

Abstract
The mechanistic relationship between the widely used monocrotaline model of primary pulmonary hypertension and altered TGFβ family signaling due to genetic defects in the Bone Morphogenetic Protein type II receptor in affected humans has not been investigated. In this study we use fluorescent microscopy to demonstrate nuclear translocation of Smad 4 in human pulmonary arterial endothelial cell (HPAEC) cultures treated with monocrotaline pyrrole (MCTP), Bone Morphogenetic Protein (BMP) and TGFβ. While MCTP induced transient nuclear accumulation of phosphorylated Smad 1 (P-Smad 1) and phosphylated Smad 2 (P-Smad 2), only expression of P-Smad 1 was significantly altered in western blots. P-Smad 1 expression significantly increased 30 minutes following treatment with MCTP correlating with P-Smad 1 and Smad 4 nuclear translocation. Although a modest, but significant decrease in P-Smad 1 expression occurred 1 hr after treatment, expression was significantly increased at 72 hr. Evaluation of components of the signal and response pathway at 72 hours showed decreased expression of the BMP type II receptor (BMPrII), no change in TGFβ Activin Receptor-like Kinase 1 (Alk 1), no change in Smad 4 but increase in the inhibitory Smad 6, decrease in the alternate BMP signaling pathway p38MAPK but no change in the psmad1 response element ID 1. Our results suggest transient activation of Smad signaling pathways in initial MCTP endothelial cell toxicity, and a persistent dysregulation of BMP signaling. Electron microscopy of cell membrane caveoli revealed a dramatic decrease in these structures after 72 hrs. Loss of these structural elements, noted for their sequestration and inhibition of receptor activity, may contribute to prolonged alterations in BMP signaling.
Introduction
The TGFβ superfamily, comprised of TGFβs, activins, and the bone morphogenetic proteins (BMPs), has critical roles in embryonic development and maintenance of the vascular system. Signal transduction within the TGFβ superfamily is initiated through the formation of a heteromeric complex upon ligand stimulation, composed of type I and type II receptors, with the subsequent phosphorylation of Smad secondary messengers (Moustakas, Lin et al. 1993). BMP receptor activated Smads are Smads 1, 5, and 8, while Smads 2 and 3 are restricted to the TGFβ pathway (Nakao, Imamura et al. 1997; Miyazono 1999; Mehra, Attisano et al. 2000; Wrana and Attisano 2000). Phosphorylated Smads from either pathway form a signaling complex with the common Smad 4 which translocates into the nucleus to DNA binding sites. Gene expression varies with cell type, ligand dose and length of exposure, and is further dependent on the recruitment and interaction with transcription cofactors, which are also highly regulated. In addition, preferential signaling through MAP kinases may be designated by alternative receptor oligomerization patterns (Nohe, Hassel et al. 2002; Derynck and Zhang 2003; de Caestecker 2004; Nohe, Keating et al. 2004). Understanding of these signaling pathways within the vasculature has been enhanced by the creation of mice null for receptors and Smads, which have defective pulmonary development and vasculogenesis of distinct phenotypes (Mehra and Wrana 2002; Lebrin, Deckers et al. 2005).
TGFβ cytokine and receptor expression has been extensively studied in the context of pulmonary hypertension (PH), a disease that is characterized by vascular remodeling and endothelial cell injury. TGFβ, normally growth restrictive in the vasculature, is invariably up-regulated in PH, and expression is marked within endothelial lesions (Botney, Bahadori et al. 1994; Voelkel and Tuder 1995). TGFβ has both pro- and anti-angiogenic properties that only recently were shown to be mediated through different type I receptors that vary as to cell population. Endothelial cells within the pulmonary vasculature express both receptor types, Alk1 (pro-angiogenic) and Alk5 (anti-angiogenic) (Goumans, Valdimarsdottir et al. 2002; Lebrin, Deckers et al. 2005). Mutations in TGFβ family receptors have been described in idiopathic forms of PH (Yeager, Halley et al. 2001; van den Driesche, Mummery et al. 2003).
More recent interest has focused on the specific role BMP signal transduction has in the pulmonary vasculature system with the discovery that polymorphic germline mutations in the BMP type II receptor (BMPrII) are present in familial PH (Deng, Morse et al. 2000; Machado, Pauciulo et al. 2001; Newman, Wheeler et al. 2001). Patients have reduced expression of this receptor. Although it is not known how mutations contribute to the pathogenesis (Atkinson, Stewart et al. 2002), we are beginning to elucidate the function of BMP signal transduction within the vasculature. While most studies have focused on smooth muscle response to BMP cytokines, we have demonstrated that endothelial cells abundantly express BMPrII (Ramos, Lame et al. 2006), suggesting a role for these mutations within the endothelium in the pathogenesis of PH.
The monocrotaline pyrrole (MCTP) model of PH is well established. MCTP is a pyrrole derivative of a pyrrizolidine alkaloid and an alkylating agent that forms DNA and protein adducts ultimately leading to cell cycle arrest (Thomas, Lame et al. 1996; Wilson, Lame et al. 1998; Mathew, Huang et al. 2004; Shah, Patel et al. 2005). Altered expression of TGFβ ligand isoforms (Botney, Bahadori et al. 1994; Tanaka, Bernstein et al. 1996) and transcripts (Arcot, Lipke et al. 1993) have been reported for MCTP PH. We were interested in examining the effects of MCTP on Smad signal transduction pathways, and hypothesized that Smad signal transduction pathways are activated in MCTP endothelial injury. Based on previous demonstrations of persistent alterations in endothelial cell cycle associated with binding of MCT metabolites to macromolecules (Thomas, Lame et al. 1996), we also evaluated effects of MCTP treatment on components of the BMP signal at later timepoints.
In this study we show that MCTP induces transient nuclear accumulation of Smad 4, P-Smad 1 and P-Smad 2 in HPAEC, suggesting activation of both TGFβ and BMP signaling, but induces altered signaling after prolonged treatment suggesting dysregulation in BMP pathways. Structural loss of cell membrane caveolae over a corresponding time period by MCTP treatment may contribute to BMP signal dysfunction.
Materials and Methods
Materials
HPAEC from a five year old female and supporting media were obtained from Clonetics (San Diego, CA). HEK293H cells and supporting media Dulbecco’s modified eagle medium (DMEM) were obtained from Gibco BRL (Rockville, MD). Antibodies to Smad 4 used in western blot (cat. #7966) and immunocytochemistry (ICC) (cat. #7154) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to Cav-1 was obtained from BD Transduction Laboratories (San Diego, CA). Antibodies to p-Smad-1 were obtained from both Upstate (Lake Placid, NY) and Cell signaling (Danvers, MA), the later for the 30 min western analysis, p-Smad-2 (Chemicon International, Temecula, CA); those used in cytology were obtained from Cell Signaling Technology (Beverly,MA). TGFβ1 was obtained from PeproTech Inc (Rocky Hill, NJ) while BMP2 and BMP4 were obtained from R&D Systems (Minneapolis, MN).
