

SUMMARY
PPARγ is a member of the nuclear receptor family for which agonist ligands have antigrowth effects. However, clinical studies using PPARγ ligands as a monotherapy failed to show a beneficial effect. Here we have studied the effects of PPARγ activation with chemotherapeutic agents in current use for specific cancers. We observed a striking synergy between rosiglitazone and platinum-based drugs in several different cancers both in vitro and using transplantable and chemically induced “spontaneous” tumor models. The effect appears to be due in part to PPARγ-mediated downregulation of metallothioneins, proteins that have been shown to be involved in resistance to platinum-based therapy. These data strongly suggest combining PPARγ agonists and platinum-based drugs for the treatment of certain human cancers.
INTRODUCTION
Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors (Michalik et al., 2004; Rosen and Spiegelman, 2001). Ligands for PPARγ include natural compounds such as fatty acids and their derivatives and synthetic agents such as the antidiabetic drugs rosiglitazone (Avandia) and pioglitazone (Actos) (Forman et al., 1995; Lehmann et al., 1995). Although a central role for PPARγ has been demonstrated in the differentiation of adipose cells, PPARγ has also been shown to regulate the growth, differentiation, and gene expression of a number of different cancer cells (Altiok et al., 1997; Barak et al., 1999; Gupta and Dubois, 2002; Kubota et al., 1999; Michalik et al., 2004; Rosen et al., 1999; Rosen and Spiegelman, 2001; Sporn et al., 2001; Tontonoz et al., 1994, 1997). Based on the antigrowth and prodifferentiation properties of PPARγ, several clinical studies have been performed with PPARγ ligands in human cancer. Treatment of patients with pleomorphic/myxoid round-cell liposarcomas with troglitazone showed a significant effect on tumor differentiation and decreased cell proliferation (Demetri et al., 1999). However, small clinical trials involving several more common advanced epithelial malignancies showed no beneficial effect using PPARγ ligands as a monotherapy (Burstein et al., 2003; Kulke et al., 2002; Smith et al., 2004). Taken together, these studies suggest that, despite their promise, the use of PPARγ ligands as a monotherapy in advanced disease may not be beneficial.
In the context of human cancer, it is important to note that PPARγ ligands are relatively nontoxic and well tolerated. This is evidenced by the fact that approximately 5 million people in the US are currently taking Actos or Avandia for long-term control of type 2 diabetes. Therefore, we have undertaken studies to explore the adjuvant use of PPARγ with a variety of agents that are currently in use in the cancer clinic. We demonstrate in this manuscript a striking synergy between PPARγ and several members of the platinum family of drugs on cultured cell growth and tumor growth in vivo. Our data also suggest a likely mechanism for this synergy: PPARγ activation reduces expression of multiple members of the metallothionein gene family. These studies offer an exciting therapy to enhance primary use of platinum-based cancer drugs, and also suggest a method for overcoming platinum-drug resistance of tumors.
SIGNIFICANCE
Platinum-based drugs are used extensively in the cancer clinic. Dose-limiting toxicities and resistance remain significant hurdles in the use of these drugs. In this manuscript we describe a powerful synergy between PPARγ agonists and carboplatin. PPARγ agonists are already in use for the treatment of type II diabetes and have a relatively low toxicity profile. In addition, the mechanisms appear to be mediated in part via a pathway that has been shown to play a role in the resistance of a number of cancers to platinum-based therapy. These data suggest the use of PPARγ ligands and platinum-based drugs not only to achieve better cancer control but also for use in cancers that have acquired resistance to platinum-based therapy.
RESULTS
PPARγ and Carboplatin Synergize to Inhibit Cell Growth
PPARγ agonists are in wide use clinically for type II diabetes and have a very good toxicity profile in comparison to most anticancer drugs. To investigate the ability of PPARγ agonists to modify the response to chemotherapeutic drugs already in common use in the cancer clinic, we examined several epithelial-derived cancers in which PPARγ has been shown to have an antiproliferative effect. Low doses of the PPARγ ligand rosiglitazone were used in combination with several different chemotherapeutic agents. In most of the cancers examined, PPARγ ligand and the drugs used in treating those cancers failed to demonstrate additive or synergistic effects. However, there was a dramatic effect on a non-small-cell lung cancer (NSCLC) cell line when a PPARγ ligand was combined with carboplatin, a platinum-based drug used to treat lung cancer (Figure 1A) (Cosaert and Quoix, 2002). Treatment of the NSCLC cell line with a relatively low dose (0.5 μM) of rosiglitazone reduced cell growth by about 10%. When this dose of rosiglitazone was combined with 2.5 μM carboplatin, growth was reduced by 50%. Enhanced growth inhibition by rosiglitazone was also observed using 10 μM and 25 μM carboplatin. For example, while 10 μM carboplatin alone reduced growth to about 40%, the combination of rosiglitazone and 10 μM carboplatin reduced growth to 15%, an almost 70% reduction in cell growth versus carboplatin alone.
Figure 1. PPARγ Activation Enhances Carboplatin Growth Inhibition in A549 Lung Adenocarcinoma Cells.
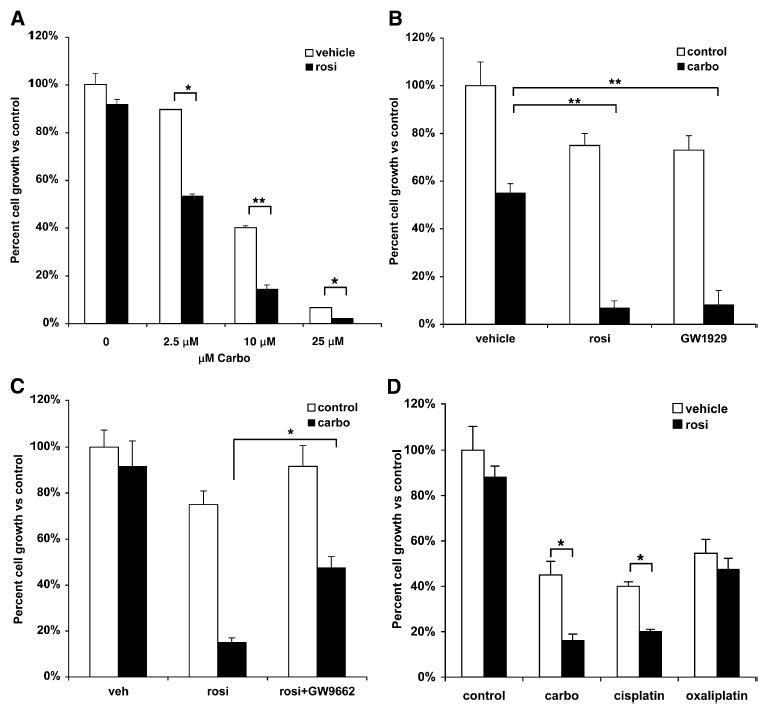
(A) A549 cells treated with 0.5 μM of rosiglitazone and indicated doses of carboplatin alone or in combination.
(B) A549 cells were treated with 1 μm rosiglitazone, 250 nM GW1929, or carboplatin, alone or in combination.
(C) A549 cells were treated with 0.5 μM rosiglitazone alone or in combination with 250 nM GW9662, with and without carboplatin.
