

Abstract
Although the phase I Coxiella burnetii cellular vaccine is completely efficacious in humans, adverse local and systemic reactions may develop if immune individuals are inadvertently vaccinated. The phase I chloroform-methanol residue (CMRI) vaccine was developed as a potentially safer alternative. Human volunteers with no evidence of previous exposure to C. burnetii received a subcutaneous vaccination with the CMRI vaccine in phase I studies under protocol IND 3516 to evaluate the safety and immunogenicity of the vaccine. This clinical trial tested escalating doses of the CMRI vaccine, ranging from 0.3 to 60 μg, followed by a booster dose of 30 μg, in a placebo-controlled study. Although priming doses of the CMRI vaccine did not induce a specific antibody detectable by enzyme-linked immunosorbent assay, booster vaccination stimulated the production of significant levels of anti-C. burnetii antibody. Peripheral blood cells (PBCs) of vaccinees responded to C. burnetii cellular antigen in vitro in a vaccine dose-dependent manner. After the booster dose, PBCs were activated by recall antigen in vitro, regardless of the priming dose. These findings suggest that vaccination with the CMRI vaccine can effectively prime the immune system to mount significant anamnestic responses after infection.
The development of Q fever vaccines for human use has been an evolving process, comprising roughly 7 decades, since the discovery in 1937 that Coxiella burnetii is the etiologic bacterial agent (7, 8). A variety of C. burnetii cellular or subunit Q fever vaccines have been produced and tested clinically. Clinical testing of formalin-inactivated phase II (31) and phase I (18, 26) cellular vaccines, live-attenuated vaccines (11, 13, 14), and soluble-component vaccines (5, 20, 36) led to refined production methods. Formalin-inactivated phase I cellular vaccines are for personnel working in laboratories (25) and in occupations related to the meat industry, where the greatest number of exposures occur per annum. The Henzerling phase I cellular vaccine has been shown to protect rhesus monkeys (Macaca mulatta) (22, 23), cynomolgus monkeys (Macaca fascicularis) (15), and humans (28) against aerosol infection with virulent phase I C. burnetii. More recently, a phase I cellular vaccine (Q-Vax) provided protection against Q fever for at least 5 years in human volunteers exposed to C. burnetii in an abattoir setting (1).
Although the phase I cellular vaccines are efficacious, they have not been widely used because of the necessity of determining preexisting immunity before vaccination (17). Historically, animals and humans sensitized to phase I C. burnetii by either infection or vaccination are at risk of developing adverse immunopathological reactions, including severe local abscesses and granulomas, after subcutaneous (s.c.) vaccination with phase I cellular vaccines (3, 27, 37). In human populations where prevaccination screening was not instituted, the incidence of severe adverse reactions after primary inoculation was 7.3% (4). Booster inoculations of individuals previously vaccinated with phase I cellular vaccine are not recommended because repetitive vaccination may result in an increase in the rate of adverse reactions, from 0.37/1,000 vaccinees after the primary inoculation to 36/1,000 vaccinees after the ninth injection (4). Dose-escalating clinical studies have shown that with doses of phase I cellular vaccine of >30 μg, adverse local reactions and immunopathological reactions render the vaccine unsafe (19). Collectively, these studies have shown that prevaccination screening for correlates of immunity effectively precludes vaccinating individuals who may develop severe granulomas and sterile abscesses at the injection site (17).
The development of a subunit Q fever vaccine for use without adjuvants was thwarted by the lack of suitable laboratory methods to separate the bacterial factors responsible for the induction of immunopathologic reactions from the efficacious immunogenic properties of phase I whole cells. The development of a chloroform-methanol residue subunit of phase I (CMRI) C. burnetii for vaccinating humans against Q fever was initiated in 1983, after the recognition that an Ohio strain CMRI vaccine was safe and effective in mice and guinea pigs (34, 38, 42). Although dose escalation of phase I cellular vaccine and CMRI in mice (38) and guinea pigs (42) indicated that the induction of specific anti-C. burnetii antibodies was slightly more effective with phase I cellular vaccine, low-dose priming of mice with CMRI or phase I cellular vaccine indicated that CMRI was safer because it did not induce splenomegaly, hepatomegaly, or immunosuppression (38, 39). This was the first indication that the pathogenic properties of the phase I cellular vaccine could be extracted while the residue retained an immunogenic phase I subunit which also protected animals against virulent challenge. In addition, this particulate CMRI vaccine did not require adjuvant to stimulate the immune response. Subsequently, the more benign nature of CMRI than that of the phase I cellular vaccine was shown in goats (42) and sheep (6).
Because immunogenicity and safety results with laboratory-prepared CMRI vaccine indicated that vaccination of immune guinea pigs with phase I cellular vaccine, but not with the CMRI vaccine, induced undesirable tissue reactions (37), we proposed that prior sensitization of humans to C. burnetii by infection or prevaccination may not preclude vaccination with CMRI. We tested a phase I Henzerling strain CMRI vaccine in American volunteers who were unscreened for preexisting immunity to Q fever but who had no previous medical history of either overt Q fever or vaccination with phase I cellular vaccine (12). Although this CMRI vaccine was safe at doses ranging from 30 μg to 240 μg, we hypothesized that a booster dose could improve rates of seroconversion and lymphoproliferation. We therefore undertook a phase I clinical trial designed as a placebo-controlled, dose (from 0.3 μg to 60 μg)-escalating study, followed by a booster dose of 30 μg, to determine the safety and immunogenicity of the CMRI vaccine in human volunteers screened for existing immunity to C. burnetii. The presence of specific immune responses and the absence of adverse reactions after the primary vaccination and booster could enhance the possibility of eventual licensure of this product.
