest pH values and declined in intensity as the pH rose. For the hydrazine-trapped state, the intensity of the EPR signal rose to a maximum at pH 7.5 and then declined in intensity at higher pH values. The microwave power dependence on the diazene- and hydrazine-derived EPR signal intensities was also determined, and found to be nearly identical (see Supporting Information, Figure S3).
15N, 1H-ENDOR
To establish if the diazene-trapped state contained a diazene-derived species bound to FeMo-cofactor, Q-band 15N pulsed Mims ENDOR was used on nitrogenase with 15N-labeled diazene trapped (Figure 9A). 15N2H2 was prepared by first synthesizing biurea (3 in Figure 2) from 15N-hydrazine (1 in Figure 2). The biurea (3) was oxidized to azocarboamide (4), which in turn could be readily converted to azodiformate (5). The 15N-azodiformate was used to generate 15N-diazene as outlined above, and the α-70Ala/α-195Gln MoFe protein was freeze-trapped during turnover with this substrate. To test whether this state is the same as the hydrazine-derived state, the α-195His/α-70Val MoFe protein also was trapped with 14,15N2H4
Figure 9. ENDOR spectra.
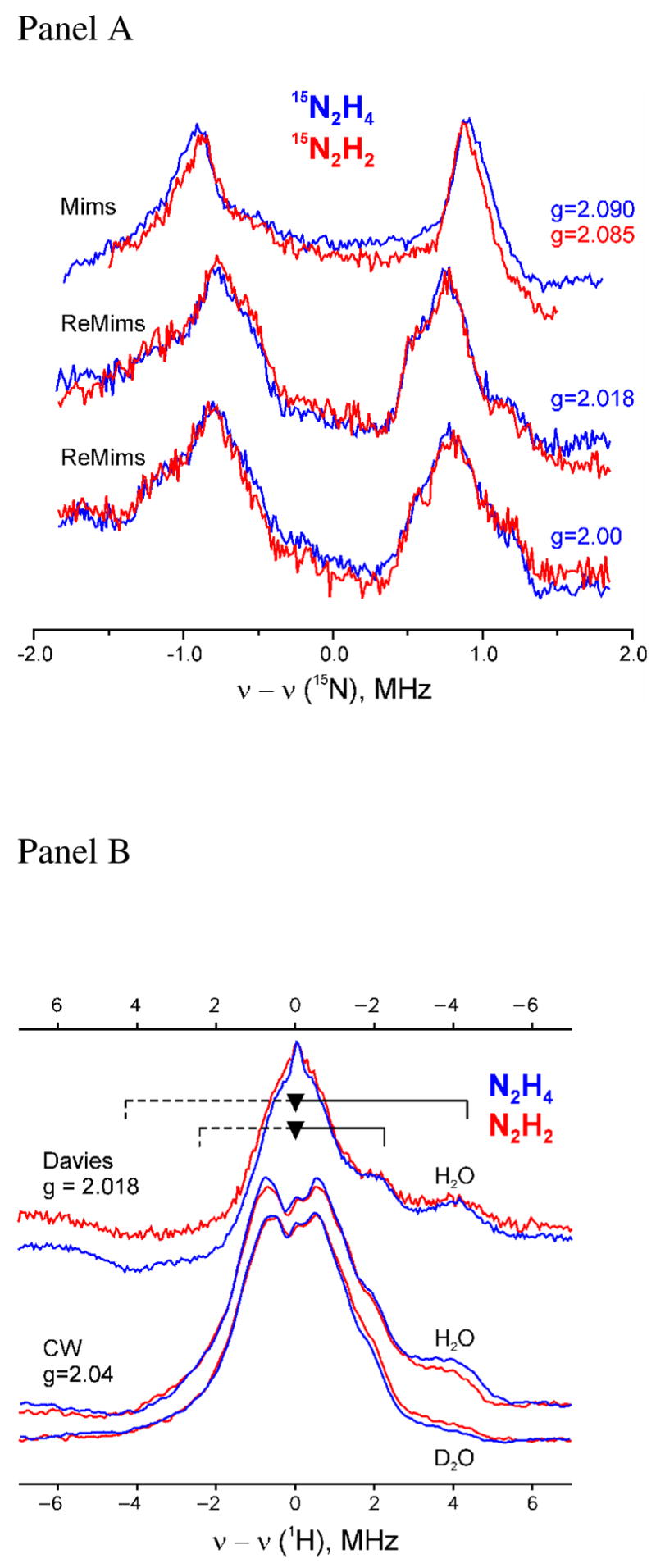
(Panel A) Comparison of pulsed 15N-ENDOR spectra for α-70Ala/α-195Gln MoFe protein trapped during turnover with diazene (red) or hydrazine (blue). Conditions: Mims sequence; π/2 = 52ns, τ= 300ns, RF 20μs, 50shots/point, 10scans, 2K, (N2H4) g = 2.09, 34.776 GHz, 20ms repetition rate, and (N2H2) g = 2.085−34.702 GHz, 10ms repetition rate. ReMims sequence; π/2 = 32ns, tau;1= 224ns, RF 20μs, 50 shots/point, 10ms repetition rate, 2K, (N2H4) 34.776 GHz, 20 scans, and (N2H2) 34.768 GHz, 40 scans. (Panel B) Comparison of pulsed and CW 1H ENDOR spectra for α-70Ala/α-195Gln MoFe protein trapped during turnover with diazene (red) or hydrazine (blue) in H2O and D2O buffers. Resolved hyperfine couplings to exchangeable (8.5 MHz) and non-exchangeable (4.5 MHz) proton(s) are indicated by braces; the asymmetry of the spectra reflect relaxation effects. Conditions: Davies: g = 2.018, π/2 =40ns, τ = 560ns, RF 40μs, 50 shots/point, 10ms repetition rate, (N2H4) 34.712GHz, 18 scans and (N2H2) 34.730GHz, 30 scans; CW ENDOR: 35.071 – 35.129GHz, modulation amplitude = 1.3 G, time constant = 64ms, RF sweep speed = 1 MHz/sec, bandwidth of RF broadened to 100kHz, 2K.