(D) Interaction of rosiglitazone with different platinum-based drugs. A549 cells treated with 0.5 μM rosiglitazone, 10 μM carboplatin, 1 μM cisplatin, or 1 μM oxaliplatin alone or in combination. Cell number was determined after 7–10 days and expressed as a percent of control cells. Representative experiments, n = 3, mean ± SD. *p < 0.05, **p < 0.01.
PPARγ-independent effects have (rarely) been observed for the TZD family of PPARγ agonists (Chawla et al., 2001; Koeffler, 2003). However, to rule out a PPARγ-independent effect, A549 cells were treated with GW1929, a high-affinity non-TZD PPARγ ligand (Henke et al., 1998). Treatment of cells with 1 μM rosiglitazone or 250 nM GW1929 reduced growth to about 80% compared to control (Figure 1B). Whereas 10 μM carboplatin reduced growth to 55%, both GW1929 and rosiglitazone in combination with carboplatin reduced growth to less than 10% compared to control cells. To further demonstrate the PPARγ-dependent synergistic effect of rosiglitazone with carboplatin, we treated cells with the PPARγ antagonist GW9662 (Huang et al., 1999). As expected, combining rosiglitazone with carboplatin dramatically increased growth inhibition compared to carboplatin alone (Figure 1C). However, the synergistic effect was significantly blunted in the presence of GW9662. The effect of the combination on growth was reduced by only 45% using 2.5 μM carboplatin and rosiglitazone (compared to 80% for the combination versus carboplatin alone). These data strengthen the argument that the synergy between rosiglitazone and carboplatin is due to PPARγ-dependent effects.
Carboplatin is a member of the platinum family of chemotherapy agents. Carboplatin and its parent compound, cisplatin, are two of the most commonly used platinum drugs for the treatment of lung cancer (Cosaert and Quoix, 2002). There are also third-generation platinum-based drugs designed to treat cancers that have become platinum resistant, such as oxaliplatin, which is used to treat other cancers (Chawla et al., 2001; Raymond et al., 2002). To determine if the effect of PPARγ with carboplatin was a class effect or specific for carboplatin, A549 cells were treated with carboplatin, cisplatin, or oxaliplatin alone and in combination with rosiglitazone. Doses of the three platinum drugs that had roughly equipotent effects alone were used, reducing growth to about 40%–55% (Figure 1D). Treatment of A549 cells with rosiglitazone had a minimal effect on its own, but the combination with carboplatin or cisplatin reduced growth to 15% and 20%, respectively. In contrast, the combination of rosiglitazone and oxaliplatin did not have any additive or synergistic effects on cell growth.
Non-small-cell lung cancer (NSCLC) is the most common lung cancer and is comprised of different histological subtypes, of which adenocarcinoma is the most common (this includes the A549 cell line). We therefore evaluated the effect of rosiglitazone and carboplatin in additional NSCLC cell lines. Rosiglitazone had no effect on the growth of the H23 adenocarcinoma cell line (Figure 2A). While 10 μM carboplatin reduced growth to 40%, combining carboplatin with rosiglitazone reduced growth to 15%, representing more than a 70% reduction in growth versus carboplatin alone. In another adenocarcinoma cell line, H1650, rosiglitazone reduced growth to 70% of controls and carboplatin reduced growth to 40% (Figure 2B). However, the combination of rosiglitazone and carboplatin dramatically reduced growth to 6% of controls. This represents an 85% reduction in growth versus carboplatin alone.
Figure 2. Rosiglitazone and Carboplatin Synergize to Suppress Growth in Multiple Cell Types.
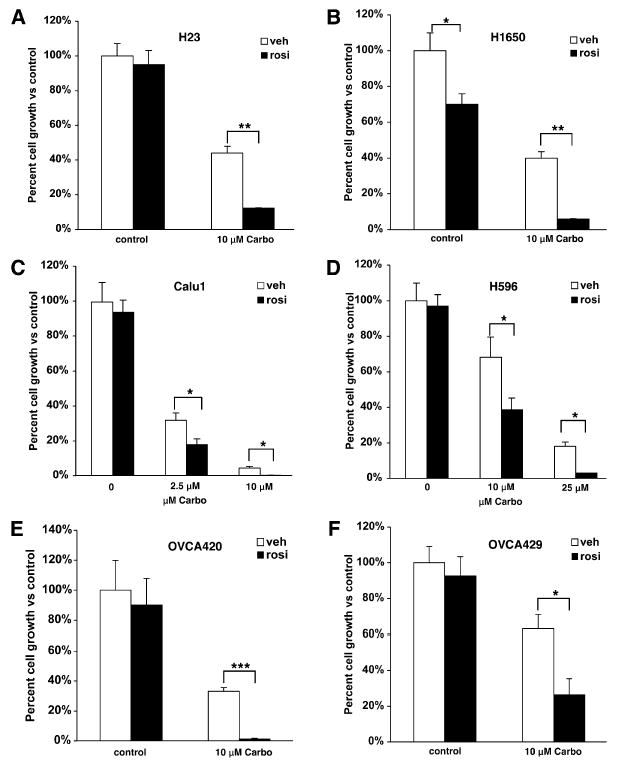
Cells were treated with the indicated concentrations of carboplatin and 0.5 μM rosiglitazone or (B) 0.2 μM rosiglitazone. (A and B) NSCLC adenocarcinoma, (C) squamous cell carcinoma, (D) adenosquamous cell carcinoma, (E) OVCA420, or (F) OVCA429 ovarian epithelial cancer cell lines were treated with 1 μM rosiglitazone and 10 μM carboplatin alone or in combination. Cell number was determined after 7–10 days and expressed as a percent of control cells. Representative experiments, n = 3 mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
We next examined the effects of rosiglitazone in combination with carboplatin in two nonadenocarcinoma NSCLC cell lines. Rosiglitazone had no effect on growth in the squamous cell carcinoma cell line Calu1 or the adenosquamous carcinoma cell line H596 (Figures 2C and 2D). The Calu1 cells were relatively sensitive to carboplatin, with 2.5 μM carboplatin alone reducing growth over 60%. However, growth was reduced an additional 50% when rosiglitazone was added. An even greater effect was observed with 25 μM carboplatin where the combination reduced growth to about 1%. Similar effects were seen in the H596 cell line using 10 μM and 25 μM carboplatin. Combining rosiglitazone with 10 and 25 μM carboplatin increased growth inhibition 50% and 80%, respectively (Figures 2C and 2D).
Carboplatin is also used as a main line therapy in the treatment of ovarian cancer. While many patients initially respond to carboplatin, most eventually develop resistance (Cannistra, 2004). Therefore, we investigated the effect of carboplatin and rosiglitazone on two ovarian cancer cell lines that are considered to be relatively resistant to platinum-drug therapy (Hagopian et al., 1999). Figures 2E and 2F show that a relatively low dose of rosiglitazone had very little effect on the growth of the ovarian cancer cell lines OVCA420 and OVCA429. However, combining carboplatin with rosiglitazone dramatically enhanced growth inhibition versus carboplatin alone. This was particularly striking for the OVCA420 cell line, where combining carboplatin with rosiglitazone reduced growth over 90%. These data demonstrate that the synergistic effect between PPARγ activation and carboplatin is observed for cells derived from multiple cancers where platinum drugs are used.