Rabbit antibody to BMPrII was generated in our laboratory according to previously described methods (Gullick, Downward et al. 1985; Rosenzweig, Imamura et al. 1995). Briefly, a peptide corresponding to amino acid sequence 185–202 of human BMPrII was coupled to keyhole limpet hemocyanin. Male rabbits were immunized according to standard methods, and the presence of antibody verified by an Elisa assay. Rabbit serum was purified in two successive steps on keyhole limpet hemocyanin agarose (Alpha Diagnostics International, Inc; San Antonio, TX) and thiophilic gel (Pierce) using manufactures’ suggested protocol. Final protein concentration was 7.4 ug/ul.
Alexa 488 fluorescent-tagged secondary antibodies were obtained from Molecular Probes (Eugene, OR). ECL detection kit and HRP conjugated antibodies (anti-rabbit and anti-mouse) used in western analysis were acquired from Amersham Biosciences (UK). Additional supplies include Superblock (Pierce) and PermaFluor mounting media (Immunon; Pittsburgh, PA).
Moncrotaline (MCT; Trans World Chemicals, Rockville. MD) was converted to monocrotaline pyrrole (MCTP) using tetrabromo-1,2-benzoquinone (Aldrich, Milwaukee, WI) (Mattocks, Jukes et al. 1989). The product was recrystallized from hexane:diethyl ether and stored in N,N-dimethylformamide (DMF) at −80°C prior to use.
Cell Culture
HPAEC (passage 6–9) used in western analysis were grown to confluence in 175 cm² flasks in supplemented media. HPAEC and HEK293H cells (passage 4) used in immunocytochemistry (ICC) were grown on fibronectin-coated coverslips to 60–90% confluence.
Immunocytochemistry of Smad nuclear translocation
Supplemented media was replaced with basal media and allowed to equilibrate for 1 hour prior to treatments. Treatments consisted of 60ug MCTP/1 ml of media and vehicle controls, 1uL DMF/ml of media. Positive controls included 100 ng/ml BMP4, 300 ng/ml BMP2, or 20ng/ml TGFβ1. Superblock containing 0.05% saponin was used for initial blocking of non-specific antigens and for primary and secondary antibody incubations; a 10% dilution was used in all washes. At specific time intervals the media was aspirated and replaced with 1% PFM at room temperature for 30 minutes. Cells were rinsed twice with PBS, blocked for 30 minutes, followed by incubation with the primary antibody at 1:100 for 1 hour (Smad 4) or overnight at 4°C (P-Smad 1 and P-Smad 2). Cells were washed 5x, followed by incubation with the secondary antibody (1.5 ul/ml) for 30 minutes. Cells were washed again 5x, fixed 10 minutes in 1% PFM, washed 2x in PBS, and finally mounted on slides with PermaFluor. Slides were examined on a Provis 1X70 fluorescent microscope (Olympus; New Hyde Park, NY) and images were digitally captured with AxioCam software (Carl Zeiss Inc; Thornwood, NY). For quantification 3 replicates per treatment (MCTP, DMF vehicle, TGFβ2, BMP2, BMP4, and non-treated control) were used to score HPAEC cells for nuclear translocation of Smad 4. Ten fields were randomly selected at 60X, and cells containing strong nuclear signal were counted and compared to total cells present per field. Significance of treatment effect was determined by two-tail hypothesis testing at p< 0.05. Cells were counted as positive for nuclear translocation when the fluorescent signal was predominantly within the nucleus. Cells containing fluorescent signal predominantly in the cytoplasm or evenly distributed throughout both the nucleus and cytoplasm were counted as negative for translocation. Baseline nuclear translocation of Smad 4 was generally less than 10% in HPAEC.
Construction of GFP-SMAD-4 plasmid and transfection in 293-H cells
The method in brief is as follows: cDNA derived from m-RNA was obtained from HPAEC’s initially extracted with TRIzol (according to manufactures recommendations, Invitrogen; Carlsbad, CA). The aqueous layer containing the total RNA was further purified using an RNeasy kit (Qiagen); mRNA was then extracted using an Oligotex mRNA kit (Qiagen) and converted to c-DNA using a high capacity cDNA archive kit (Applied Biosystems). c-DNA was amplified using forward primer 5′-ATG-GAC-AAT-ATG-TCT-ATT-ACG-AAT-ACA-CCA-3′, reverse primer 5′-TCA-GTC-TAA-AGG-TTG-TGG-GTC-TG-3′, and Expanded Long Template PCR Taq (buffer system1) (Roche). The PCR product was separated on a 1% agarose gel and the band of appropriate mass extracted with S.N.A.P. gel purification kit (Invitrogen). Material was then inserted into a pcDNA3.1/NT-GFP-TOPO vector (Invitrogen) followed by transformation of chemically competent E. coli. Positive clones were identified by PCR, expanded and the plasmid extracted using QIAfilter plasmid kit (Qiagen). Sequencing of the resulting construct revealed no deviations from the reported sequence (NCBI accession number NM_005359). The plasmid was then tested for the actual fusion protein by expression in HEK 293-H cells transfected in the presence of Lipofectamine 2000 (Invitrogene) according to manufacture’s recommendation. Cells were monitored for GFP production with fluorescent microscopy, extracted with RIPA 72 hours post transfection, and the lysate separated by SDS-PAGE, blotted to PVDF and the membrane probed with rabbit anti-GFP (Invitrogen) (1/5000 dilution), secondary donkey anti-rabbit conjugated with HRP (1/5000 dilution). Control cells, plasmid absent, were negative on western blot for a band at ~ 100 kDa that reacted with the GFP antibody while HEK 293-H cells containing the plasmid produced a robust band at this mass. Development of western blots with either Rabbit anti-Smad 4 (1/200 dilution, #sc-7154, Santa Cruz) or mouse anti-Smad 4 (1/400 dilution, #sc-7466, Santa Cruz) and the corresponding HRP conjugated secondary (1/5000), donkey anti-rabbit or sheep anti-mouse (Amersham Pharmacia, Piscataway, NJ) produced a corresponding band.