Selected 15N-ENDOR spectra for the diazene-dependent state and for the hydrazine-dependent state are shown at fields near to the common principal g-values (Figure 9A). Spectra collected over a wider frequency range (not shown) revealed no additional signals from 15N with larger couplings for either sample; neither were additional signals detected with smaller hyperfine couplings through lengthening of the interpulse spacing to τ = 800 ns, which accentuates smaller couplings (see Materials and Methods).
The single-crystal-like spectra for the two states, collected at a field corresponding to g|| = 2.09, show a doublet from a single (type of) 15N, centered at the 15N Larmor frequency and split by the 15N hyperfine coupling, A = 1.80(4) MHz. The individual peaks broaden and show additional resolved features as the field is increased, a consequence of an anisotropic contribution to the hyperfine tensor. As the spectra for the two states are indistinguishable at all fields, we conclude that the hyperfine tensors of the 15N that give rise to the signals are identical for the substrate-derived species bound to FeMo-cofactor in the two intermediates.
A combination of 15N and 1H-ENDOR measurements showed that the hydrazine-derived species binds to FeMo-cofactor through an [-NHx] moiety (36, 49). To complete the comparison between the diazene and hydrazine-derived states, each was prepared in H2O or D2O solvents and CW and Davies pulsed 1H ENDOR spectra were collected at g = 2.04 and g2 = 2.018, respectively (Figure 9B). These were collected under conditions yielding higher resolution than the original CW spectra for hydrazine. The spectra for both states reveal an unresolved, non-exchangeable (unchanged in D2O buffer) matrix 1H peak at the proton Larmor frequency, as well as resolved shoulders from non-exchangeable proton(s) with coupling, A ~ 4.5 MHz. In addition, the spectra showed thesignal seen earlier from an exchangeable proton(s) with A(g1) ~ 8.5 MHz. As discussed, this coupling is most plausibly assigned to an [-NHx] moiety bound directly to the FeMo-cofactor.