PPARγ Agonist and Carboplatin Synergize in Colon Cancer
To further extend these studies, we asked if rosiglitazone could make carboplatin effective even in a cancer where it is not used clinically. For these experiments the Moser human colon cancer cell line was treated with each agent alone or in combination. A very low dose of rosiglitazone (50 nM) or 2.5 μM carboplatin alone reduced growth about 15%–20%. However, the combination of both drugs led to a 70% reduction in cell growth compared to control cells (See Figure S1 in the Supplemental Data available with this article online). Therefore, combining carboplatin with PPARγ ligands reduced cell growth even in cancers where platinum-based drugs are not commonly used.
Mechanisms of Synergy between PPARγ Agonists and Platinum Drugs
The mechanism(s) operating in this drug interaction was studied using cell-cycle analysis and global transcriptional profile analysis. The A549 cells were treated with rosiglitazone or carboplatin alone or in combination for 3 days, and cell-cycle analysis was performed. Carboplatin alone led to an increase in the fraction of cells in the G2-M phase of the cell cycle from 5% to 12% (Figure 3A). This is consistent with the known effects of carboplatin on the cell cycle (Eastman, 1999). Although the low dose of rosiglitazone had no effect on the distribution of cells in the cell cycle, combining rosiglitazone with carboplatin caused an approximate doubling of cells in the G2-M phase compared to carboplatin alone.
Figure 3. Combination Treatment Increases G2-M Arrest and Apoptosis.
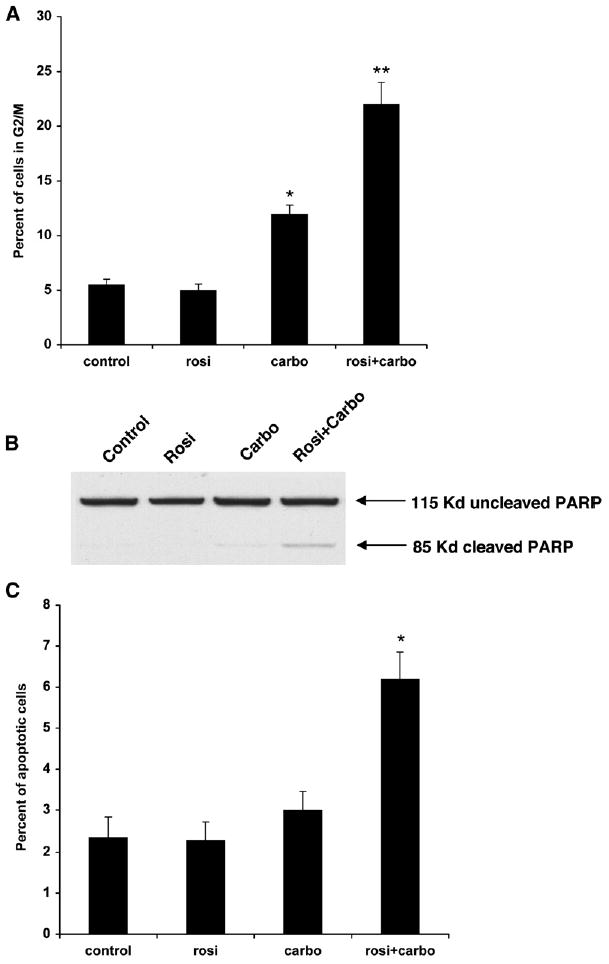
A549 cells were treated with 0.5 μM rosiglitazone or 10 μM carboplatin alone or in combination.
(A) Cell-cycle analysis was determined by PI staining using FACS. n = 3 ± SD.
(B) Immunoblotting for cleaved PARP-1. 115 kDa, uncleaved PARP1; 85 kDa, cleaved PARP1.
(C) Percentage of apoptotic cells as determined by annexin V-positive cells using FACS analysis. Representative experiments, n = 3 mean ± SD. *p < 0.05, **p < 0.01.
There is evidence that the induction of G2-M arrest by carboplatin leads to apoptotic death (DiPaola, 2002; Eastman, 1999; Gonzalez et al., 2001). We therefore investigated the effect of the drug combination treatment on apoptosis by analyzing several molecular markers of apoptosis; cleavage of the poly(ADP-ribosylating) enzyme (PARP1) and annexin V-positive cells. At the doses used, rosiglitazone alone failed to induce significant PARP1 cleavage, and carboplatin alone led to only a small increase in PARP1 cleavage (Figure 3B). However, the combination of rosiglitazone and carboplatin led to significant induction of PARP1 cleavage, suggesting increased apoptotic cell death. Annexin V staining indicated that there is a low percentage (~2%) of cells undergoing apoptosis in both control and rosiglitazone-treated cells. The percentage of apoptotic cells increased slightly to a little over 3% following treatment with carboplatin alone (Figure 3C). However, the combination doubled the percentage of apoptotic cells to 6%. These data indicate that PPARγ activation is capable of increasing carboplatin-mediated G2-M arrest and apoptosis.
To investigate the molecular interaction between the PPARγ agonist/platinum-drug synergy, cDNA microarray analysis was performed on the RNAs after drug treatment. We then performed cluster analysis for changes in gene expression, focusing on pathways associated with platinum-drug resistance/sensitivity. Interestingly, we observed reduced expression for five members of the metallothionein (MT) gene family (Figure 4A). These included MT1G, MT1H, MT1L, MT1X, and MTII. Metallothioneins are heavy metal-binding proteins best known for their ability to protect against heavy metal toxicity, and numerous studies have suggested a critical role for metallothioneins in platinum-drug resistance. Real-time PCR confirmed that rosiglitazone reduced metallothionein gene expression by 40–70% for the individual family members (Figure 4B). In order to demonstrate that downregulation of metallothionein gene expression by rosiglitazone was PPARγ dependent, we again used the non-TZD PPARγ agonist GW1929. Figure 4C shows that GW1929 also reduced expression of the four metallothioneins in a similar fashion to rosiglitazone. We next wanted to determine if metallothionein protein levels were altered by PPARγ activation using rosiglitazone or GW1929. Since there is no subtype-specific antibody, we used a panmetallothionein antibody. As shown in Figure 4D, total metallothionein protein expression was reduced by both rosiglitazone and GW1929. This demonstrates that PPARγ decreases the mRNA expression of specific metallothioneins, which correlates with a reduction in total metallothionein protein expression.
Figure 4. Rosiglitazone Suppresses Several Members of the Metallothionein Gene Family.