Nuclear translocation of GFP-SMAD-4 after MCTP exposure
HEK 293-H cells were grown to 100% confluence on human fibronectin coated 22 mm square cover slips. Cells were then transfected by using 4 μg of plasmid and 10 μL of Lipofectamine 2000 in a total volume of 2.5 mls. Cells were allowed to equilibrate overnight followed by a media change. After 24 hr cells were exposed to either 60 μg/ml MCTP or DMF (1 μL/1mL) in 2 mls of DMEM with 5 % FBS. Media was then removed and the cells fixed in isotonic 1% PFM for 1 hr before mounting with PermaFluor and subsequent observation by fluorescent microscopy. Digital images were analyzed for nuclear pixel intensity (Lehr, Mankoff et al. 1997).
Western Blot analysis
Confluent cell cultures were harvested and lysed in RIPA buffers containing protease inhibitors at 4°C. Lysates were loaded on 4% stacking/11% running SDS gels at 50–100 ug of protein per lane, separated and then transferred to PVDF membranes (Bio-Rad Laboratories; Hercules, Ca). Membranes were blocked with 5% non-fat milk in TBS containing 0.05% Tween-20, and incubated overnight at 4°C with the primary antibodies at 1:1000. Bands were identified through enhanced chemiluminescence using the ECL detection system to HRP-conjugated secondary antibodies used at 1:10,000. Scanned images were inverted and quantified using ImageQuant software. Samples were considered statistically significant at p< 0.05, by two-tail hypothesis testing.
Electron Microscopy (EM) of Cell Membrane Caveoli
HPAEC were grown on LabTek 8-well permanox chamber slides (Nalge Nunc International Corp; Naperville, IL) until confluent, treated with 60 μg/ml of MCTP or an equal volume of DMF vehicle in basal media, which was exchanged for supplemented media following 2 hr of treatment. Duplicate slides were made for each time point, in addition to non-treated controls. Cultures were maintained for 24, 48, or 72 hr, followed by fixation in Karnovsky’s solution until processed. Slides were treated with OsO4, dehydrated through a graded series of alcohols and embedded in acrylic resin; ultra-thin sections were cut and grids stained with uranyl acetate and lead citrate (Venable and Coggeshall 1965). Grids were examined on a Phillips EM 400 or 120 TEM (Hillsboro, OR) at 20,000x magnification, and images captured on film.
Results
MCTP induces nuclear translocation of Smad 4 in HPAEC
Members of the TGFβ family of receptors signal via the formation of complexes, incorporating the common second messenger Smad 4 which then alters its normal flux between cytoplasm and nucleus in favor of nuclear accumulation. Smad 4 was generally observed within the cytoplasm of unstimulated cells (fig. 1, A). MCTP consistently induced marked nuclear translocation of Smad 4 in HPAEC within 15 min (fig. 1, B), and generally peaked at 30–45 min, observed as intense fluorescent green nuclei (fig. 1, C). In the results shown, translocation ranged from 33–41% of cells. In comparison, baseline levels were less than 10%. Nuclear signal generally diminished 1–2 hours post-treatment to basal levels (fig. 1, D). At 72 hr Smad 4 was predominantly cytoplasmic, similar to vehicle treated controls (results not shown).
Figure 1.
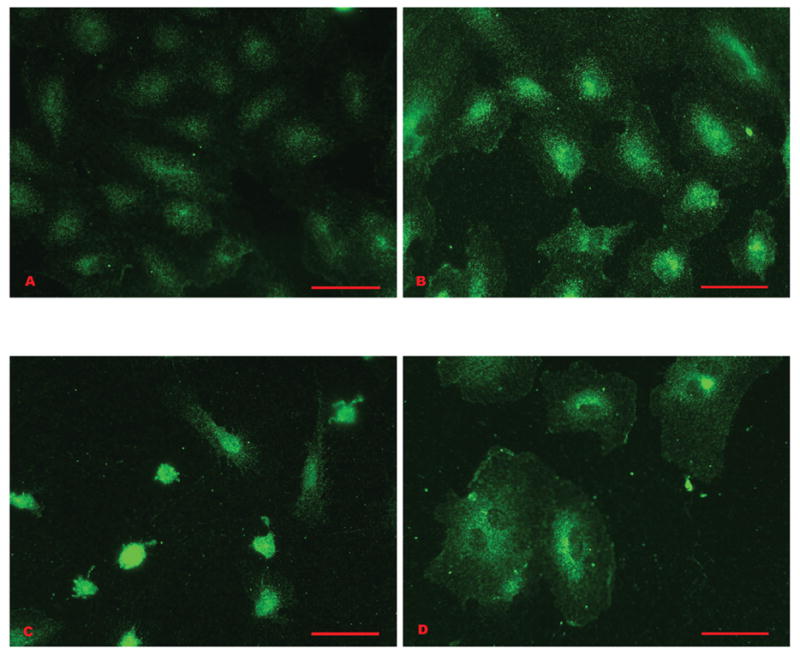
Smad 4 nuclear translocation in HPAEC treated with 60 μg/ml MCTP: A) Non-treated control cells. Smad 4 is represented by fluorescent label that is scattered predominantly throughout the cytoplasm. The nuclei appear as open vacuoles, containing little signal. B) Cells treated 15 min. There is an increase in fluorescence around and within the nuclei, while the peripheral cytoplasm contains scattered fluorescent signal. C) Cells treated 30 min. Smad 4 is concentrated within the nuclei, which appear as intensely fluorescent green balls. D) Cells treated for 60 min. The signal is predominantly cytoplasmic in the majority of the cells. Bars = 50 μm.
In contrast BMP2, BMP4, and TGFβ ligands obtained from commercial sources induced modest translocation that was generally less striking, and frequently ambiguous, compared with that of MCTP (fig. 2). Nuclear translocation was generally higher in BMP4 treated cells (fig. 2, A) compared to BMP2 or TGFβ (fig. 2 B and C, respectively). Based on prior reports of peak times for ligand-induced Smad phosphorylation/translocation (Valdimarsdottir, Goumans et al. 2002; Yang, Long et al. 2005; Yang, Long et al. 2005), we performed counts of cellular responses (fig 2 D) which show significant (p< 0.001) increase in translocation for all groups at 1 hour post treatment. There was also modest translocation induced in cells that were vehicle DMF treated.
Figure 2.