Discussion
The rapid in situ generation of diazene directly in a nitrogenase reaction solution permitted an assessment of the interaction of this unstable compound with nitrogenase. Using this strategy, it was possible to demonstrate that diazene is a substrate for nitrogenase, being reduced to ammonia. Given the instability of diazene in solution (32, 41), it was essential to establish that diazene was the principal substrate rather than a breakdown product. In fact, a fraction of the total ammonia observed in a nitrogenase turnover reaction in the presence of diazene can be ascribed to non-enzymatic diazene decomposition to ammonia in addition to nitrogenase reduction of diazene decomposition products (N2 and hydrazine). However, the significant additional ammonia production can only be assigned to nitrogenase reduction of diazene to ammonia. Importantly, N2 as a significant source for this ammonia production is ruled out because N2 was quantified in the assay vials and the concentration was far too low to account for any substantial ammonia production. Likewise, it was possible to rule out the possibility that ammonia was produced by nitrogenase reduction of hydrazine as a diazene break-down product. Again, hydrazine concentrations were determined in the diazene assay and were found to be too low to account for the ammonia production by nitrogenase. Further, hydrazine reduction is not inhibited by H2, whereas the ammonia production in the diazene assay was inhibited by H2. These results provide strong evidence that diazene itself is a substrate for nitrogenase.
Mechanistic implications of diazene as a substrate
The simplest way to explain the behavior of diazene as a nitrogenase substrate is that diazene enters the normal N2 reduction reaction pathway at a step associated with a semi-reduced diazene-species bound to FeMo-cofactor (Eq 2). Several observations presented here support this model. First is the observation that nitrogenase reduces diazene to ammonia at rates that are comparable to the rate of N2 reduction itself. The estimated specific activity found here for diazene reduction by nitrogenase of 400 nmol NH3/min/mg MoFe protein is comparable to the specific activity of 600 nmol NH3/min/mg MoFe protein observed for N2 reduction (35). Importantly, this value for diazene reduction represents a lower limit, as the actual concentration of diazene in solution is lower than the azodiformate concentration because of loss through decomposition. Thus, the true specific activity for diazene reduction will be higher than 400 nmol NH3/min/mg MoFe protein. The observed specific activity for diazene reduction is further minimized by the competition for binding and electrons coming from the diazene breakdown products hydrazine and N2. Likewise, the value, Km ~ 4.5 mM, estimated for diazene clearly is an upper limit, with the true value being lower for the same reasons put forward above.
A second important observation that implies that diazene enters the normal N2 reduction pathway at an early step is the inhibition of diazene reduction by H2. Proton reduction yielding H2 (called H2 evolution) is an essential reaction in the nitrogenase mechanism (43). H2 is a competitive inhibitor of N2 reduction by nitrogenase, but does not inhibit the reduction of any other substrate (except N2O) (55). This inhibition of N2 reduction by H2 indicates a unique relationship between H2 and N2. The special role of H2 in the nitrogenase mechanism is further reflected by the fact that in the absence of another substrate, nitrogenase directs all electron flux to the reduction of protons, yielding H2. The addition of N2 to the reaction competes for electron flux going to proton reduction, thus dramatically lowering H2 evolution rates. However, H2 evolution by nitrogenase cannot be eliminated by increasing amounts of N2, with a minimum stoichiometry of 1 H2 evolved to 1 N2 reduced (eqn 1), even at high N2 concentrations (56). This has been interpreted to indicate that when N2 binds to FeMo-cofactor it must displace a bound H2, thus accounting for the fixed stoichiometry (43). Further connection between the interactions of N2 and H2 with nitrogenase comes from the so-called “HD exchange reaction”. Nitrogenase will catalyze the formation of HD when presented with D2, but only if N2 is also present (57).
Different models haven been put forward (1) to explain these intricate interactions between N2 and H2 and they share the common feature that H2 and N2 interact with FeMo-cofactor at early steps along the N2 reaction pathway (eqn 2). The fact that H2 does not inhibit the reduction of hydrazine by nitrogenase is consistent with such models if hydrazine is presumed to enter the nitrogenase N2-reduction reaction pathway at a late stage (Eq 2). An alternative explanation would be that diazene and N2 are reduced at the same specific site on FeMo-cofactor, whereas the reduction of other substrates (e.g., hydrazine and acetylene) might occur at a different specific site (e.g., a different metal) on FeMo-cofactor (19, 58). However, taken together, the present findings that diazene is reduced to ammonia by nitrogenase and that both diazene and N2 reduction are inhibited by H2 indicate that diazene and N2 are reduced at the same location and that diazene enters the normal nitrogenase reaction pathway at an early step (Eq 2).