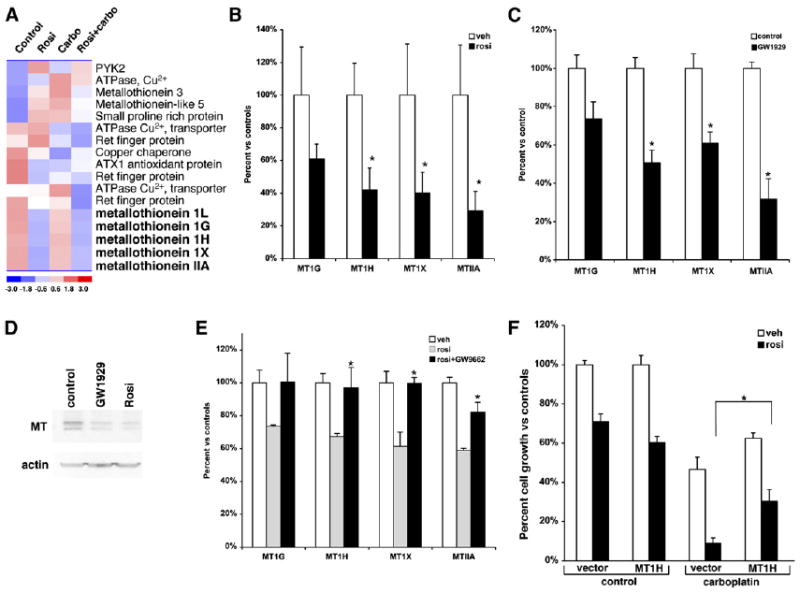
(A) Heat diagram of cluster analysis for heavy metal binding proteins from microarray data of RNA following treatment with rosiglitazone, carboplatin, or a combination of the two.
(B) Real-time PCR for the expression of metallothioneins 1G, 1H, 1X, and IIA following treatment of A549 cells with 1 μM rosiglitazone for 24 hr.
(C) Real-time PCR for the expression of metallothioneins 1G, 1H, 1X, and IIA following treatment of A549 cells with 250 nM GW1929 for 24 hr.
(D) Metallothionein protein expression in A549 cells following treatment with PPARγ agonists GW1929 (250 nM) or rosiglitazone (1 μM) for 24 hr.
(E) A549 cells were treated with 1 μM rosiglitazone alone or in combination with the PPARγ antagonist GW9662 for 24 hr, and real-time PCR was carried out for MT1G, MT1H, MT1X, and MTIIA. *p < 0.05 rosi + GW9662 versus rosiglitazone alone.
(F) Ectopic expression of MT1H blunts the synergistic effect of rosiglitazone and carboplatin. A549 cells stably transduced with a control or retrovirus expressing MT1H were treated with 0.5 μM rosiglitazone or 10 μM carboplatin alone or in combination, and the cell number was determined using a hemocytometer after 7 days. Representative experiment, n = 3 mean ± SD. *p < 0.05.
To more fully evaluate the PPARγ dependence of metallothionein regulation, we treated cells with rosiglitazone alone or in combination with the PPARγ antagonist GW9662. Rosiglitazone alone reduced metallothionein expression as expected, while GW9662 alone did not (Figure 4E and data not shown). However, cotreatment with GW9662 abrogated the downregulation of the metallothioneins by rosiglitazone (Figure 4E). These data demonstrate that not only is the synergistic effect of rosiglitazone and carboplatin on cell growth PPARγ dependent, but also the downregulation of metallothioneins.
To determine whether the ability of PPARγ to reduce metallothionein expression affected the interdrug synergy, a retrovirus expressing MT1H was used to elevate the levels of this protein in the A549 cell line. Ectopic expression of MT1H raised MT1H mRNA levels almost 45-fold (Figure S2). We then treated the cells with rosiglitazone or carboplatin alone and in combination. As expected, the combination of carboplatin and rosiglitazone caused a significant reduction in cell growth compared to carboplatin alone in the control cells containing the empty vector (Figure 4F). Growth was reduced almost 50% by carboplatin alone compared to about 10% for the combination. In contrast, the synergistic effect of rosiglitazone and carboplatin on cell growth was distinctly blunted in cells where MT1H was expressed ectopically. In cells overexpressing MT1H, combining carboplatin and rosiglitazone reduced growth to 30% compared to 60% for carboplatin alone. This strongly suggests that PPARγ is mediating its synergistic effects with carboplatin at least in part via downregulation of metallothioneins.
PPARγ and Carboplatin Synergize in Suppressing Tumor Growth In Vivo
It was critical to determine if the synergistic effect between PPARγ ligands and carboplatin could be observed on tumor growth in vivo. A549 tumor cells were injected subcutaneously into the flank of nude mice, and once tumors had formed (~50–75 mm3), mice were treated with rosiglitazone, carboplatin, or the combination of the drugs. Previous studies have shown that, separately, rosiglitazone and carboplatin are effective antineoplastic agents in this type of xenograft model (Keshamouni et al., 2004; Sirotnak et al., 2000). To investigate synergy, we first used low doses of both rosiglitazone (5 mg/kg) and carboplatin (10 mg/kg). As shown in Figure 5, these doses of carboplatin and rosiglitazone had a minimal effect on growth of the tumors, with tumors increasing in size 11-fold over the course of the experiment (Figure 5A). However, when rosiglitazone was combined with carboplatin, there was a significant reduction in tumor growth, with tumors from these mice being only one-third the size of controls.
Figure 5. Rosiglitazone and Carboplatin Synergize In Vivo to Reduce Tumor Growth.
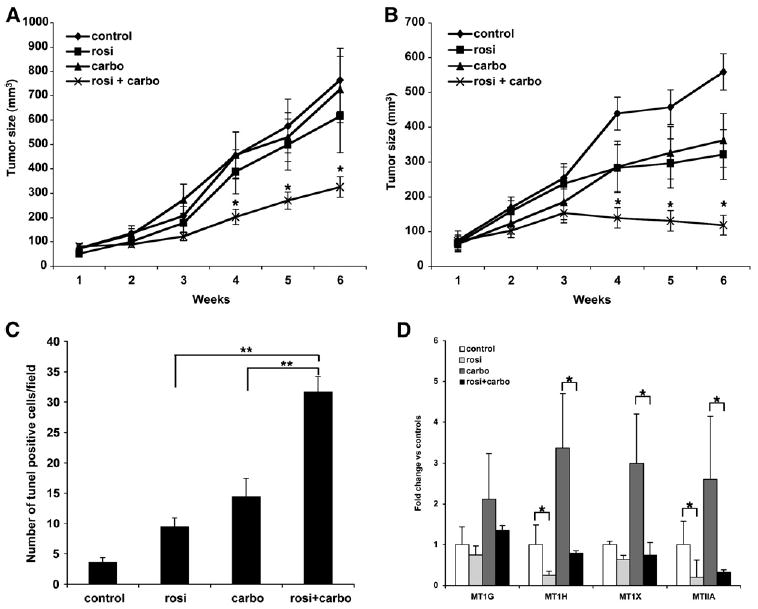
1 × 107 A549 cells were injected subcutaneously into the flank of nude mice. Once the tumor reached 50–75 mm3, treatments were initiated. (A) Control chow, chow containing 5 mg/kg/day rosiglitazone, 10 mg/kg carboplatin IP two times per week or in combination. (B) Control chow, chow containing 20 mg/kg rosiglitazone/day, 50 mg/kg carboplatin IP two times per week or in combination. Tumor growth was measured two times per week. n = 10 mice per group, mean ± SD. *p < 0.05. (C) Average TUNEL-positive cells per field from paraffin-embedded sections of animals treated with either drug alone or in combination. n = 5–8 tumors per group, 4 fields per tumor section, mean ± SD. (D) PPARγ suppresses MT gene expression in vivo. RNA was isolated from tumors of mice treated with rosiglitazone and carboplatin and expression of the indicated metallothioneins determined. n = 6–9 tumors per group, mean ± SD. *p < 0.05, **p < 0.0005.