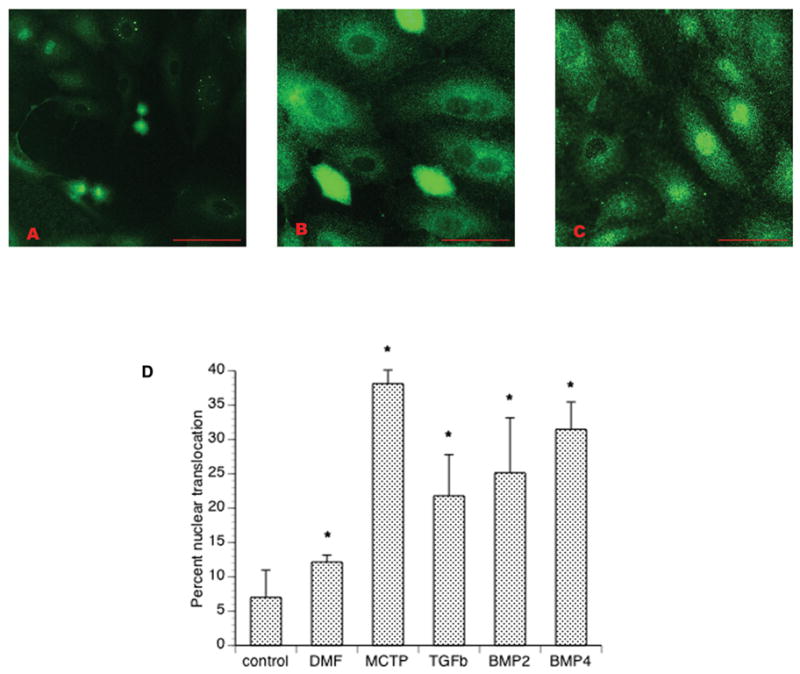
Smad 4 nuclear translocation in HPAEC treated for 1 hour with recombinant ligands: A) Cells treated with 100 ng/ml BMP4. B) Cells treated with 300 ng/ml BMP2. C) Cells treated with 20 ng/ml TGFβ-1. D) Results of nuclear counts show intense nuclear signal was less frequently observed in ligand-treated cells than cells treated with MCTP. Bars = 50 μm.
As an alternative approach evaluating the selectivity of our immunologic probe and the specificity of endothelial cells in these responses we evaluated translocation of both endogenous Smad 4 and GFP–Smad 4 constructs in HEK293H cells. Using antibody probes, baseline nuclear accumulation of endogenous Smad 4 in these cells was 60%, but significantly increased to 89 % (p< 0.001) after 1 hour treatment with MCTP (fig. 3, A and B, respectively). In comparison nuclear translocation of endogenous Smad 4 in HEK293H cells treated with BMP4 (fig. 3, C) was 84% (p< 0.001). To quantify translocation of GFP-Smad 4 we used digital images and photoshop-based image analysis to determine nuclear pixel intensity of GFP fluorescence (Lehr, Mankoff et al. 1997). Pixel intensity, correlated to GFP-Smad 4 nuclear translocation, was significantly increased (p= 0.018) in cells treated with MCTP for 1 hour, further validating this assay (fig. 4).
Figure 3.
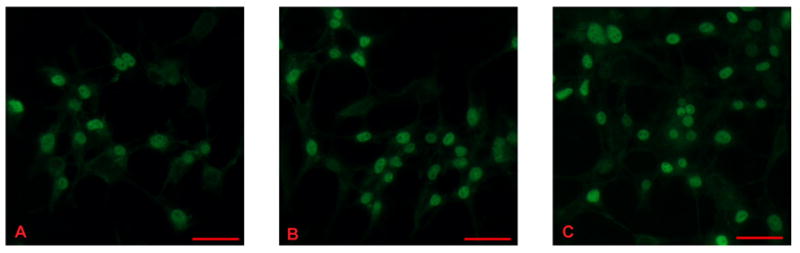
Translocation of endogenous Smad 4 in HEK293H cells in cells treated with MCTP or BMP4 1 hr: A) Non-treated cells. B) Cells treated with 60 μg/ml MCTP. C) Cells treated with 100 ng/ml BMP4. Nuclear translocation was greater in treated cells. Bars = 50 μm.
Figure 4.
Translocation of transfected GFP-Smad 4 in HEK293H cells treated with 60μg/ml MCTP: A) Non-treated cells, and B) Cells treated for 1 hr. Bars = 50 μm. Images were converted to grey-scale, and qualitative analysis of nuclear translocation C) was based on pixel intensity, which was greater in MCTP-treated cells (* p<.05).
Because Smad 4 may translocate to the nucleus independent of receptor-activated Smads, we also evaluated the nuclear accumulation of P-Smad 1 (fig. 5) and P-Smad 2 (not shown) by fluorescent microscopy. Enhanced nuclear accumulation of phosphorylated Smads would suggest direct receptor activation and subsequent phosphorylation of the receptor activated Smads was occurring (Inman, Nicolas et al. 2002; de Caestecker 2004). Peak nuclear signal occurred 15–30 min after treatment, suggesting receptor activation of these Smads. The similarity in response suggests activation of receptors in both BMP and TGFβ pathways. Similar to Smad 4, nuclear accumulation of both P-Smad 1 and P-Smad 2 diminished after one hr.
Figure 5.
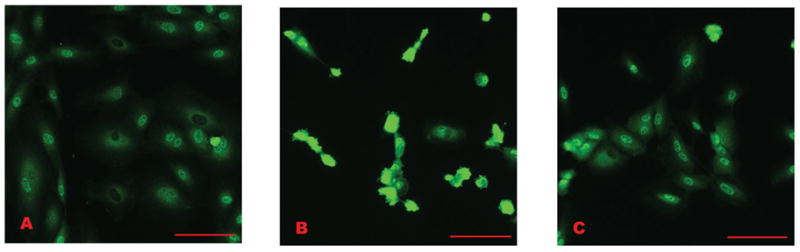
Nuclear translocation of P-Smad 1 in HPAEC treated with MCTP: A) Non-treated control cells. Signal is scattered throughout the cytoplasm, and is present within the nuclei of most cells. B) Cells treated 30 min with MCTP. In contrast to the cells pictured in “A”, the intensity of the signal is concentrated within the nuclei of most cells present. C) Cells after 60 min. Nuclear signal has diminished. (Similar results were obtained with P-Smad 2.) Bars = 50 μm.
Western Blot Analysis of MCTP Activation of Smad Signaling
Smad Phosphorylation
To further validate receptor activation of Smad signaling, we examined confluent cultures of HPAEC treated with MCTP at 3 time points by western blot analysis for changes in phosphorylation of Smad 1 and Smad 2 (fig. 6, A&B). At 30 minutes, correlating with peak nuclear translocation, P-Smad 1 expression was significantly increased (p= 0.008). At 1 hour, P-Smad 1 was decreased in MCTP treated cells (p= 0.05), however expression of P-Smad 1 was significantly increased at 72 hours (p= 0.01). In contrast there was no significant change in P-Smad 2 expression at all time points.