We found that the Henzerling strain CMRI vaccine was safe and immunogenic and that, unlike the phase I cellular vaccine, it could be administered safely in a booster regimen. Although low doses of the CMRI vaccine in human volunteers did not induce the immune system to produce detectable antigen-specific antibody, B cells were not tolerized, because a booster inoculation effectively produced significant levels of phase I antigen-specific antibody. Low-dose priming also activated T cells to significantly respond in vitro to recall antigen in a dose-dependent manner. After the booster, the T cells were significantly activated in vitro by recall antigen, regardless of the priming dose, and the T cells responded significantly more vigorously to native antigenic epitopes of phase I and phase II C. burnetii than they did to the CMRI vaccine antigen. Inferentially, the CMRI vaccine effectively primed the human immune system to recognize the native antigenic structures of both phase I and phase II cells. These findings suggest that vaccinating volunteers with CMRI could effectively prime the immune system to mount significant anamnestic responses after infection, thereby producing both significant amounts of antigen-specific antibody and vigorous cellular immune T-cell responses.
MATERIALS AND METHODS
Diagnostic antigens.
Coxiella burnetii strains used to prepare diagnostic antigens were the virulent Nine Mile phase I, clone 7 strain with smooth-type lipopolysaccharide (LPSI) (NMIS); avirulent Nine Mile phase II, clone 4 strain with rough-type LPS (NMIIR); virulent Nine Mile strain RSA-514, with semi-rough-type LPS (NMISR); and a virulent phase I Henzerling strain with smooth-type LPS (HENIS). Coxiella burnetii was propagated in the yolk sac cells of embryonated chicken eggs and separated from host components by Renografin density gradient centrifugation (41). Whole cells were suspended in phosphate-buffered saline (PBS with 0.25 M sucrose) and frozen at −70°C. Contaminated preparations were discarded.
Preparation of cellular antigens for serologic tests.
Purified viable C. burnetii was inactivated by exposure to 2.1 megarads of gamma (60Co) irradiation (GammaCell 40) at −79°C (24) and then thawed, and formalin was added to a final concentration of 0.1% per mg dry weight with continuous stirring at 4°C for 48 h. Formalin and buffer components of <3,500 Da were removed from the cellular antigens by dialysis against three changes of demineralized sterile water. A standard curve, which related the number of microorganisms and their dry weights to absorbance at 420 nm, was used to adjust the whole cells to 1 mg (dry weight) per ml of sterile water (41). Whole cells were distributed as 1-ml aliquots in cryovials and frozen at −70°C until used in the enzyme-linked immunosorbent assay (ELISA) described below.
Preparation of LPSI antigen for serologic tests.
LPSI was extracted from gamma-irradiated and formalin-treated NMIS cells as described previously (2).
Preparation of native whole cells for lymphoproliferative assays.
The native cellular antigens of C. burnetii (NMIS, NMIIR, and HENIS) were used as recall antigens in lymphoproliferative assays. After purification of viable whole cells, the microorganisms were inactivated by exposure to 2.1 megarads of gamma (60Co) irradiation at −79°C. These inactivated microorganisms were thawed and suspended in PBS-0.25 M sucrose to give a final concentration of 1 mg/ml. Preparations were distributed as 1-ml aliquots in cryovials and frozen at −70°C until used in the lymphoproliferative assays. The CMRI vaccine and native whole cells were diluted to 55 μg/ml in RPMI 1640 (Sigma Chemical Company, St. Louis, MO) before use in the lymphoproliferative assays.
Preparation of CMRI vaccine.
Because phase I cellular vaccines elicit greater protection in animals and humans than phase II cellular vaccines do (21), the phase I character of the Henzerling vaccine strain was ensured by infecting guinea pigs intraperitoneally with a seed stock used to prepare cellular vaccines for human use. Infected spleens from febrile guinea pigs that were shown to be free of autonomously growing bacteria were pooled and passaged successively three times in guinea pigs. After the third passage, infected spleens were pooled and a suspension was used to infect the yolk sac cells of fertile White Leghorn chicken eggs. After 7 to 8 days of incubation, a yolk sac seed stock was prepared to amplify phase I C. burnetii in yolk sac cells. The CMRI vaccine was produced essentially as described by Williams and Cantrell (38), by scale-up of laboratory production methods at The Salk Institute, Swiftwater, PA.
The CMRI vaccine was suspended to a final concentration of 600 μg/ml in 1% lactose solution, lyophilized, and stored at −20°C. The placebo preparation consisted of 1% lactose. Vaccine was reconstituted on the vaccination day in sterile bacteriostatic saline (McGaw Pharmaceuticals, Irvine, CA) containing 1% benzyl alcohol at dilutions which permitted delivery of the desired CMRI dosages in a volume of 0.5 ml. The placebo was prepared at the same dilutions and volumes.
ELISA.
The levels of human serum antibodies to C. burnetii antigens were determined by an ELISA developed to measure Coxiella-specific antibodies in the sera of animals (43). Conditions for the ELISA described in this study were standardized for human serum antibody testing, and the assay was performed as previously described (30, 32).
Lymphoproliferative assay.
Lymphoproliferative responses in the absence or presence of specific C. burnetii antigens or phytohemagglutinin (PHA) were assayed using freshly obtained peripheral blood mononuclear cells. The assay was performed as previously described (35). Stimulation indices (SI) were calculated using the formula SI = mean cpm in antigen- or PHA-treated wells/mean cpm in medium control wells.
Polyacrylamide gel analysis.
Coxiella burnetii phase I and phase II cellular antigens were compared by polyacrylamide gel electrophoresis (PAGE). Briefly, antigens were suspended in sample buffer at a concentration of 1 mg (dry weight)/ml and boiled for 5 min. Antigens were loaded onto a polyacrylamide gel (5% stacking gel, 12.5% separating gel), and bacterial components were separated electrophoretically. Banding patterns were visualized after silver staining (29).
Clinical trial study population.
The vaccine trial was conducted under a protocol approved by the Human Subjects Research Review Board of the U.S. Army Medical Research Institute of Infectious Diseases and the Office of the Surgeon General and reviewed by the Center for Biologics Research and Evaluation of the United States Food and Drug Administration. Healthy young adults of both sexes between the ages of 20 and 27 (mean, 23.5) years old with no evidence of any underlying disease, immune impairment, or history of Q fever and with no immunologic signs of previous Q fever, as measured by the presence of antigen-specific antibodies and lymphoproliferative responses to C. burnetii recall antigens, were selected for entry into this study. By informed consent, 20 volunteers, including 17 males and 3 females, agreed to participate in this dose-escalating and booster trial.