Trapping a diazene-derived state
To gain insights into the nature of diazene reduction by nitrogenase, we sought to capture and characterize a state with a diazene-derived species bound to FeMo-cofactor. Our earlier work localized a single Fe-S face in the middle of FeMo-cofactor (Fe atoms 2, 3, 6, and 7) as a site for interaction with both alkyne and nitrogenous substrates (Figure 1) (10, 36, 44, 59). Two observations from those studies are important to the present effort. First, the size of substrates gaining access to FeMo-cofactor can be controlled by the size of the side-chain of the MoFe protein α-70 amino acid residue (54). That the wild-type Val at this position significantly limits larger substrates access to the active site is shown by the result that substitution of α-70Val by the smaller side chain amino acid, Ala, allows substantially larger molecules to act as substrates for nitrogenase. Thus, propyne and hydrazine become much better substrates in the α-70Ala MoFe protein when compared to the wild-type (α-70Val) MoFe protein (35, 54). A second important observation relevant to trapping a diazene-derived species on FeMo-cofactor is the critical role of α-195His (located near the same FeS face, Figure 1) in delivery of protons during the reduction of nitrogenous substrates. This is indicated by the observation that substitution of this residue by glutamine significantly lowers N2- (52) and hydrazine- (36) reduction rates, while essentially leaving proton and acetylene reduction rates unchanged. Further, it was also shown that hydrazine and methyldiazene reduction is interrupted in the α-195Gln MoFe protein, allowing an intermediate state to be trapped by rapidly freezing this MoFe protein variant during turnover with these substrates (27, 49).
When the α-195Gln MoFe protein is rapidly frozen during turnover using diazene as substrate, a partial conversion of the resting state FeMo-cofactor S = 3/2 EPR signal to a new S = ½ spin state signal is observed. Prompted by the earlier findings with the α-70Ala MoFe protein, we then used the doubly substituted MoFe protein (α-70Ala/α-195Gln), and found that it was possible to freeze-trap a state that contains a diazene-derived species bound to FeMo-cofactor at relatively high concentration.
The EPR spectrum of this intermediate shares many similarities with EPR spectra observed for the hydrazine-trapped state: similar g-values, and dependences of EPR signal intensity on microwave power and temperature (36). Overlaying the X- and Q-band EPR spectra for the hydrazine- and diazene-dependent intermediates suggests that they represent the same intermediate, with slightly different populations of two conformational substates. More definitively, 15N and 1H-ENDOR spectra of the diazene-and hydrazine-dependent states show that they contain substrate-derived [-NHx] species bound to FeMo-cofactor whose characteristics are identical. Each shows a 15N-ENDOR signal from a single (type of) 15N and the spectra match precisely (Figure 9A). Likewise the 1H ENDOR spectra match precisely (Figure 9B), with each showing a resolved non-exchangeable signal with A(g1) ~ 4 MHz, and an exchangeable signal with A(g1) ~ 8–9 MHz.
The equivalence of the EPR and 15N, 1H-ENDOR spectra for the diazene- and hydrazine-dependent states can be explained in three different ways: (i) Diazene could first decompose to hydrazine, and then the resulting hydrazine could react with the MoFe protein, being trapped in the same state as accumulates when hydrazine is used as a substrate; (ii) diazene could be reduced by nitrogenase to the same intermediate state that is trapped when hydrazine is presented as the substrate; and (iii) the diazene- and hydrazine-dependent states could represent different species bound to FeMo-cofactor, but with such similar properties for the bound [-NHx] fragment that these differences are not reflected in the EPR or 15N-ENDOR spectra.
Possibility (i) is ruled out because the majority of the EPR signal observed for nitrogenase trapped during turnover with diazene can be assigned to diazene rather than to a diazene breakdown product (Figure 7). If alternative (ii) applies, it would mean that the diazene-dependent intermediate is generated by enzymatic reduction of diazene, not, for example, merely by binding of diazene in a possibly non-productive fashion. In other words, the enzyme has reduced the diazene substrate enough that it has ‘caught up’ to the hydrazine-derived species trapped during turnover with hydrazine. As hydrazine, and now diazene, are bona fide substrates that are reduced to NH 3, this would then mean that both of these substrates in fact access the same, productive, mechanistic pathway to NH3that is used by N2, thereby validating the initial hypothesis about reaction intermediates (eqn 2), which implies that nitrogenase functions through an alternating mechanism, rather than a distal mechanism (27).