We then treated the mice with doses of both carboplatin (50 mg/kg) and rosiglitazone (20 mg/kg) at doses that have each been shown to reduce tumor growth in vivo by each agent alone (Keshamouni et al., 2004; Sirotnak et al., 2000). Although tumor growth was reduced in mice treated with each agent alone, tumors still increased in size by 3-fold (Figure 5B). In striking contrast, growth of the tumors in animals treated with the combination was completely arrested. It is important to note that the combination did not appear to have an overall increased toxic effect, since a difference in weights of the mice was not observed (Figure S3). These data demonstrate that combining the PPARγ ligand rosiglitazone with carboplatin dramatically reduces tumor growth in vivo.
Our in vitro data indicate that the synergistic effect between rosiglitazone and carboplatin on cell growth is in part via increased apoptosis. We performed TUNEL staining on paraffin-embedded sections from the xenografted tumors of mice treated with rosiglitazone and carboplatin. Figure 5C shows that rosiglitazone and carboplatin alone increased the number of TUNEL-positive cells compared to sections from control-treated tumors. However, the combination dramatically increased the number of TUNEL-positive cells compared to either treatment alone.
We also performed an additional xenograft study using a different cancer type where carboplatin is used clinically: ovarian epithelial cancer. Following inoculation of ES2 ovarian cancer cells and establishment of tumors (~150 mm3), mice were treated with rosiglitazone or carboplatin alone or in combination for 6 weeks. While rosiglitazone and carboplatin alone did not have a significant effect on tumor growth, there was a significant reduction in tumor size in mice treated with the combination (Figure S4). Tumors were roughly half the size in mice treated with a combination of agents versus control mice or mice treated with a single agent alone. Indeed, due to IACUC guidelines on tumor size, many of the control and single-drug-treated mice had to be euthanized, compared to none of the combination-treated mice.
We next extracted RNA from the A549 lung cancer-derived tumors that grew in control mice or mice treated with the low doses of carboplatin, rosiglitazone, or both drugs together and examined changes in metallothionein gene expression. As shown in Figure 5D, rosiglitazone alone significantly lowered MT1H and IIA gene expression in tumors by 70%–80% compared to control tumors. MT1G and MT1X gene expression was reduced as well, although it was not statistically significant. Interestingly, expression of all four metallothioneins was elevated 2- to 3-fold in tumors from animals treated with carboplatin. Importantly, in the animals receiving carboplatin and rosiglitazone, the induction of MT1H, MT1X, and MTIIA by carboplatin was blunted significantly (MT1G expression was also reduced, but it was not statistically significant). Expression of all three metallothioneins from tumors of mice treated with both rosiglitazone and carboplatin was down over 70% compared to animals receiving carboplatin alone. These data demonstrate that the ability of rosiglitazone to synergize with carboplatin in vivo is associated with a reduction in metallothionein expression in the tumors themselves.
Xenograft experiments are useful as an in vivo model for tumor growth. However, they are limited in that they do not truly represent a tumor that develops “spontaneously” such as is the case in the human condition. We used the colon-specific carcinogen azoxymethane to induce spontaneous colonic tumors in mice. Mice were treated with AOM once a week for 6 weeks. We waited an additional 12 weeks to begin treating mice in order to establish carcinogenesis. Mice were treated with rosiglitazone, carboplatin, or a combination of the two for 6 weeks. We then examined mice for incidence of polyp formation and multiplicity of tumors. As shown in Figure 6A, the incidence of polyp formation was reduced in mice treated with rosiglitazone and carboplatin compared to control or single treatments. Only 25% of the combination-treated animals developed polyps compared to almost half or more of the rosiglitazone or carboplatin treatments alone. The number of polyps per mouse was also down more than 75% compared to single treatment or controls (Figure 6B). These data demonstrate the effect of carboplatin and rosiglitazone in different cancer types and against “spontaneous” tumor formation.
Figure 6. Rosiglitazone and Carboplatin Synergize In Vivo in a Spontaneous Tumor Model.
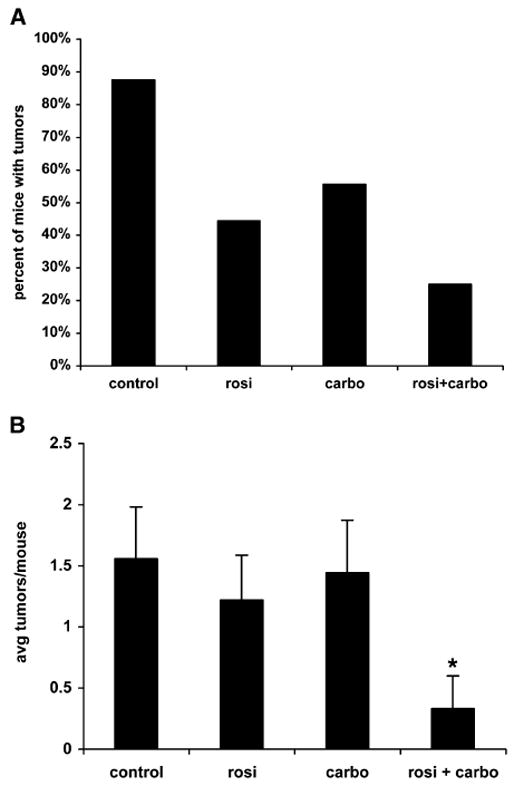
Mice were treated with 10 mg/kg AOM once a week for 6 weeks. Following an additional 12 weeks, mice were treated as described in the Experimental Procedures for 6 weeks. Mice were then euthanized and examined for tumor incidence (A) or number of polyps per mouse (B). n = 8–9 mice per group, mean ± SD. *p < 0.05 versus control, rosiglitazone alone, or carboplatin alone.
DISCUSSION
Numerous reports over the last 10 years have documented the antiproliferative effects of PPARγ ligands (Gupta and Dubois, 2002; Michalik et al., 2004; Rosen and Spiegelman, 2001; Sporn et al., 2001). Genetic studies have also indicated that PPARγ functions as a tumor suppressor in a variety of tissues, including breast, prostate, and colon (Girnun et al., 2002; Mueller et al., 2000; Nicol et al., 2004; Sabatino et al., 2005; Sarraf et al., 1999). Together, these studies offered the potential of cancer control with agonist agents that are generally well tolerated and relatively nontoxic compared to current chemotherapy. Unfortunately, with the exception of a small trial on liposarcomas, clinical trials have indicated that PPARγ ligands may not be useful as a monotherapy in advanced disease (Burstein et al., 2003; Demetri et al., 1999; Kulke et al., 2002; Smith et al., 2004). Because current cancer therapy invariably utilizes a combination of chemotherapeutic drugs, we investigated whether PPARγ ligands could be used as an adjuvant therapy with agents already in use for specific cancers. We describe here that agonist activation of PPARγ dramatically increases the growth-inhibitory effect of the platinum-based drugs cisplatin and carboplatin in several different types of cancers where platinum drugs are currently used. For example, in lung adenocarcinoma cells, a low concentration of rosiglitazone increased the efficacy of carboplatin 4-fold. Notably, we observed a similar synergistic effect on growth inhibition between carboplatin and rosiglitazone in a number of different non-small-cell lung cancer subtypes and ovarian cancer cells. Our data also demonstrate that the synergistic effect of rosiglitazone and carboplatin on cell growth may very well be applicable to cancers where carboplatin is not currently in clinical use, such as colon cancer.