Clinical trial study design.
Each of the volunteer's geometric mean titer baseline values for serum antibody responses and lymphoproliferative responses were established before vaccination. The study was carried out in two phases, the priming dose phase and the booster dose phase. The initial subcutaneous priming dose was 0.3 μg (dry weight) of CMRI vaccine per group, with escalating doses of 3 μg, 15 μg, 30 μg, and 60 μg per inoculation. The subsequent dose was given no sooner than 7 days after the previous group was vaccinated to prevent adverse vaccination reactions in volunteers scheduled to be given higher vaccine doses. Two volunteers received 0.3 μg, two volunteers received 3.0 μg, four volunteers received 15 μg, four volunteers received 30 μg, and eight volunteers were vaccinated with 60 μg of CMRI vaccine. Each volunteer served as his or her own control, with the upper deltoid region of one arm being injected s.c. with diluent while the other arm received the appropriate dose of CMRI vaccine. Arms were selected for injection in a randomized and blinded fashion. The actual dose of vaccine was not blinded, as the initial groups were given the low doses and the doses progressed to higher levels in each successive group. All inoculations were given with 23-gauge, 3/8-inch needles in a volume of 0.5 ml.
Between 3 and 6 months after primary vaccination (Fig. 1), a booster dose of 30 μg was given to each individual in the arm previously inoculated with diluent. Because there were no reactions to the diluent after the priming dose, no diluent was administered to volunteers receiving the booster vaccination. The time between the priming dose and the booster dose inoculations varied between 82 and 160 days. This lack of consistency in administering the booster dose was unavoidable because of volunteer compliance with the dose-escalating aspect of the primary vaccination protocol.
FIG. 1.

Schematic representation of CMRI vaccine priming and booster dose study design. Twenty healthy volunteers with no evidence of exposure to C. burnetii were inoculated with 0.3, 3.0, 15, 30, or 60 μg of CMRI and boosted according to the schedule shown. Blood was drawn for serologic and cellular immune response assays on the days indicated. Numbers signify the day or range of days (d) on which blood was drawn or booster immunization was given and are relative to the primary vaccination (d0).
After primary and booster vaccination, volunteers were examined daily for 7 days, weekly thereafter for 4 weeks, and at 6 weeks. Oral temperatures were taken, and the sites of inoculation were examined. Any erythema or induration present was measured and recorded in square millimeters (length in mm by width in mm). Between days 0 and 42, blood samples were also taken to determine specific antibodies, T-cell proliferative responses, complete blood counts, sedimentation rates, chemistry screening panels, and urinalyses. Serologic and cellular immune response assays were performed 3, 7, 14, 21, 28, or 42 days after the primary dose. The same schedule, with no testing done on day 3, was followed after boosting.
Statistical methods.
The group rise in geometric mean titer was assessed by a general linear model procedure which compared escalating doses against the time after vaccination. The results are expressed as the number of volunteers per group who had at least two consecutive significant increases in the geometric mean titer. The within-subject profile of SI was assessed by SAS Proc GLM repeated-measures analysis of variance (SAS 9.1; SAS Institute, Cary, NC). Group rise is expressed as the number of volunteers per dose who had at least two successive significant rises in the SI, based on the CONTRAST transformation output, which compared successive times to baseline. Significant differences from baseline were determined using t tests with step-down Bonferroni adjustment for multiple comparisons.
RESULTS
Characterization of CMRI vaccine.
The results from tests performed on the final vaccine product are presented in Table 1. The final container had 0.74% moisture, a reconstituted pH of 5.6, a reconstituted osmolality of 392 mosM, and an endotoxin activity by Limulus amoebocyte lysate assay of ≤20 endotoxin units/μg dry weight. Identity and potency testing carried out with mice and guinea pigs indicated that temporal anti-phase II followed by anti-phase I antibodies were induced by the CMRI vaccine and that the CMRI vaccine protected mice against aerosol challenge with phase I C. burnetii (34). Both the antibody response profile and the protective efficacy were characteristic of vaccination with a phase I cellular vaccine (21). Because LPSI is an important protective antigen in C. burnetii vaccines (40), the phase (I or II) of the vaccine was tested by SDS-PAGE (2) to verify the LPSI profile in the CMRI vaccine. This analysis indicated that the CMRI vaccine LPS banding profile was similar to that of phase I chemotypes (Fig. 2). Although a rough-type LPS band was detected (Fig. 2, lane 1) for the CMRI vaccine, the predominant profile was that of LPSI. The ratios of protein to heptose and protein to 3-deoxy-d-manno-2-octulosonic acid (KDO) were used to estimate the proportion of LPSI in the CMRI vaccine (2). This determination indicated that the CMRI vaccine was about 5% (by weight) LPSI. Studies have shown that the percentages of LPSI in CMRI and unextracted phase I whole cells are similar (2).
TABLE 1.
Composition of phase I Henzerling strain CMRI vaccinea
| Compound | Concn (ng/μg dry wt) |
|---|---|
| Total carbohydrate | 76.0 |
| Total nitrogen | 124.7 |
| Nonprotein nitrogen | 2.6 |
| Total protein | 366.7 |
| Heptose | 10.6 |
| Neutral sugar | 81.7 |
| KDOb | 9.5 |
Bulk CMRI vaccine was diluted to a final concentration of 600 μg/ml in 1% lactose solution, lyophilized, and stored at −20°C.
3-Deoxy-d-manno-2-octulosonic acid.
FIG. 2.