We consider (iii) to be possible only if the bound [–NHx,] has the same value of xfor both intermediates. If x were different for the two intermediates, then the bound N would be hybridized differently: for example, sp2 if diazene itself were bound in the diazene-dependent intermediate; sp3 if hydrazine were bound in the hydrazine intermediate. This would mean the nitrogen orbitals involved in binding to M have different orbital compositions in the two states (1/3 2s for sp2, ¼ 2s for sp3). However, this would be expected to lead to different hyperfine couplings, contrary to the results presented here. While further experiments are required to definitively distinguish between alternatives (ii) and (iii), possibility (ii) seems the most likely explanation.
In summary, through in situ generation of diazene we have demonstrated that wild-type nitrogenase reduces diazene to ammonia. The reductions of both N2 and diazene are inhibited by H2, indicating that N2 and diazene are reduced at the same specific site on FeMo-cofactor and that diazene enters at an early step in the same reaction pathway. An EPR-active state has been trapped during turnover of an α-70Ala/α-195Gln MoFe protein variant with diazene and 15N, 1H-ENDOR have established that this state incorporates an [-NHx] moiety bound to FeMo-cofactor that is indistinguishable from the [-NHx] moiety bound to FeMo-cofactor in the hydrazine-state trapped during turnover with hydrazine. An important consequence of the studies presented here is that diazene is highly likely to join the normal N2-reduction pathway, and thus the diazene-and hydrazine-trapped states are likely to represent intermediates in the normal reduction of N2, as postulated by alternating mechanisms for reduction of N2 by nitrogenase. Further characterization of these two trapped states is expected to provide insights into their nature and thus to shed further light on the nitrogenase N2-reduction mechanism.
Supplementary Material
Additional figures illustrating the microwave power dependence of the X- and Q-band EPR spectra and the pH dependence of the EPR spectra are available. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Dr. Graham Palmer for guidance in the in situ synthesis of diazene.
Abbreviations
- FeMo-cofactor
Iron-molybdenum cofactor
- EPR
Electron paramagnetic resonance
- ENDOR
Electron nuclear double resonance
Footnotes
This work was supported by grants from the National Institutes of Health (R01-GM59087 to LCS and DRD; HL13531 to BMH), the National Science Foundation (MCB-0316038 to BMH) and the United States Department of Agriculture Postdoctoral Fellowship program (2004-35318-14905 to BMB).
Throughout, all reference to the quantity of diazene added actually represents the corresponding quantity of azodiformate added. The actual quantity of diazene present will be lower.
References
- 1.Burgess BK, Lowe DJ. The mechanism of molybdenum nitrogenase. Chem Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Howard JB, Rees DC. Structural basis of biological nitrogen fixation. Chem Rev. 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]
- 3.Smil V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. MIT Press; Cambridge, MA: 2001. [Google Scholar]
- 4.Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science. 1992;257:1653–1659. doi: 10.1126/science.1529353. [DOI] [PubMed] [Google Scholar]
- 5.Seefeldt LC, Dean DR. Role of nucleotides in nitrogenase catalysis. Acc Chem Res. 1997;30:260–266. [Google Scholar]
- 6.Howard JB, Rees DC. Nitrogenase: A nucleotide-dependent molecular switch. Annu Rev Biochem. 1994;63:235–264. doi: 10.1146/annurev.bi.63.070194.001315. [DOI] [PubMed] [Google Scholar]