Figure 6.
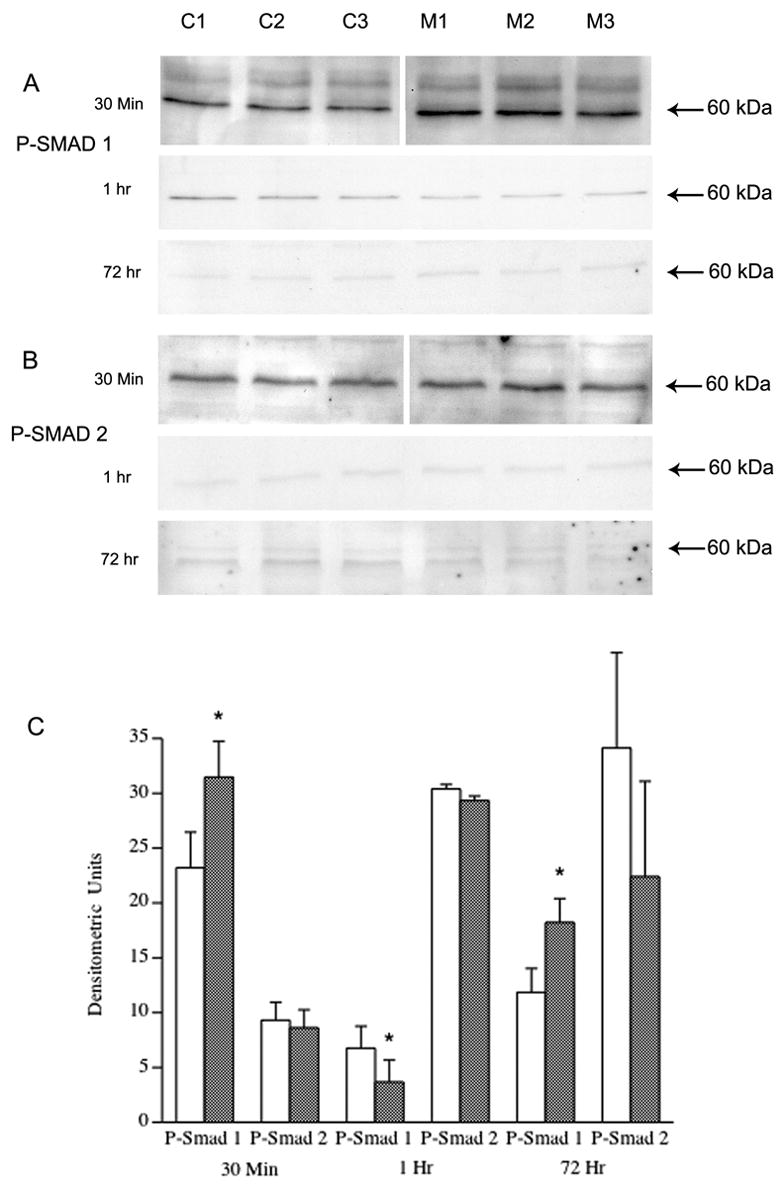
Western Blot of P-Smad 1 and P-Smad 2 in DMF vehicle (C1–C4) vs. 60 μg/ml MCTP (M1–M4) treated cells, 30 min: A) P-Smad 1 is significantly increased in cells treated for 30 min. with MCTP (p= 0.008) By 1 hr, P-Smad 1 is significantly decreased in MCTP treated cells but by 72 hr. P-Smad 1 is again significantly increased (p=0.018). B) There was no significant difference in expression of P-Smad 2 at any time point. All lanes contain 50 μg protein. C) Densitometric analysis of western blots (* p<.05).
Signaling Proteins at 72 hr
Since altered P-Smad1 expression was evident 72 hours after initial MCTP treatment and we and others have previously shown persistent effects of MCTP on endothelial cell cycle and morphology,, we evaluated the expression of several proteins that associate with either the BMP or TGFβ pathway by western blotting (fig 7 A–G) with densitometric analysis (fig 7H). There was no significant difference in Smad 4 (p= 0.18) at 72hr in MCTP treated HPAEC compared to control cells (fig. 7 C). Expression of BMPrII was marginally decreased (p= 0.05). There was upregulation of Smad 6 (p < .05), a member of this family that inhibits receptor Smad activation. There was also downregulation of p-38MAPK (p< .02), an alternate second messenger in BMP responses. We found no change in the putative pSmad 1 response element ID 1 nor was there a change in expression of caveolin -1. Because Smad 1 may be phosphorylated through either BMP or TGFβ pathways, (Goumans, Valdimarsdottir et al. 2002; de Caestecker 2004) we also evaluated the expression of Alk1, a TGFβ type I receptor prevalent on endothelium and found no significant difference in expression (p=0.07; fig. 7, B).
Figure 7.