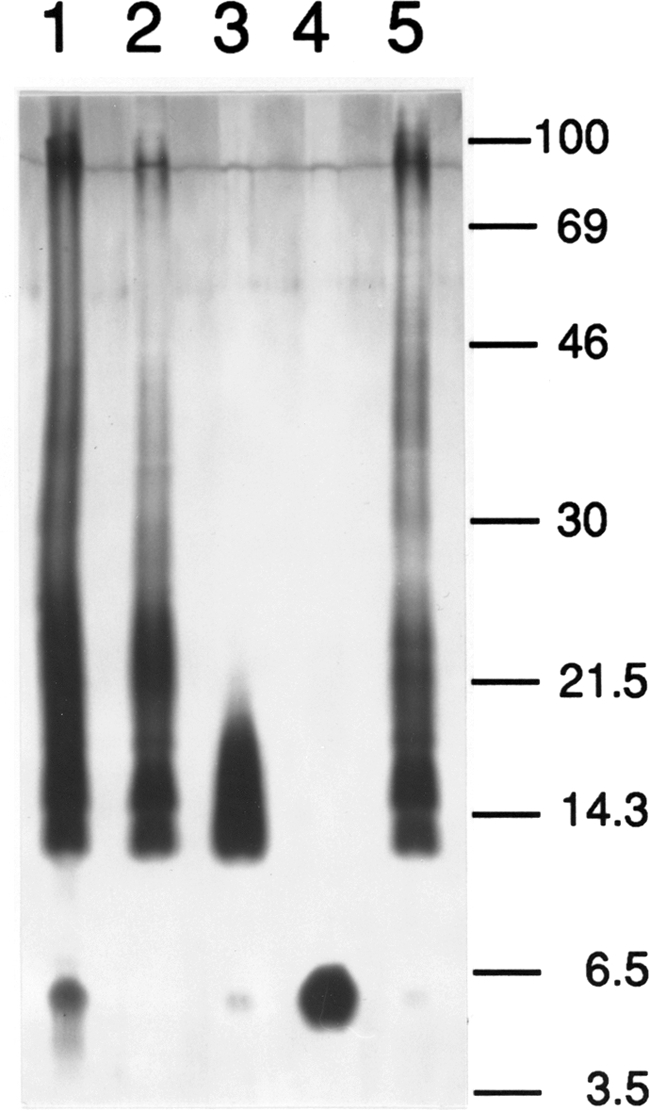
Comparison of LPSs from four strains of C. burnetii by SDS-polyacrylamide gel electrophoresis followed by silver staining. Lane 1, CMRI; lane 2, NMIS; lane 3, NMIISR; lane 4, NMIIR; lane 5, HENIS. Molecular masses are in kDa.
Clinical tolerance of the priming dose.
Fifteen of 20 volunteers receiving primary vaccination doses of 3.0 μg through 60 μg demonstrated a local erythematous response, and 6 volunteers receiving similar doses also had palpable induration after the priming dose (Table 2). The peak responses usually occurred on days 2 and 3. The correlation coefficient comparing the priming dose of vaccine and the area of erythema (0.366; P = 0.11) was not significant (P > 0.05), nor was the correlation coefficient comparing the priming dose and the area of induration (0.247; P = 0.29), which would support a dose-dependent response. No priming dose resulted in a loss of arm function. A single volunteer had a transient fever of 101°F 36 h after inoculation with a 60-μg dose of CMRI vaccine. This volunteer also experienced brief malaise and loss of energy. Recovery was complete within 12 h of the onset of symptoms. No volunteers exhibited significant biochemical or hematological abnormalities attributed to the priming dose of vaccine (data not shown).
TABLE 2.
Local reactions at the inoculation site in 20 volunteers primed with escalating doses of CMRI vaccine and boosted with a single 30-μg dose
| Primary vaccine dose (μg of dry wt) | Day of maximum response (P/B)a | Area (mm2)b
|
|
|---|---|---|---|
| Erythema | Induration | ||
| 0.3 | 2/3 | 0/6,300 | 0/4,200 |
| 3/3 | 0/0 | 0/0 | |
| 3.0 | 2/3 | 500/0 | 225/0 |
| 2/3 | 500/0 | 0/0 | |
| 15.0 | 2/3 | 0/0 | 0/0 |
| 3/3 | 750/60 | 0/0 | |
| 2/1 | 0/460 | 0/0 | |
| 2/3 | 0/0 | 0/0 | |
| 30.0 | 2/2 | 1,400/1,350 | 0/500 |
| 1/2 | 1/100 | 0/0 | |
| 2/2 | 4,500/2,925 | 1,600/1,600 | |
| 3/2 | 1,600/3,300 | 625/980 | |
| 60.0 | 3/3 | 6,000/3,600 | 0/900 |
| 4/2 | 2,000/625 | 0/0 | |
| 2/2 | 9/80 | 0/0 | |
| 1/3 | 1/0 | 0/0 | |
| 2/2 | 9/6,300 | 0/0 | |
| 3/3 | 5,580/8,625 | 1,500/2,475 | |
| 2/3 | 400/0 | 1,200/0 | |
| 3/3 | 4,950/400 | 4,950/625 | |
Day of maximum induration and erythema after primary dose (P) or booster (B).
Area of erythema or induration (mm2) on day of maximum response after primary dose/booster.
Clinical tolerance of the booster dose.
After the 30-μg booster dose, 13 of 20 volunteers had local erythema and 7 volunteers also had palpable induration (Table 2). With one exception, all volunteers who demonstrated induration also had erythema after the priming and booster doses. The reverse was not the case. There was no significant correlation between the priming dose and the local reactions after the booster dose of 30 μg. The correlation coefficient comparing erythema to the priming dose was 0.37 (P = 0.133), and that for induration versus the priming dose was 0.128 (P = 0.589). The peak days of response were days 2 and 3. There was no loss of arm function after the booster dose. There was also no difference in the sizes of local reactions in comparing the priming dose and the booster dose (for erythema, P = 0.867; and for induration, P = 0.590). Reactions in all recipients of the CMRI vaccine were self-resolving, and no reaction persisted for 7 days.
No volunteer had fever or exhibited any systemic response, such as malaise or loss of energy, after the booster dose. No vaccine-related biochemical or hematological abnormalities occurred during the vaccination portion of the study (data not shown).
Humoral immune response to the priming and booster doses.