- 7.Hageman RV, Burris RH. Nitrogenase and nitrogenase reductase associate and dissociate with each catalytic cycle. Proc Natl Acad Sci USA. 1978;75:2699–2702. doi: 10.1073/pnas.75.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orme-Johnson WH, Münck E. On the prosthetic groups of nitrogenase. In: Coughlan MP, editor. Molybdenum and molybdenum containing enzymes. Pergamon Press; Oxford: 1980. pp. 427–438. [Google Scholar]
- 9.Seefeldt LC, Dance I, Dean DR. Substrate interactions with nitrogenase: Fe versus Mo. Biochemistry. 2004;43:1401–1409. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 10.Dos Santos PC, Igarashi RY, Lee HI, Hoffman BM, Seefeldt LC, Dean DR. Substrate Interactions with the nitrogenase active site. Acc Chem Res. 2005;38:208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 11.Thorneley RNF, Eady RR, Lowe DJ. Biological nitrogen fixation by way of an enzyme-bound dinitrogen-hydride intermediate. Nature. 1978;272:557–558. [Google Scholar]
- 12.Dance I. Theoretical investigations of the mechanism of biological nitrogen fixation at the FeMo cluster site. J Biol Inorg Chem. 1996;1:581–586. [Google Scholar]
- 13.Hinnemann B, Nørskov JK. Chemical activity of the nitrogenase FeMo cofactor with a central nitrogen ligand: Density functional study. J Am Chem Soc. 2004;126:3920–3927. doi: 10.1021/ja037792s. [DOI] [PubMed] [Google Scholar]
- 14.Rod TH, Hammer B, Nørskov JK. Nitrogen adsorption and hydrogenation on a MoFe6S9 complex. Phys Rev Lett. 1999;82:4054–4057. [Google Scholar]
- 15.Stavrev KK, Zerner MC. Studies on the hydrogenation steps of the nitrogen molecule at the Azotobacter vinelandii nitrogenase site. Inter J Quant Chem. 1998;70:1159–1168. [Google Scholar]
- 16.Deng H, Hoffmann R. How N2 might be activated by the FeMo-cofactor in nitrogenase. Angew Chem Int Ed Engl. 1993;32:1062–1065. [Google Scholar]
- 17.Kastner J, Hemmen S, Blochl PE. Activation and protonation of dinitrogen at the FeMo cofactor of nitrogenase. J Chem Phys. 2005;123:074306. doi: 10.1063/1.2008227. [DOI] [PubMed] [Google Scholar]
- 18.Durrant MC. An atomic-level mechanism for molybdenum nitrogenase. Part 1. Reduction of dinitrogen. Biochemistry. 2002;41:13934–13945. doi: 10.1021/bi025623z. [DOI] [PubMed] [Google Scholar]
- 19.Huniar U, Ahlrichs R, Coucouvanis D. Density functional theory calculations and exploration of a possible mechanism of N2 reduction by nitrogenase. J Am Chem Soc. 2004;126:2588–2601. doi: 10.1021/ja030541z. [DOI] [PubMed] [Google Scholar]
- 20.Fryzuk MD, MacKay BA. Dinitrogen coordination chemistry: On the biomimetic borderlands. Chem Rev. 2004;104:385–401. doi: 10.1021/cr020610c. [DOI] [PubMed] [Google Scholar]
- 21.Chatt J, Dilworth JR, Richards RL, Sanders JR. Chemical evidence concerning the function of molybdenum in nitrogenase. Nature. 1969;224:1201–1202. doi: 10.1038/2241201a0. [DOI] [PubMed] [Google Scholar]
- 22.Chatt J, Dilworth JR, Richards RL. Recent advances in the chemistry of nitrogen fixation. Chem Rev. 1978;78:589–625. [Google Scholar]
- 23.Pickett CJ. The Chatt cycle and the mechanism of enzymic reduction of molecular nitrogen. J Biol Inorg Chem. 1996;1:601–606. [Google Scholar]
- 24.Yandulov DV, Schrock RR. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 25.Schrock RR. Catalytic reduction of dinitrogen under mild conditions. Chem Commun. 2003:2389–2391. doi: 10.1039/b307784p. [DOI] [PubMed] [Google Scholar]
- 26.Schrock RR. Catalytic reduction of dinitrogen to ammonia at well defined single metal sites. Phil Trans R Soc A. 2005;363:959–969. doi: 10.1098/rsta.2004.1541. [DOI] [PubMed] [Google Scholar]