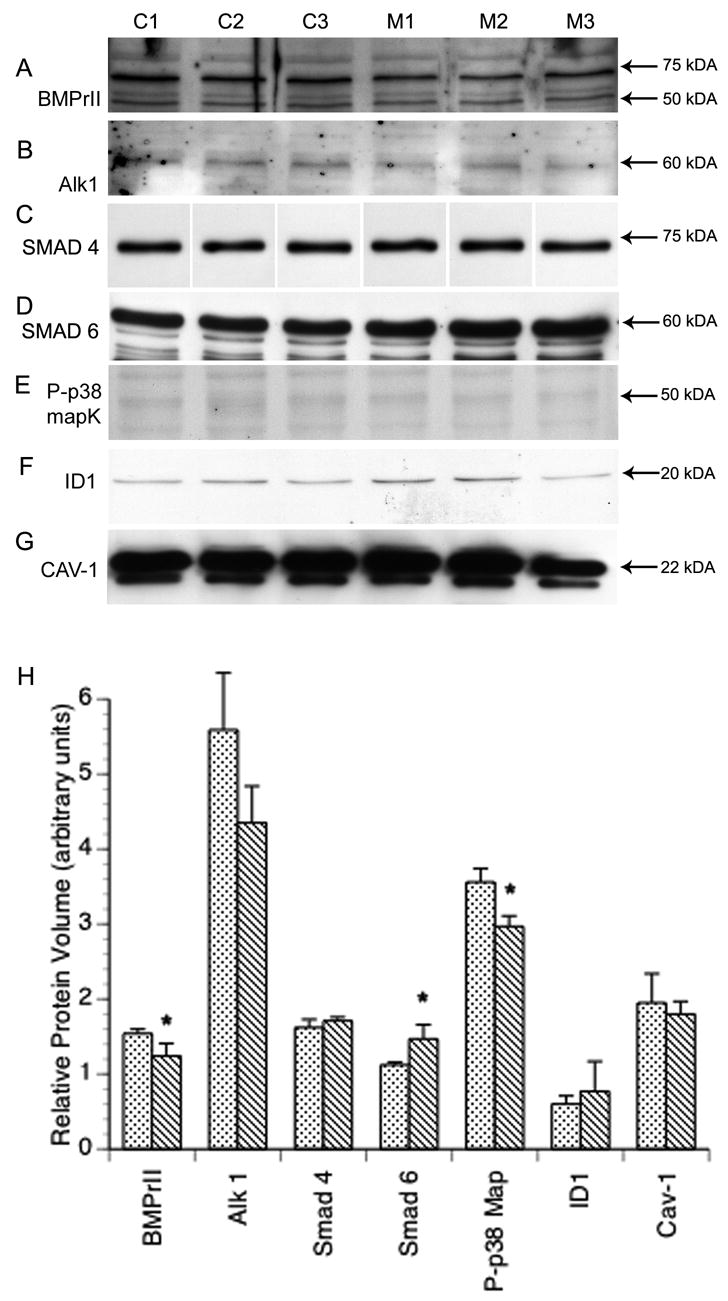
Western Blot of HPAEC 72 hr treatment; DMF vehicle (C1–C3) vs. 60 μg/ml MCTP (M1–M3): A) Expression of BMPrII is significantly decreased in MCTP treated cells (p= 0.05). No significant change was present in treated cells for B) Alk1 (p= 0.07) or C) Smad 4. D) Smad 6 expression is significantly increased (p= 0.035) while E) Phospho-p38 MAPK is significantly decreased (p= 0.012). No change was evident in F) ID 1 (p= 0.51) or G) Cav-1 (p= 0.58). All lanes contain 50 μg protein. G) Densitometric analysis of 72 hr. western blots (* p<.05).
The effects of MCTP on Caveolae
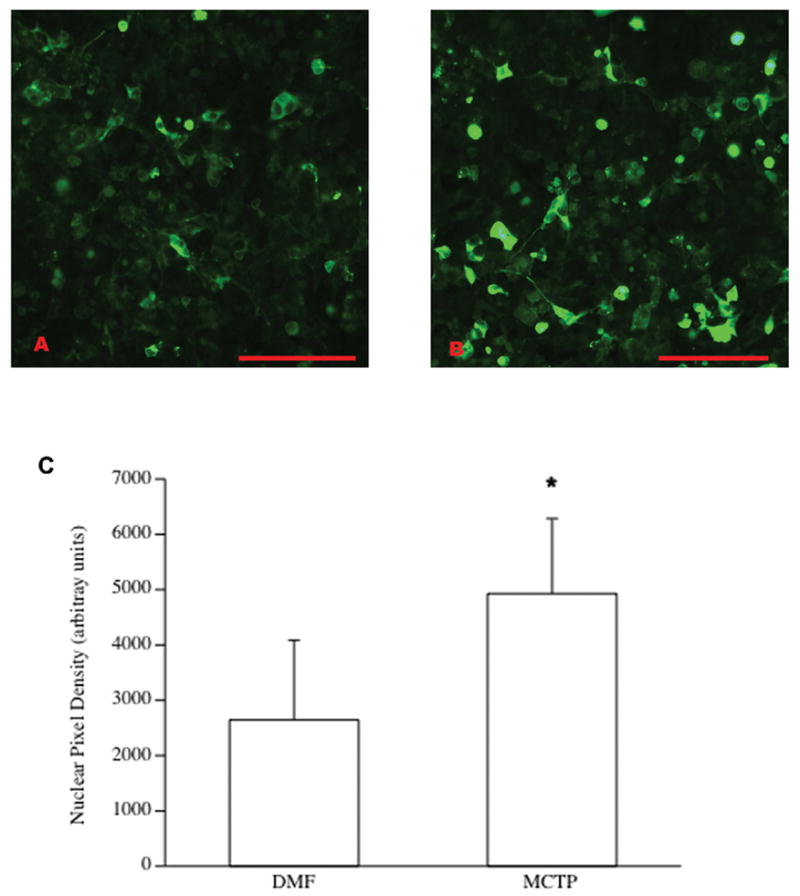
Because of our previous demonstrations of associations of BMPrII with caveolae (Ramos, Lame et al. 2006) and a potential role for disrupted caveolar-receptor interactions in the pathogenesis of PH, we examined Caveolae formation structurally by EM. The presence of caveolae on the cell membrane, was significantly altered with MCTP treatment through time. Numerous caveolae are present on both the apical and basal membrane of non-treated HPAEC (fig. 8, A). Caveolae are present, but less abundant after a 48 hr treatment with MCTP (fig. 8, B). In contrast, the membrane surface is often devoid of caveolae in cells treated for 72 hr (fig. 8, C, D, &E), but are abundant in DMF vehicle treated cells (fig. 8, F).
Figure 8.