Most importantly, the synergistic interaction between rosiglitazone and carboplatin could also be observed on tumor growth in vivo with transplantable human cancers. Initially, we used doses of rosiglitazone and carboplatin that were five and ten times lower, respectively, than what has been published as being inhibitory toward tumor growth in order to demonstrate synergy for particular lung adenocarcinoma cell line. Indeed, using these low doses, we saw no effect of either drug on its own, but a dramatic growth-inhibitory effect on tumors was observed when carboplatin and rosiglitazone were combined. This effect was also seen at doses where the individual drugs had some mild tumor-inhibitory effects; the combination at these higher concentrations essentially led to stasis if not tumor shrinkage, which appears to be accompanied by increased apoptosis. Using these higher doses on an ovarian cancer cell line, we observed no effect of either drug alone, but a significant reduction in tumor growth by the combination of rosiglitazone and carboplatin. Importantly, the synergistic effect of the combination was observed using a chemically induced “spontaneously” growing tumor. Tumor incidence and multiplicity were significantly lower than control or either rosiglitazone or carboplatin alone. In addition, since the effect was observed in a cancer type (colon) where carboplatin is not typically used, it strengthens the possibility of adding a new chemotherapeutic regimen to the treatment of colorectal cancer.
The combination of carboplatin and the PPARγ agonist did not appear to increase systemic toxicity since the body weights and overall appearance of mice being given the combination of drugs were not different from controls or the mice receiving only one drug. In addition, a recent study showed that PPARγ ligands are actually protective against platinum-drug toxicity in mice (Lee et al., 2006). Thus, our finding that PPARγ activation dramatically increases the efficacy of carboplatin, potentially causing tumor stasis or even tumor shrinkage, could represent a significant advance in chemotherapy.
Platinum-based drugs are known to induce G2-M phase arrest, followed by apoptosis (DiPaola, 2002; Eastman, 1999; Gonzalez et al., 2001). Many studies have shown that the growth-inhibitory effects of PPARγ ligands occur via effects on cell-cycle arrest, as well as through increased apoptosis (Altiok et al., 1997; Chang and Szabo, 2000; Keshamouni et al., 2004; Koeffler, 2003). These previous studies were performed using doses of rosiglitazone (or equivalent compounds) that were five to ten times greater than the doses we used. Using low doses of rosiglitazone, we did not observe effects on cell cycle or apoptosis. However, these low concentrations of rosiglitazone significantly increased the cell-cycle arrest and apoptosis induced by carboplatin. Furthermore, TUNEL staining of sections from tumors of mice following treatment with either drug alone or drug combination demonstrated increased apoptosis as a result of the combination treatment. Therefore, these studies demonstrate a dual effect of combining rosiglitazone with rosiglitazone: decreased proliferation and increased apoptosis. In addition to direct effects on tumor cell apoptosis and cell-cycle arrest, the effects of rosiglitazone in vivo have also been attributed to certain antiangiogenic properties of PPARγ ligands; such additional effects on angiogenesis in vivo cannot be ruled out here (Panigrahy et al., 2002).
The mechanism(s) of how platinum-based drugs exert their cytotoxic effects has been extensively studied. Many pathways have been studied that are believed to play a role in the resistance to platinum-based drugs (Akiyama et al., 1999; Chu, 1994; Perez, 1998). These pathways can be classified into two functional categories. One is the response to and repair of platinum-adducted DNA and the other is limiting/inactivating platinum-drug activity. Our data demonstrate that PPARγ suppresses the expression of several members of the metallothionein family, a family of small (~60 amino acids) zinc- and cysteine-rich proteins that are best characterized by their ability to bind to and detoxify heavy metals (Coyle et al., 2002). Many studies have shown that metallothioneins play a role in the resistance of certain cancers to platinum-based drugs (Akiyama et al., 1999; Chu, 1994; Perez, 1998). This is highlighted by the observation that metallothionein expression is elevated in many platinum-resistant cell lines and tumors (Hishikawa et al., 1997). In addition, chemically or genetically increasing metallothionein levels has been shown to induce resistance to platinum drugs (Andrews et al., 1987; Cheng et al., 2006; Hishikawa et al., 1997; Kasahara et al., 1991; Kelley et al., 1988). Therefore, the ability of rosiglitazone to reduce the expression of multiple members of the metallothionein family shown here probably renders cells more sensitive to platinum therapy. We observed increased metallothionein expression in tumors of mice following treatment with carboplatin, which was reduced when combined with rosiglitazone. This effect correlated with enhanced growth suppression of these tumors; ectopic expression of a metallothionein reduced the synergy between the rosiglitazone and carboplatin in cultured cells. These data together suggest strongly that the ability of PPARγ ligands to suppress metallothionein levels both in vitro and in vivo may, in part, explain the synergy observed with carboplatin.
Although PPARγ is generally considered a transcription factor that increases gene expression, PPARγ has also been shown to repress gene expression. Studies by a number of other labs have demonstrated that PPARγ can repress gene expression via transrepression of AP1 (Delerive et al., 2001; Ricote et al., 1998). Activation of AP1 is antiapoptotic and associated with increased proliferation (Eferl and Wagner, 2003; Karin et al., 2002). In addition, metallothioneins have been shown to be regulated by AP1 (Abate et al., 1990; Lee et al., 1987). Therefore, the repression of metallothioneins we observe by PPARγ activation may be mediated via transrepression of AP1.
Though these studies support the suppression of metallothionein expression as a plausible mechanism by which rosiglitazone may augment platinum action, there may well be other important mechanisms of synergy. Indeed, the fact that adding back MT1H partially blunts the synergy suggests that other pathways may be involved. The partial effect may reflect the fact that we are ectopically expressing only MT1H, whereas rosiglitazone represses several additional metallothionein family members. Other platinum resistance pathways such as the DNA repair gene ERCC1, the DNA-binding gene HMGB1, or the glutathione pathway may also be involved (Akiyama et al., 1999; Chu, 1994; Perez, 1998). We examined our DNA microarray data for changes in expression of these genes by rosiglitazone. However, significant changes in expression were not observed (Figure S1). We are currently exploring additional members of the metallothionein family and other resistance pathways to more fully explain the synergistic effect.