- 27.Barney BM, Lukoyanov D, Yang TC, Dean DR, Hoffman BM, Seefeldt LC. A methyldiazene (HN=N-CH3) derived species bound to the nitrogenase active site FeMo-cofactor: implications for mechanism. Proc Natl Acad Sci (USA) 2006;103:17113–17118. doi: 10.1073/pnas.0602130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis LC. Hydrazine as a substrate and inhibitor of Azotobacter vinelandii nitrogenase. Arch Biochem Biophys. 1980;204:270–276. doi: 10.1016/0003-9861(80)90033-8. [DOI] [PubMed] [Google Scholar]
- 29.Malinak SM, Simeonov AM, Mosier PE, McKenna CE, Coucouvanis D. Catalytic reduction of cis-dimethyldiazene by the [MoFe3S4](3+) clusters. The four-electron reduction of a N=N bond by a nitrogenase-relevant cluster and implications for the function of nitrogenase. J Am Chem Soc. 1997;119:1662–1667. [Google Scholar]
- 30.McKenna CE, Simeonov AM, Eran H, Bravo-Leerahhandh M. Reduction of cyclic and acyclic diazene derivatives by Azotobacter vinelandii nitrogenase: diazirine and trans-dimethyldiazene. Biochemistry. 1996;35:4502–4514. doi: 10.1021/bi950964g. [DOI] [PubMed] [Google Scholar]
- 31.Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR. Catalytic and biophysical properties of a nitrogenase apo-MoFe protein produced by a nifB-deletion mutant of Azotobacter vinelandii. Biochemistry. 1998;37:12611–12623. doi: 10.1021/bi981165b. [DOI] [PubMed] [Google Scholar]
- 32.Liao GL, Palmer G. Diazene- a not so innocent ligand for the binuclear center in cytochrome c oxidase. Biochemistry. 1998;37:15583–15592. doi: 10.1021/bi981476m. [DOI] [PubMed] [Google Scholar]
- 33.Audrieth DC, Mohr EB. Biurea. Inorg Syntheses. 1953;4:26–28. [Google Scholar]
- 34.Hristova-Kazmierski MK, Kepler JA. Synthesis of [14C]azodicarbonamide. J Labelled Comp and Radiopharm. 1999;42:203–206. [Google Scholar]
- 35.Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC. Substrate interaction at an iron-sulfur face of the FeMo-cofactor during nitrogenase catalysis. J Biol Chem. 2004;279:53621–53624. doi: 10.1074/jbc.M410247200. [DOI] [PubMed] [Google Scholar]
- 36.Barney BM, Laryukhin M, Igarashi RY, Lee HI, Dos Santos PC, Yang TC, Hoffman BM, Dean DR, Seefeldt LC. Trapping a hydrazine reduction intermediate on the nitrogenase active site. Biochemistry. 2005;44:8030–8037. doi: 10.1021/bi0504409. [DOI] [PubMed] [Google Scholar]
- 37.Davoust CE, Doan PE, Hoffman BM. Q-band pulsed electron spin-echo spectrometer and its application to ENDOR and ESEEM. J Magn Reson. 1996;119:38–44. [Google Scholar]
- 38.Mims WB. Pulsed ENDOR experiments. Proc Royal Soc Lond. 1965;238:452–457. [Google Scholar]
- 39.Doan PE, Hoffman BM. Making hyperfine selection in Mims ENDOR independent of deadtime. Chem Physics Lett. 1997;269:208–214. [Google Scholar]
- 40.Hoffman BM, DeRose VJ, Doan PE, Gurbiel RJ, Houseman ALP, Telser J. Metalloenzyme active-site structure and function through multifrequency CW and pulsed ENDOR. In: Berliner LJ, Reuben J, editors. Biol Magn Reson. Plenum Press; New York: 1993. pp. 151–218. [Google Scholar]
- 41.Stanbury DM. Kinetic behavior of diazene in aqueous solutions. Inorg Chem. 1991;30:1293–1296. [Google Scholar]
- 42.Guth JH, Burris RH. Inhibition of nitrogenase catalyzed NH3formation by H2. Biochemistry. 1983;22:5111–5122. doi: 10.1021/bi00291a010. [DOI] [PubMed] [Google Scholar]
- 43.Thorneley RNF, Lowe DJ. Kinetics and mechanisms of the nitrogenase enzyme system. In: Spiro TG, editor. Molybdenum Enzymes. Wiley; New York: 1985. pp. 221–284. [Google Scholar]
- 44.Benton PMC, Laryukhin M, Mayer SM, Hoffman BM, Dean DR, Seefeldt LC. Localization of a substrate binding site on FeMo-cofactor in nitrogenase: trapping propargyl alcohol with an α-70-substituted MoFe protein. Biochemistry. 2003;42:9102–9109. doi: 10.1021/bi034595x. [DOI] [PubMed] [Google Scholar]