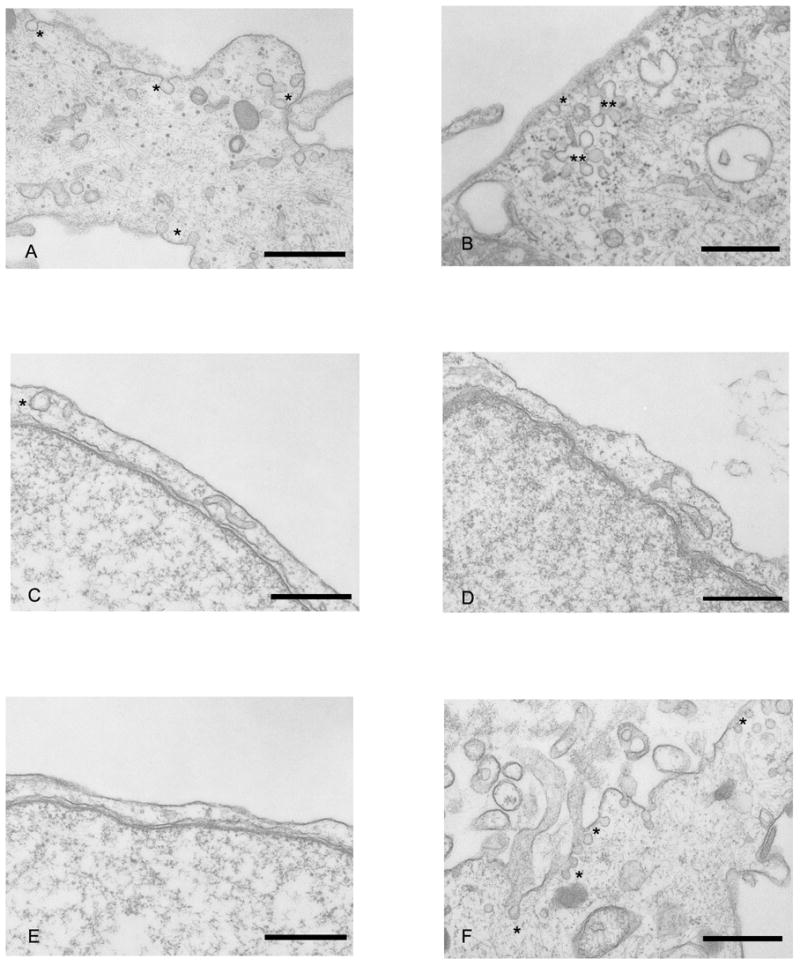
EM of HPAEC depicting caveolae, indicated by an asterisk: A) Non-treated control cell. Caveolae are present on both apical and basal surfaces of the peripheral edge of this cell. B) Cell treated 48 hr with MCTP. Caveolae are located at both the membrane and in clusters just below the surface. C, D, & E) Cells treated for 72 hr with MCTP. Caveolae are sparsely distributed on the cell membrane. F) Cell treated 72 hr with DMF vehicle. Numerous caveolae are present both on the cell membrane and within the cytoplasm. Bars = 500 nm.
Discussion
We have shown that both TGFβ and BMP signal transduction pathways are transiently activated in cultured human endothelial cells treated with MCTP. Consistent with previous reports (Pierreux, Nicolas et al. 2000; Xiao, Watson et al. 2001; Inman, Nicolas et al. 2002; Nicolas, De Bosscher et al. 2004) we found Smads predominantly located within the cytoplasm of unstimulated HPAEC. By fluorescent microscopy we observed marked nuclear accumulation of the common Smad 4, P-Smad 1 and P-Smad 2 within 5–15 minutes of MCTP treatment. Peak nuclear accumulation of Smads generally occurred at 30 min, and diminished thereafter. Phosphorylation and enhanced Smad nuclear translocation are canonical processes that are indicative of receptor activation (Inman, Nicolas et al. 2002; de Caestecker 2004; Nicolas, De Bosscher et al. 2004). Rapid accumulation of Smads within the nucleus is speculated to occur both due to enhanced import (Smad 4, P-Smad 1 and P-Smad 2) and decreased export (Smad 4 and P-Smad 1) (Pierreux, Nicolas et al. 2000; Xiao, Watson et al. 2001). Our results reflect the kinetics of ligand-receptor activation and are similar to other reports demonstrating the nuclear accumulation of P-Smad 1 (Langenfeld and Langenfeld 2004) and P-Smad 2 (Inman, Nicolas et al. 2002) in response to BMP2 and TGFβ respectively. Although exaggerated, nuclear translocation of Smad 4 in response to MCTP paralleled what we observed with recombinant ligand treatment of HPAEC. Smad nuclear accumulation reflects both the strength and duration of the signal mechanism (Inman, Nicolas et al. 2002). MCTP consistently induced nuclear accumulation of Smad 4 at a higher percentage than that of BMP2, BMP4, or TGFβ. Our studies were conducted in un-supplemented basal media exchanged one hr prior to treatment. It is unlikely that receptor activation by endogenous ligands would account for our observations, given the rate and sequence at which we observed these events immediately following MCTP treatment.
Collaborative evidence of transient signal transduction was additionally observed by western blot analysis. Although we saw no change in P-Smad 2 at any time points examined, a significant increase in P-Smad 1expression occurred 30 min. following treatment with MCTP. A modest but significant decrease in P-Smad 1 expression was present following 1 hr of MCTP treatment. Rapid de-phosphorylation in the nucleus is thought to promote nuclear export of receptor activated Smads, terminating the signal response in absence of continual receptor activation (Inman, Nicolas et al. 2002; Nicolas, De Bosscher et al. 2004). In addition, ubiquitization and proteasome degradation of phosphorylated Smads may further contribute to rapid diminished expression and down-regulation of the signaling response (Mehra and Wrana 2002; Panopoulou, Gillooly et al. 2002; Penheiter, Mitchell et al. 2002). The observed change in P-Smad 1 by cytology and western blot suggest receptor activation associated with MCTP treatment with subsequent down-regulation of BMP signal transduction, consistent with these concepts.
In addition to transient activation of signal transduction, we also observed significant alterations in BMP signaling with prolonged MCTP treatment. Expression of P-Smad 1 was significantly increased 72 hr following treatment suggesting recurrent and/or prolonged receptor activity. Interestingly, no changes in expression were observed for P-Smad 2 suggesting persistent signaling was limited to the BMPrII like pathway.
We also examined BMP and TGFβ specific receptor expression. Conventionally, Smad 1 has been considered a BMP specific receptor activated Smad, however several recent studies have shown that Alk1, a type I TGFβ receptor, can also signal through Smad 1. There was no change in Alk1 expression in this study. We did however observe a modest, but significant decrease in BMPrII. This suggests a correlate between MCTP induced endothelial injury and the downregulation of BMPrII seen in many PPH patients with BMPrII mutations. Diminished receptor expression may occur through proteosome/ubiquitization mechanisms similar to that of receptor activated Smads (Panopoulou, Gillooly et al. 2002). We also examined the p38MAPK alternate pathway thought to be activated in patients with PPH but found downregulation of phospho-p38 at the 72 hour time point. One explanation for this is the upregulation of the inhibitory Smad 6 which is reported to inhibit the activation of p38MAPK by BMPrII-TAK1 (Nohe et al. 2004). While not conclusive, our results collectively suggest MCTP induced dysregulation of BMP signal transduction.