The ability of PPARγ activation to synergize with platinum-based drugs may be important for the treatment of cancers, where they are used as standard therapy. This includes two of the cancers we examined in our studies, lung and ovarian cancer. Lung cancer is the third most common malignancy in the United States, behind prostate and breast cancer, with over 170,000 cases diagnosed each year (of which almost 90% are NSCLC). Despite multimodality therapies, last year more than 160,000 deaths were reported in the United States alone (ACS, 2006). Ovarian cancer is the second most common gynecological cancer, with over 20,000 cases diagnosed each year, resulting in over 15,000 deaths in the United States each year (ACS, 2006). While most women with ovarian cancer initially respond to platinum-drug therapy, many tumors eventually develop resistance (Cannistra, 2004). Therefore the ability to increase the effectiveness of platinum drugs, as well as the ability to overcome resistance, would represent a significant advance in the treatment of these cancers. In addition, dose-limiting toxicities often prevent the use of platinum drugs for other cancers. While there are some conflicts regarding the antigrowth properties of PPARγ agonists and even some rare reports of increased tumorigenesis by PPARγ agonists in murine models of cancer, the vast majority of data indicate that PPARγ is antineoplastic (Koeffler, 2003; Lefebvre et al., 1998; Saez et al., 1998; Sarraf et al., 1998). Therefore, while caution should be used with PPARγ agonists, our data strongly suggest that combining PPARγ ligands with carboplatin may enable the expansion of platinum-based drugs into cancers where they are not currently used.
EXPERIMENTAL PROCEDURES
Cell Culture
H23, H1600, Calu1, and H596 NSCLC cell lines were obtained from ATCC. A549 NSCLC, OVCA 420, OVCA429, and ES2 ovarian cancer cell lines were kind gifts from Drs. Barrett Rollins and Ronald Drapkin (DFCI). Moser human colon cancer cells have been described previously (Sarraf et al., 1998). Rosiglitazone, GW1929, and GW9662 were obtained from ALEXIS Biochemicals. Carboplatin, cisplatin, and oxaliplatin were obtained from Sigma-Aldrich. Cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin. Cells were seeded at a density of 1000 to 10,000 cells/well in 6-well plates and treated with the indicated drugs as described in the figure legends. Cell number was determined using a hemocytometer.
Cell Cycle and Analysis of Apoptosis
Cell-cycle analysis, PARP1 cleavage, and annexin V staining were performed following treatment as indicated. Propidium iodide staining was used for cell-cycle analysis. Annexin V staining was performed according to manufacturer’s protocol (BD Biosciences). FACS analysis for cell-cycle and annexin V staining was performed by the Dana-Farber Cancer Institute’s Flow Cytometry Core. For immunoblotting, proteins from control and treated cells were separated by PAGE and transferred to nitrocellulose as previously described (Drori et al., 2005). PARP1 antibody recognizing uncleaved and cleaved PARP1 was used at 1:2000 (BD Bioscience). Paraffin embedding, sectioning, and TUNEL staining of tumors were performed by the Dana-Farber/Harvard Cancer Center Rodent Histopathology Core.
RNA Isolation and Real-Time PCR/Protein Analysis of Metallothionein Expression
RNA was isolated from cells and tissues using Trizol reagent according to the manufacturer’s protocol (Invitrogen). cDNA was synthesized using iScript reverse transcriptase reagent from 1 μg of total RNA (Bio-Rad). For real-time PCR using Sybr-Green, the following primers were used: MT1G, forward, CTCCTGCAAGTGCAAAGAGTGCAA, reverse, ATTTGTACTTGGGAGCAGGGCTGT; MT1H, forward, AGTCTCACCTCGGCTTGCAATGGA, reverse, GCTCTTCTTGCAGGAGGTG CATTT; and MT1X, forward, TCTGCAAAGGGACGTCAGACAAGT, reverse, TGTAGCAAACGGGTCAGGGTTGTA, and 18S as previously described (Drori et al., 2005). Taqman PCR was performed using Applied Biosystems Assays on Demand for MTIIA. Real-time PCR reactions were carried as previously described on an ABI 7300 system (Applied Biosystems).
Following treatment with PPARγ agonists, cells were harvested and proteins were separated and transferred to nitrocellulose as previously described (Drori et al., 2005). A panmetallothionein antibody (Abcam) was used at 1:750 or β-actin antibody (1:5000) in TBST-5% milk overnight, followed by HRP-conjugated anti-mouse secondary antibody (Jackson Immuno) at 1:15000. Membranes were developed by chemiluminescence (Pierce, Supersignal West Pico).
Microarray Analysis
A549 cells were treated for 24 hr with vehicle control, or 0.5 μM rosiglitazone, 10 μM carboplatin alone, or in combination. Total RNA was isolated using an RNeasy Mini Kit (Qiagen Inc.) and used for global expression analysis. Affymetrix array hybridization and scanning were performed by the Core Facility at Dana-Farber Cancer Institute using human genome U133A chip (Affymetrix). Array data were analyzed with a d-CHIP array analysis program (Li and Wong, 2001). Array data are deposited in the Gene Expression Omnibus (GEO) repository.
Xenograft Studies
Male nude mice were obtained from Taconic Farms. Eight- to ten-week-old mice were inoculated subcutaneously into the right flank with 1 × 107 A549 or ES2 cells in media. Tumors were measured weekly with calipers in two dimensions, and tumor volume was calculated using the formula V = (π*length) × (width2)/6. Treatments were initiated when tumors reached 50–75 mm3 using 8 to 12 mice per group. For the low-dose experiment, mice were given chow containing 5 mg/kg rosiglitazone per day, 10 mg/kg carboplatin by intraperitoneal injection two times per week, alone or in combination. For the experiment with higher dose of drugs and for the ES2 cells, mice were given chow containing 20 mg/kg rosiglitazone per day, 50 mg/kg carboplatin by intraperitoneal injection two times per week, alone or in combination. Control mice were given control chow or injected with saline. All studies were conducted in compliance with the Dana-Farber Cancer Institute IACUC guidelines. Once tumors reached IACUC guidelines, mice were euthanized.
Colon Carcinogenesis Studies
Twelve-week-old mice were injected IP once a week for 6 weeks with 10 mg/kg azoxymethane. Mice were followed out for 12 weeks, after which time mice with mice were given control chow or chow containing 20 mg/kg rosiglitazone per day, 50 mg/kg carboplatin by intraperitoneal injection two times per week, alone or in combination. After 6 weeks, mice were euthanized, and colons were removed and flushed with PBS, cut longitudinally, and fixed in 10% buffered formalin (Fisher). Colons were stained with methylene blue, and polyps were counted blindly under a dissecting microscope.
MT1H Overexpression
MT1H expression clone was obtained from ATCC. MT1H was subcloned into the pMSCV-neo-retroviral vector (BD Bioscience). Retrovirus expressing MT1H was generated as previously described. A549 cells were infected with control retrovirus or retrovirus expressing MT1H, and cells were selected with G418. Expression of MT1H was determined by real-time PCR as described above. Control and MT1H-overexpressing cells were treated as indicated.
Microarray Data
The microarray data set has been deposited in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/), accession no. GSE7035.
Acknowledgments
We extend our appreciation to the Dana-Farber Cancer Institute Microarray Core for expert processing of RNA samples, the Flow Cytometry Core for FACS analysis, and the Pathology Core at Brigham and Women’s Hospital for processing immunohistochemistry of mice samples. We thank Adah Levens for administrative assistance, Eliezer Zimble for technical assistance, and Derick E. Carpenter and William Bautista for maintenance and assistance with animal experiments. We also thank Dr. Srikripa Devarakonda for helpful discussions and careful reading of the manuscript and Eric Smith for graphics assistance. This work was supported by NIH grants R37DK31405 and RO1DK57670 (B.M.S.) and DK064685 (G.D.G.) and the Madeline Franchi Ovarian Cancer Award (G.D.G.).