No priming dose of CMRI induced an increase in specific antibody during the 42-day observation period (data not shown). Boosting after primary doses of 0.3 and 3.0 μg was likewise unable to stimulate rises in mean antibody titers (data not shown), although both recipients primed with 3 μg of CMRI had two successive twofold or greater increases (i.e., fourfold or greater rise) in specific immunoglobulin M (IgM) antibody titers to the NMIIR and NMISR antigens. There were no increases in specific IgA to any recall antigen and no rise in antibody responses to LPSI. The magnitudes of the IgM and IgG antibody responses after boosting were correlated with escalating primary doses. For the 15-μg dose, four of four volunteers had successive increases in specific IgM to the NMIIR antigen and increases in IgG directed against the NMIS, NMIIR, CMRI, and NMISR antigens. For the 30-μg dose, four of four volunteers had at least two successive increases in specific IgM titers to the NMIS and CMRI antigens and in specific IgG titers to NMIS, NMIIR, CMRI, and NMISR antigens. For the 60-μg dose, eight of eight volunteers had at least two successive increases in specific IgM titers to the CMRI antigen and in specific IgG titers to the NMIIR, NMIS, CMRI, and NMISR antigens. Collectively, after booster vaccination, we noted significant increases (P < 0.05) in the IgG antibody responses to CMRI and NMISR in volunteers previously inoculated with doses of 30 μg and 60 μg and in the antibody response to NMIS in volunteers sensitized with 60 μg of CMRI between 14 and 42 days after boosting (Fig. 3).
FIG. 3.

Humoral immune response (IgG) to C. burnetii antigens at various times after booster vaccination with CMRI (expressed as 2n). Mean geometric antibody titers to NMIS, CMRI, and NMISR after a 30-μg (open diamonds, open squares, and open triangles, respectively) or 60-μg (closed diamonds, closed squares, and closed triangles, respectively) primary dose and 30-μg boost were measured before boosting and 7, 14, 21, 28, and 42 days after boosting. Anti-CMRI mean titers measured 14 and 21 days after boosting (30-μg primary dose) were significantly (P < 0.05) elevated from baseline titers. Anti-NMISR mean titers measured 7, 14, 21, 28, and 42 days after boosting (30-μg primary dose) were significantly (P < 0.05) elevated from baseline titers. Anti-NMIS mean titers measured after boosting (30-μg primary dose) were not significantly (P ≥ 0.05) elevated from baseline titers. All mean titers (anti-NMIS, anti-CMRI, and anti-NMISR) measured 14, 21, 28, and 42 days after a 60-μg primary CMRI dose and boosting were significantly elevated (P < 0.05) compared to corresponding baseline (day 0) titers.
Cellular immune response to the priming and booster doses.
The ability of the CMRI vaccine (1 μg/ml and 5 μg/ml) to stimulate sensitized peripheral blood cells in a lymphocyte proliferation assay using either autologous serum or pooled human AB (Phab) serum was investigated. There were no statistically significant differences between the SI for these two concentrations of CMRI vaccine in the test wells. Therefore, in our presentation of the data, the SI for the CMRI vaccine antigen are reported as the mean SI for both 1-μg and 5-μg test wells.
In addition, the ability of the native antigenic structures of NMIS, NMIIR, and HENIS to perform as recall antigens in the presence of autologous or Phab serum was tested at 5 μg/ml. No statistically significant differences between the SI for these three recall antigens were observed during the course of these experiments (data not shown). Therefore, to simplify the presentation of results for these three antigens, the mean SI for these antigens are presented as native antigen results. Moreover, because the results generated using Phab serum were similar to those generated using autologous sera, we chose to present data obtained for peripheral blood cultures incubated in the presence of Phab serum.
After primary vaccination, the magnitude of the cell-mediated immune response, as measured by SI, could be correlated with the vaccination dose (Fig. 4). Mean SI after primary inoculation doses of 0.3 and 3 μg were not significantly different (P > 0.05) from baseline values and are not shown. Responses using native antigen and CMRI were virtually indistinguishable. The time to the maximum proliferative response after administration of the 60- and 30-μg vaccination doses was inversely correlated with the dose, and the maximum proliferative response occurred as early as 7 days after vaccination (60-μg dose).
FIG. 4.

Responses of human peripheral blood monocytes cultured in vitro in the presence of Phab and C. burnetii antigens at various times after vaccination with CMRI. Mean SI were determined for volunteers vaccinated with 15, 30, or 60 μg of CMRI. Peripheral blood monocytes were cultured in the presence of native (open circles, open squares, and x's, respectively) or CMRI (closed circles, closed squares, and plus signs, respectively) antigens.
SI for the native or CMRI recall antigens were significantly (P < 0.05) higher than corresponding baseline values for volunteers given the 15-μg dose and examined 3 to 28 days later and also significantly (P < 0.05) higher on days 7 through 42 after vaccination for volunteers given the 30- and 60-μg doses (except for the day 7 response to native antigen in vaccinees given 30 μg of CMRI). All volunteers in groups vaccinated with 15, 30, or 60 μg of CMRI had at least two successive significant rises in SI after in vitro stimulation of peripheral blood lymphocytes (PBL) with native antigen or CMRI (data not shown). We did not observe depressed proliferative responses to PHA after vaccination, as might be seen if the CMRI vaccine induced a nonspecific immunosuppressive effect (data not shown).
Although the booster dose was given at various times after the primary inoculation, we noted a rise in lymphoproliferative responses to the recall antigens (CMRI and native antigens) for all doses of the CMRI vaccine (Fig. 5). In addition, SI for these recall antigens were significantly (P < 0.05) greater than corresponding baseline values at all vaccine doses for PBL collected on days 7 through 42 after the booster, with the exception of the day 7 response to CMRI in volunteers given the 15-μg dose. While volunteers given the 60-μg primary dose generally exhibited the greatest responses to native antigen in vitro, neither the magnitude of the response nor the time to peak response could be correlated directly with the primary vaccine dose. All volunteers in all vaccine groups tested 7 to 42 days after boosting displayed at least two successive significant rises in SI after in vitro stimulation of PBL with native antigen or CMRI (data not shown).
FIG. 5.

Responses of human peripheral blood monocytes cultured in vitro in the presence of Phab and C. burnetii antigens at various times after booster vaccination with CMRI. Mean SI were determined for volunteers vaccinated with 0.3, 3, 15, 30, or 60 μg of CMRI and boosted with 30 μg of CMRI. Peripheral blood monocytes were cultured with native (open diamonds, open triangles, open circles, open squares, and x's, respectively) or CMRI (closed diamonds, closed triangles, closed circles, closed squares, and plus signs, respectively) antigens.