- 45.Davis LC, Henzl MT, Burris RH, Orme-Johnson WH. Iron-sulfur clusters in the molybdenum-iron protein component of nitrogenase. Electron paramagnetic resonance of the carbon monoxide inhibited state. Biochemistry. 1979;18:4860–4869. doi: 10.1021/bi00589a014. [DOI] [PubMed] [Google Scholar]
- 46.Pollock CR, Lee H-I, Cameron LM, DeRose VJ, Hales BJ, Orme-Johnson WH, Hoffman BM. Investigation of CO bound to inhibited forms of nitrogenase MoFe protein by 13C ENDOR. J Am Chem Soc. 1995;117:8686–8687. [Google Scholar]
- 47.Sørlie M, Christiansen J, Dean DR, Hales BJ. Detection of a new radical and FeMo-cofactor EPR signal during acetylene reduction by the α-H195Q mutant of nitrogenase. J Am Chem Soc. 1999;121:9457–9458. [Google Scholar]
- 48.Lee HI, Sørlie M, Christiansen J, Song R, Dean DR, Hales BJ, Hoffman BM. Characterization of an intermediate in the reduction of acetylene by nitrogenase α-Gln195 MoFe protein by Q-band EPR and 13C, 1H ENDOR. J Am Chem Soc. 2000;122:5582–5587. [Google Scholar]
- 49.Barney BM, Yang TC, Igarashi RY, Dos Santos PC, Laryukhin M, Lee HI, Hoffman BM, Dean DR, Seefeldt LC. Intermediates trapped during nitrogenase reduction of N2, CH3-N=NH, and H2N-NH2. J Am Chem Soc. 2005;127:14960–14961. doi: 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- 50.Smith BE, Lowe DJ, Bray RC. Nitrogenase of Klebsiella pneumoniae: Electron-paramagnetic-resonance studies on the catalytic mechanism. Biochem J. 1972;130:641–643. doi: 10.1042/bj1300641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith BE, Lowe DJ, Bray RC. Studies by electron paramagnetic resonance on the catalytic mechanism of nitrogenase of Klebsiella pneumoniae. Biochem J. 1973;135:331–341. doi: 10.1042/bj1350331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim CH, Newton WE, Dean DR. Role of the MoFe protein alpha-subunit histidine-195 residue in FeMo-cofactor binding and nitrogenase catalysis. Biochemistry. 1995;34:2798–2808. doi: 10.1021/bi00009a008. [DOI] [PubMed] [Google Scholar]
- 53.Fisher K, Dilworth MJ, Newton WE. Differential effects on N2binding and reduction, HD formation, and azide reduction with alpha-195His- and alpha-191Gln-substituted MoFe proteins of Azotobacter vinelandii nitrogenase. Biochemistry. 2000;39:15570–15577. doi: 10.1021/bi0017834. [DOI] [PubMed] [Google Scholar]
- 54.Mayer SM, Niehaus WG, Dean DR. Reduction of short chain alkynes by a nitrogenase alpha-70Ala-substituted MoFe protein. J Chem Soc, Dalton Trans. 2002;5:802–807. [Google Scholar]
- 55.Jensen BB, Burris RH. N2O as a substrate and as a competitive inhibitor of nitrogenase. Biochemistry. 1986;25:1083–8. doi: 10.1021/bi00353a021. [DOI] [PubMed] [Google Scholar]
- 56.Simpson FB, Burris RH. A nitrogen pressure of 50 atmoshperes does not prevent evolution of hydrogen by nitrogenase. Science. 1984;224:1095–1097. doi: 10.1126/science.6585956. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Burris RH. Influence of pN2 and pD2 on HD formation by various nitrogenases. Biochemistry. 1983;22:4472–4480. doi: 10.1021/bi00288a019. [DOI] [PubMed] [Google Scholar]
- 58.Dance I. The mechanism of nitrogenase. Computed details of the site and geometry of binding of alkyne and alkene substrates and intermediates. J Am Chem Soc. 2004;126:11852–11863. doi: 10.1021/ja0481070. [DOI] [PubMed] [Google Scholar]
- 59.Igarashi RY, Dos Santos PC, Niehaus WG, Dance IG, Dean DR, Seefeldt LC. Localization of a catalytic intermediate bound to the FeMo-cofactor of nitrogenase. J Biol Chem. 2004;279:34770–34775. doi: 10.1074/jbc.M403194200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional figures illustrating the microwave power dependence of the X- and Q-band EPR spectra and the pH dependence of the EPR spectra are available. This material is available free of charge via the Internet at http://pubs.acs.org.