Footnotes
Supplemental Data
The Supplemental Data include four supplemental figures and one supplemental table and can be found with this article online at http://www.cancercell.org/cgi/content/full/11/5/395/DC1/.
Accession Numbers
The microarray data set has been deposited in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo/), accession no. GSE7035.
References
- Abate C, Luk D, Gentz R, Rauscher FJ, 3rd, Curran T. Expression and purification of the leucine zipper and DNA-binding domains of Fos and Jun: Both Fos and Jun contact DNA directly. Proc Natl Acad Sci USA. 1990;87:1032–1036. doi: 10.1073/pnas.87.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ACS. Cancer Facts and Figures. Atlanta: American Cancer Society; 2006. [Google Scholar]
- Akiyama S, Chen ZS, Sumizawa T, Furukawa T. Resistance to cisplatin. Anticancer Drug Des. 1999;14:143–151. [PubMed] [Google Scholar]
- Altiok S, Xu M, Spiegelman BM. PPARgamma induces cell cycle withdrawal: Inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 1997;11:1987–1998. doi: 10.1101/gad.11.15.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PA, Murphy MP, Howell SB. Metallothionein-mediated cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol. 1987;19:149–154. doi: 10.1007/BF00254568. [DOI] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res. 2000;60:1129–1138. [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- Cheng TC, Manorek G, Samimi G, Lin X, Berry CC, Howell SB. Identification of genes whose expression is associated with cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol. 2006;58:384–395. doi: 10.1007/s00280-005-0171-8. [DOI] [PubMed] [Google Scholar]
- Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J Biol Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- Cosaert J, Quoix E. Platinum drugs in the treatment of non-small-cell lung cancer. Br J Cancer. 2002;87:825–833. doi: 10.1038/sj.bjc.6600540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: The multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman BM, Singer S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci USA. 1999;96:3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaola RS. To arrest or not to G(2)-M Cell-cycle arrest: Commentary re: A.K. Tyagi et al. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin Cancer Res. 2002;8:3311–3314. [PubMed] [Google Scholar]
- Drori S, Girnun GD, Tou L, Szwaya JD, Mueller E, Xia K, Shivdasani RA, Spiegelman BM. Hic-5 regulates an epithelial program mediated by PPARgamma. Genes Dev. 2005;19:362–375. doi: 10.1101/gad.1240705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman A. The mechanism of action of cisplatin: From adducts to apoptosis. In: Lippert B, editor. Cisplatin Chemistry and Biochemistry of a Leading Anti-Cancer Drug. Basel, Switzerland: Wiley-VCH; 1999. pp. 111–134. [Google Scholar]
- Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci USA. 2002;99:13771–13776. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–663. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Dubois RN. Controversy: PPARgamma as a target for treatment of colorectal cancer. Am J Physiol Gastrointest Liver Physiol. 2002;283:G266–G269. doi: 10.1152/ajpgi.00486.2001. [DOI] [PubMed] [Google Scholar]
- Hagopian GS, Mills GB, Khokhar AR, Bast RC, Jr, Siddik ZH. Expression of p53 in cisplatin-resistant ovarian cancer cell lines: Modulation with the novel platinum analogue (1R, 2R-diaminocyclohexane)(trans-diacetato)(dichloro)-platinum(IV) Clin Cancer Res. 1999;5:655–663. [PubMed] [Google Scholar]
- Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, Harrington WW, Jr, Hashim MA, Hull-Ryde EA, Kaldor I, et al. N-(2-Benzoylphenyl)-L-tyrosine PPAR-gamma agonists. 1 Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J Med Chem. 1998;41:5020–5036. doi: 10.1021/jm9804127. [DOI] [PubMed] [Google Scholar]
- Hishikawa Y, Abe S, Kinugasa S, Yoshimura H, Monden N, Igarashi M, Tachibana M, Nagasue N. Overexpression of metallothionein correlates with chemoresistance to cisplatin and prognosis in esophageal cancer. Oncology. 1997;54:342–347. doi: 10.1159/000227714. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: From innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kasahara K, Fujiwara Y, Nishio K, Ohmori T, Sugimoto Y, Komiya K, Matsuda T, Saijo N. Metallothionein content correlates with the sensitivity of human small cell lung cancer cell lines to cisplatin. Cancer Res. 1991;51:3237–3242. [PubMed] [Google Scholar]
- Kelley SL, Basu A, Teicher BA, Hacker MP, Hamer DH, Lazo JS. Overexpression of metallothionein confers resistance to anticancer drugs. Science. 1988;241:1813–1815. doi: 10.1126/science.3175622. [DOI] [PubMed] [Google Scholar]
- Keshamouni VG, Reddy RC, Arenberg DA, Joel B, Thannickal VJ, Kalemkerian GP, Standiford TJ. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene. 2004;23:100–108. doi: 10.1038/sj.onc.1206885. [DOI] [PubMed] [Google Scholar]
- Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4:597–609. doi: 10.1016/s1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, Spiegelman BM, Kim H, Mayer RJ, Fuchs CS. A phase II study of troglitazone, an activator of the PPAR-gamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002;8:395–399. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- Lee W, Haslinger A, Karin M, Tjian R. Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature. 1987;325:368–372. doi: 10.1038/325368a0. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim W, Moon SO, Sung MJ, Kim DH, Kang KP, Jang YB, Lee JE, Jang KY, Park SK. Rosiglitazone ameliorates cisplatin-induced renal injury in mice. Nephrol Dial Transplant. 2006;21:2096–2105. doi: 10.1093/ndt/gfl194. [DOI] [PubMed] [Google Scholar]
- Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Li J, Wong L. Emerging patterns and gene expression data. Genome Inform. 2001;12:3–13. [PubMed] [Google Scholar]
- Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, Oh W, Demetri G, Figg WD, Zhou XP, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci USA. 2000;97:10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM, Gonzalez FJ. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RP. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer. 1998;34:1535–1542. doi: 10.1016/s0959-8049(98)00227-5. [DOI] [PubMed] [Google Scholar]
- Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. PPARgamma: A nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- Sabatino L, Casamassimi A, Peluso G, Barone MV, Capaccio D, Migliore C, Bonelli P, Pedicini A, Febbraro A, Ciccodicola A, Colantuoni V. A novel peroxisome proliferator-activated receptor gamma isoform with dominant negative activity generated by alternative splicing. J Biol Chem. 2005;280:26517–26525. doi: 10.1074/jbc.M502716200. [DOI] [PubMed] [Google Scholar]
- Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res. 2000;6:4885–4892. [PubMed] [Google Scholar]
- Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, Slovin S, Spiegelman B, Small E, Kantoff PW. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer. 2004;101:1569–1574. doi: 10.1002/cncr.20493. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Suh N, Mangelsdorf DJ. Prospects for prevention and treatment of cancer with selective PPARgamma modulators (SPARMs) Trends Mol Med. 2001;7:395–400. doi: 10.1016/s1471-4914(01)02100-1. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci USA. 1997;94:237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]