A consistent pattern of priming at all primary doses was evident from the increases in the in vitro responses of peripheral blood cells to recall antigens after the booster dose. After the booster dose, the magnitudes of the differences between the SI for the native antigens and the CMRI antigen were significant (Fig. 4). Clearly, the native recall antigens were significantly more effective than the CMRI antigen in the induction of in vitro lymphoproliferative responses for all priming doses of the CMRI vaccine following the booster.
DISCUSSION
The development of the CMRI vaccine was undertaken with the goal of vaccinating recipients without the need to prescreen for immunity to Q fever. Subsequently, this phase I clinical trial was conducted to determine if immunization induced measurable specific immune responses against C. burnetii, to assess whether the vaccine induced any detectable adverse reactions at the vaccination site, and to support eventual vaccine licensure. A booster vaccination was incorporated into the study to see if measurable specific immune responses could be improved. To date, 55 volunteers in two separate studies have been inoculated with various doses of the CMRI vaccine.
Prior sensitization of humans by either infection or vaccination has been shown to play a role in the induction of adverse immunopathological reactions after vaccination with phase I C. burnetii cellular vaccines (3, 4, 27). Prescreening recipients by an in vitro serologic assay and an in vivo skin test was shown to be effective at preventing vaccination of individuals with immunity or hypersensitivity to C. burnetii (17). In more recent studies conducted in Australia (19), over 5,000 abattoir workers having no discernible evidence of previous Q fever were safely vaccinated with a phase I cellular vaccine. This suggests that skin testing and serology before vaccination are needed before the phase I cellular vaccine can be administered safely to at-risk populations.
As reported here, a phase I C. burnetii CMR vaccine, formulated only with 1% lactose and rehydrated with bacteriostatic saline, was safely administered s.c. to 20 volunteers in a primary and booster dose regimen. Local reactions of erythema and induration were transient, generally peaking in the majority of recipients by day 2 or 3 and subsiding by day 7. Erythema and induration were not significantly correlated with either the first or second dose. The local reactions were self-limiting. Delayed-onset reactions of erythema, induration, chronic lesions, or abscesses were not observed for any primary or booster dose. Also, significant systemic reactions or limitation of function was not induced by inoculation of the CMRI vaccine. In another CMRI vaccine dose escalation study, similar self-limiting local reactions were observed after the inoculation of volunteers with vaccine doses ranging from 30 μg to 240 μg (12). The lack of induction of significant adverse reactions after the booster dose of the CMRI vaccine suggests that a study needs to be undertaken to further assess the safety of the CMRI vaccine. We are currently evaluating the safety of the CMRI vaccine in skin test-positive volunteers.
In our study, the CMRI vaccine did not induce significant antibody responses when it was administered at doses of 0.3 through 60 μg in a primary vaccination. B cells were not tolerized, however, because boosting volunteers with 30 μg of the CMRI vaccine resulted in the induction of significant antibody responses in individuals primed with 30 or 60 μg of the CMRI vaccine. Thus, significant priming of the immune system to produce antigen-specific antibodies had occurred after the primary inoculation. After the booster, seroconversion was obvious, as recipients who had received primary doses of 15 μg, 30 μg, and 60 μg of the CMRI vaccine generated successive significant rises in titer to C. burnetii antigens.
After the booster, a striking difference was found in evaluating antibody classes of specific antibody responses. Significant increases (P < 0.05) in antigen-specific IgA or IgM responses to any of the antigens were not detected. However, threefold increases in antigen-specific IgG in response to CMRI and NMISR occurred in volunteers who had received the primary inoculation of 30 μg and the booster. Moreover, recipients receiving the 60-μg primary inoculation and the booster had threefold increases in antigen-specific IgG responses to NMIS, CMRI, and NMISR. Although antibody responses to NMIIR (with a phase II cellular antigen with rough-type LPS) were detectable, there were no significant increases above baseline values. This was an unexpected finding, because after natural infection (9, 32) or inoculation of recipients with phase I cellular vaccine (39, 41, 42), antibodies to NMIIR (phase II whole cells) appear before antibodies to NMIS (phase I whole cells). The lack of detection of any antibody to LPSI (a phase I antigen, making up roughly 5% of the dry weight of the CMRI vaccine) was not surprising because this antigen is poorly immunogenic (10) and antibody to LPSI is detected mostly in the sera of patients with chronic Q fever (33). In the absence of the induction of specific antibody to LPSI after the inoculation of volunteers with the CMRI vaccine, the dominant priming antigenic determinants were proteins. Taken collectively, these results suggest that the CMRI vaccine primed B cells to produce antibodies predominately to protein epitopes on the surfaces of phase I whole cells.
In our evaluation of cell-mediated immunity, the 0.3-μg and 3-μg vaccine doses, in contrast to higher doses, were insufficient to activate T cells to proliferate in vitro in the presence of native antigen or CMRI. However, the ability to detect significant in vitro T-cell proliferation after the booster showed that antigen-specific T-cell priming had occurred in vivo after the initial inoculation. The dose escalation of the CMRI vaccine from 0.3 μg to 60 μg effectively primed T cells to be activated to proliferate in vitro by antigenic determinants available on the CMRI antigen and the native C. burnetii antigens (NMIS, NMIIR, and HENIS). For all vaccine doses, the magnitudes of proliferative responses after boosting were greater than those obtained after initial inoculation. Although the 0.3-μg and 3-μg doses effectively primed T cells to become activated by the booster, no corresponding significant increases in antigen-specific antibody were detected in the sera.
The development and assessment of Q fever vaccines have been thwarted by the lack of a suitable laboratory-based correlate of immunity. Recent studies of humoral immunity indicate that vaccination with phase I cellular vaccine, although offering complete protection, induces about 60% seroconversion (12). Dependence on cell-mediated immunity for protection against Q fever suggests that vaccines can be protective even in the absence of detectable levels of serum antibody. To date, vaccine efficacy must be assessed by clinical trials. Our data indicate that peripheral blood cells are primed by an initial exposure to antigen to respond when the antigen is again encountered.
Examination of the effects of native antigens and CMRI antigen on activation of T cells after the booster indicated that native antigens induced significantly greater T-cell proliferation than did the CMRI antigen. Presentation of the antigenic peptides may be more effective with the native surface than with the formalin-treated and chloroform-methanol-extracted CMRI antigen. Alternatively, the CMRI antigen may have selected a population of T-suppressor cells (16) that may be activated when presented in vitro with the same antigen. Still, there were no significant differences between the native recall antigens (NMIS, NMIIR, and HENIS) in the ability to stimulate lymphoproliferation in vitro. The native surfaces of phase I whole cells and phase II whole cells were equally effective at activating T cells to proliferate. Therefore, we propose that the CMRI vaccine antigen carries the appropriate protein antigens to prime the host to rapidly respond to subsequent C. burnetii challenges.
Acknowledgments
We thank David Bunner and the staff of Medical Division, USAMRIID, for recruiting human volunteers needed for this study and providing clinical monitoring after vaccination and boosting. Gene Nelson of the Biometrics and Information Management Division, USAMRIID, provided the statistical analysis.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army.
This effort was funded by Defense Threat Reduction Agency project number 5.10023_05_RD_B.
In the conduct of research where humans were the subjects, the investigators adhered to the policies regarding the protection of human subjects prescribed by 45 CFR 46 and 32 CFR 219 (Protection of Human Subjects).
Footnotes
Published ahead of print on 13 August 2008.
REFERENCES
- 1.Ackland, J. R., D. A. Worswick, and B. P. Marmion. 1994. Vaccine prophylaxis of Q fever. A follow-up study of the efficacy of Q-Vax (CSL) 1985-1990. Med. J. Aust. 160:704-708. [PubMed] [Google Scholar]
- 2.Amano, K., and J. C. Williams. 1984. Chemical and immunological characterization of lipopolysaccharides from phase I and phase II Coxiella burnetii. J. Bacteriol. 160:994-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell, J. F., D. B. Lackman, A. Meis, and W. J. Hadlow. 1964. Recurrent reaction of site of Q fever vaccination in a sensitized person. Mil. Med. 129:591-595. [PubMed] [Google Scholar]
- 4.Benenson, A. S. 1959. Q fever vaccine: efficacy and present status, p. 47-60. In J. E. Smadel (ed.), Symposium on Q fever. Walter Reed Army Institute of Medical Science publication no. 6. Government Printing Office, Washington, DC.
- 5.Brezina, R., S. Schramek, J. Kazar, and J. Urovolgyi. 1974. Q fever chemovaccine for human use. Acta Virol. 18:269. [Google Scholar]
- 6.Brooks, D. L., R. W. Ermel, C. E. Franti, R. Ruppanner, D. E. Behymer, J. C. Williams, E. H. Stephenson, and J. C. Stephenson. 1986. Q fever vaccination of sheep: challenge of immunity in ewes. Am. J. Vet. Res. 47:1235-1238. [PubMed] [Google Scholar]
- 7.Burnet, F. M., and M. Freeman. 1937. Experimental studies on the virus of “Q” fever. Med. J. Aust. 2:299-305. [DOI] [PubMed] [Google Scholar]
- 8.Derrick, E. H. 1937. Q fever, a new fever entity: clinical features, diagnosis and laboratory investigation. Med. J. Aust. 2:281-299. [DOI] [PubMed] [Google Scholar]
- 9.Dupuis, G., O. Peter, M. Peacock, W. Burgdorfer, and E. Haller. 1985. Immunoglobulin responses in acute Q fever. J. Clin. Microbiol. 22:484-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Embil, J., J. C. Williams, and T. J. Marrie. 1990. The immune response in a cat-related outbreak of Q fever as measured by the indirect immunofluorescence test and the enzyme-linked immunosorbent assay. Can. J. Microbiol. 36:292-296. [DOI] [PubMed] [Google Scholar]
- 11.Fiset, P. 1966. Vaccination against Q fever, p. 528-531. In Vaccines against viral and rickettsial diseases in man. PAHO science publication number 147. Pan American Health Organization, Washington, DC.
- 12.Fries, L. F., D. M. Waag, and J. C. Williams. 1993. Safety and immunogenicity in human volunteers of a chloroform-methanol residue vaccine for Q fever. Infect. Immun. 61:1251-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genig, V. A. 1965. Experience in mass immunization of humans with the M-44 live vaccine against Q-fever. 2. Skin and oral routes of immunization. Vopr. Virusol. 10:703-707. [PubMed] [Google Scholar]
- 14.Johnson, J. W., G. A. Eddy, and C. E. Pedersen, Jr. 1976. Biological properties of the M-44 strain of Coxiella burnetii. J. Infect. Dis. 133:334-338. [DOI] [PubMed] [Google Scholar]
- 15.Kishimoto, R. A., J. C. Gonder, J. W. Johnson, J. A. Reynolds, and E. W. Larson. 1981. Evaluation of a killed phase I Coxiella burnetii vaccine in cynomolgus monkeys (Macaca fascicularis). Lab. Anim. Sci. 31:48-51. [PubMed] [Google Scholar]
- 16.Koster, F. T., J. C. Williams, and J. S. Goodwin. 1985. Cellular immunity in Q fever: modulation of responsiveness by a suppressor T cell-monocyte circuit. J. Immunol. 135:1067-1072. [PubMed] [Google Scholar]
- 17.Lackman, D., E. J. Bell, J. F. Bell, and E. Pickens. 1962. Intradermal sensitivity testing in man with a purified vaccine for Q fever. Am. J. Public Health 52:87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luoto, L., J. F. Bell, M. L. Casey, and D. B. Lackman. 1963. Q fever vaccination of human volunteers. I. The serologic and skin test response following subcutaneous injections. Am. J. Hyg. 78:1-15. [PubMed] [Google Scholar]
- 19.Ormsbee, R., and B. Marmion. 1990. Prevention of Coxiella burnetii infection: vaccines and guidelines for those at risk, p. 225-248. In T. Marrie (ed.), Q fever. The disease, vol. I. CRC Press, Boca Raton, FL. [Google Scholar]
- 20.Ormsbee, R. A., E. J. Bell, and D. B. Lackman. 1962. Antigens of Coxiella burnetii. I. Extraction of antigens with non-aqueous organic solvents. J. Immunol. 88:741-749. [PubMed] [Google Scholar]
- 21.Ormsbee, R. A., E. J. Bell, D. B. Lackman, and G. Tallent. 1964. The influence of phase on the protective potency of Q fever vaccine. J. Immunol. 92:404-412. [PubMed] [Google Scholar]
- 22.Pollok, N. L. 1965. Mixed infections of staphylococcal enterotoxin B, VEE, and Q fever in Macaca mullata monkeys, p. 19. Technical manuscript no. 237. U.S. Army Laboratories, Fort Detrick, MD.
- 23.Pollok, N. L. 1962. Mixed infections of Venezuelan equine encephalomyelitis and Q fever in Macaca mullata monkeys, p. 26. Technical memorandum no. 28. U.S. Army Biological Laboratories, Ft. Detrick, MD.
- 24.Scott, G. H., T. F. McCaul, and J. C. Williams. 1989. Inactivation of Coxiella burnetii by gamma irradiation. J. Gen. Microbiol. 135:3263-3270. [DOI] [PubMed] [Google Scholar]
- 25.Smadel, J. E., M. J. Snyder, and F. C. Robins. 1948. Vaccination against Q fever. Am. J. Hyg. 47:71-78. [DOI] [PubMed] [Google Scholar]
- 26.Spicer, D. S., and A. N. DeSanctis. 1976. Preparation of phase 1 Q fever antigen suitable for vaccine use. Appl. Environ. Microbiol. 32:85-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoker, M. G. P. 1957. Q fever down the drain. Br. Med. J. 1:425-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tigertt, W. D. 1959. Studies on Q fever in man, p. 39. In J. E. Smadel (ed.), Symposium on Q fever, vol. 6. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- 29.Tsai, C. M., and C. E. Frasch. 1982. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119:115-119. [DOI] [PubMed] [Google Scholar]
- 30.Uhaa, I. J., D. B. Fishbein, J. G. Olson, C. C. Rives, D. M. Waag, and J. C. Williams. 1994. Evaluation of specificity of indirect enzyme-linked immunosorbent assay for diagnosis of human Q fever. J. Clin. Microbiol. 32:1560-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vivona, S., J. P. Lowenthal, S. Berman, A. S. Benenson, and J. E. Smadel. 1964. Report of a field study with Q fever vaccine. Am. J. Hyg. 79:143-153. [DOI] [PubMed] [Google Scholar]
- 32.Waag, D., J. Chulay, T. Marrie, M. England, and J. Williams. 1995. Validation of an enzyme immunoassay for serodiagnosis of acute Q fever. Eur. J. Clin. Microbiol. Infect. Dis. 14:421-427. [DOI] [PubMed] [Google Scholar]
- 33.Waag, D. M., C. R. Bolt, T. J. Marrie, and J. C. Williams. 1991. Specific responses of humans to Coxiella burnetii: specific antibody and cell-mediated responses after vaccination or infection, p. 157-173. In J. C. Williams and H. A. Thompson (ed.), Q fever: the biology of Coxiella burnetii. CRC Press, Boca Raton, FL.
- 34.Waag, D. M., M. J. England, and M. L. Pitt. 1997. Comparative efficacy of a Coxiella burnetii chloroform:methanol residue (CMR) vaccine and a licensed cellular vaccine (Q-Vax) in rodents challenged by aerosol. Vaccine 15:1779-1783. [DOI] [PubMed] [Google Scholar]
- 35.Waag, D. M., K. T. McKee, Jr., G. Sandstrom, L. L. Pratt, C. R. Bolt, M. J. England, G. O. Nelson, and J. C. Williams. 1995. Cell-mediated and humoral immune responses after vaccination of human volunteers with the live vaccine strain of Francisella tularensis. Clin. Diagn. Lab Immunol. 2:143-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wachter, R. F., G. P. Briggs, and C. E. Pedersen, Jr. 1978. Enhancement of the immunogenicity of phase I antigen of Coxiella burnetii. Infect. Immun. 22:627-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilhelmsen, C. L., and D. M. Waag. 2000. Guinea pig abscess/hypersensitivity model for study of adverse vaccination reactions induced by use of Q fever vaccines. Comp. Med. 50:374-378. [PubMed] [Google Scholar]
- 38.Williams, J. C., and J. L. Cantrell. 1982. Biological and immunological properties of Coxiella burnetii vaccines in C57BL/10ScN endotoxin-nonresponder mice. Infect. Immun. 35:1091-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams, J. C., T. A. Damrow, D. M. Waag, and K. Amano. 1986. Characterization of a phase I Coxiella burnetii chloroform-methanol residue vaccine that induces active immunity against Q fever in C57BL/10 ScN mice. Infect. Immun. 51:851-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams, J. C., T. A. Hoover, D. M. Waag, N. Banerjee-Bhatnagar, C. R. Bolt, and G. H. Scott. 1990. Antigenic structure of Coxiella burnetii. A comparison of lipopolysaccharide and protein antigens as vaccines against Q fever. Ann. N. Y. Acad. Sci. 590:370-380. [DOI] [PubMed] [Google Scholar]
- 41.Williams, J. C., M. G. Peacock, and T. F. McCaul. 1981. Immunological and biological characterization of Coxiella burnetii, phases I and II, separated from host components. Infect. Immun. 32:840-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams, J. C., M. G. Peacock, D. M. Waag, G. Kent, M. J. England, G. Nelson, and E. H. Stephenson. 1992. Vaccines against coxiellosis and Q fever. Development of a chloroform:methanol residue subunit of phase I Coxiella burnetii for the immunization of animals. Ann. N. Y. Acad. Sci. 653:88-111. [DOI] [PubMed] [Google Scholar]
- 43.Williams, J. C., L. A. Thomas, and M. G. Peacock. 1986. Identification of phase-specific antigenic fractions of Coxiella burnetii by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 24:929-934. [DOI] [PMC free article] [PubMed] [Google Scholar]