The mechanisms by which MCTP induces Smad phosphorylation and nuclear translocation were not addressed in this study. Studies show that ligand affinity and downstream Smad signal response is highly variable and dependent on receptor oligomerization patterns (Gilboa, Wells et al. 1998; Nohe, Hassel et al. 2002; Derynck and Zhang 2003). The differences in cellular receptor expression is thought to play a major role in the diversity of signal response to a limited number of ligands, as well as partially explain the organ specific phenotype of receptor or Smad mutations. Endothelial cells express a full compliment of TGFβ and BMP receptors, however current understanding of endothelial receptor oligomerization patterns or ligand preferences is limited. Endothelial cells also express ligands for these receptors, which presumably exert autocrine and/or paracrine effects on the vasculature. BMP receptors are reportedly more diverse in oligomerization pattern and ligand response than other TGFβ receptors (Gilboa, Nohe et al. 2000). In HPAEC, MCTP consistently induced a higher percentage of Smad 4 nuclear translocation than that of recombinant ligands. These differences may reflect altered cellular responses or protein expression induced by in-vitro conditions, inappropriate ligand dose or observed time point for receptor stimulation, or reduced activity of the recombinant ligands. Variation of response to ligand stimulation within endothelial sub-populations has been observed by others (Langenfeld and Langenfeld 2004).
Alternatively, MCTP could induce phosphorylation and nuclear translocation through mechanisms other than pyrrole-receptor interactions. Possible mechanisms include MCTP acting as an accessory molecule, enhancing endogenous ligand activity; disruption of activity of receptor or Smad inhibitory molecules; or activation of other kinase activity, such as MAPK/erk kinase kinase 1, or Ca/calmodulin-dependent protein kinase II (de Caestecker, Parks et al. 1998; Wicks, Lui et al. 2000; Nohe, Keating et al. 2004). Additionally, Smad 1 phosphorylation by receptor kinases other than Alk1 or BMP receptors should be considered. Such pathways have been described for Smad 2 (de Caestecker, Parks et al. 1998).
Treatments were conducted in basal media that was unchanged during the treatment phase, thus endogenous ligand production could potentially account for Smad 1 phosphorylation at 72 hrs. Pyrrole or metabolite interaction with inhibitory mechanisms of Smad phosphorylation may also have a predominate role. MCTP forms covalent bonds with cytoskeletal tropomyosin and β- or γ-actin (Lame, Jones et al. 2000). Filamin is thought to inhibit Smad phosphorylation by tethering receptor activated Smads to cytoskeletal actin (Sasaki, Masuda et al. 2001). Structural interference by MCTP with cytoskeletal proteins could thus indirectly enhance Smad phosphorylation. However, this proposed mechanism fails to justify the differences in observation of P-Smad 1 and P-Smad 2 and further suggests specificity of the BMP pathway in this model.
We also investigated the effects of MCTP on caveolae expression. Caveolae are vesicular structures that play a role in intracellular transport, hemodynamics, and the compartmentalization of signaling molecules (Anderson 1998; Mineo, Gill et al. 1999; Schlegel and Lisanti 2001; Zhao, Liu et al. 2002; Minshall, Sessa et al. 2003; Rodriguez-Pascual, Redondo-Horcajo et al. 2003). Cav-1, the predominant structural protein for caveolae, has important regulatory roles in receptor activation (Okamoto, Schlegel et al. 1998; Razani, Zhang et al. 2001; Nohe, Keating et al. 2005). Cav-1 is thought to structurally inhibit receptor activity until specific ligand stimulation (Minshall, Sessa et al. 2003). Cav-1 expression is decreased in lung tissue derived from patients with PH (Geraci, Moore et al. 2001), and Cav-1 knockout mice develop clinical symptoms of PH (Razani, Engelman et al. 2001; Zhao, Liu et al. 2002) suggesting a role for Cav-1 in PH pathogenesis. More recently a role for terminating receptor activation through caveolin-dependent endocytic pathways with proteosomal degradation have been described (Derynck and Zhang 2003; de Caestecker 2004), and demonstrated for TGFβ type 1 receptor/Smad 7 complexes (Razani, Zhang et al. 2001; Di Guglielmo, Le Roy et al. 2003). Although the total expression of Cav-1 by PCR or western blot at 72 hr did not vary between MCTP and vehicle treated cells, we have shown in this study by EM that the presence of caveolae at the cell surface markedly diminishes with time, resulting in sparsely scattered caveolae at the membrane surface at 72 hrs. Loss of cell surface Cav-1 has also been reported for MCTP treated A549 cells (Mukhopadhyay, Shah et al. 2005; Mukhopadhyay, Shah et al. 2006). MCTP disruption of Cav-1α/raft scaffolding has also been described in the rat, with Golgi block in transport as the proposed mechanism (Mathew, Huang et al. 2004). Although the functional significance is currently unknown, we have previously reported the structural association between BMPrII and caveolae (Ramos, Lame et al. 2006). Depletion of surface caveolae might indirectly contribute to an increase in Smad 1 phosphorylation in this model at later time points, either through diminished receptor endocytosis or by loss of cav-1 inhibitory function on receptor activity.
In conclusion, there are several reports that suggest a role for activated Smad signaling in lung inflammation (Rosendahl, Checchin et al. 2001; Rosendahl, Pardali et al. 2002; Zhao and Geverd 2002) or endothelial injury (Tanaka, Schuster et al. 1996; Akman, Zhang et al. 2001). Our results suggest BMP pathway specificity for MCTP endothelial cell toxicity in vitro, and demonstrates the continued usefulness of this model for elucidating the molecular pathogenesis of PH as well as toxic cell injury.
Acknowledgments
This research was supported by NIH grant #HL48411 from NHLBI. M. Ramos was additionally supported by grant #ESO70555 from NIEH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akman HO, Zhang H, et al. Response to hypoxia involves transforming growth factor-beta2 and Smad proteins in human endothelial cells. Blood. 2001;98(12):3324–31. doi: 10.1182/blood.v98.12.3324. [DOI] [PubMed] [Google Scholar]
- Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- Arcot SS, Lipke DW, et al. Alterations of growth factor transcripts in rat lungs during development of monocrotaline-induced pulmonary hypertension. Biochem Pharmacol. 1993;46(6):1086–91. doi: 10.1016/0006-2952(93)90675-m. [DOI] [PubMed] [Google Scholar]
- Atkinson C, Stewart S, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- Botney MD, Bahadori L, et al. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol. 1994;144(2):286–95. [PMC free article] [PubMed] [Google Scholar]
- de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15(1):1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- de Caestecker MP, Parks WT, et al. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998;12(11):1587–92. doi: 10.1101/gad.12.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Morse JH, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, et al. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5(5):410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Geraci MW, Moore M, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88(6):555–62. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- Gilboa L, Nohe A, et al. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell. 2000;11(3):1023–35. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L, Wells RG, et al. Oligomeric structure of type I and type II transforming growth factor beta receptors: homodimers form in the ER and persist at the plasma membrane. J Cell Biol. 1998;140(4):767–77. doi: 10.1083/jcb.140.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, et al. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21(7):1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullick WJ, Downward J, et al. Antibodies to the autophosphorylation sites of the epidermal growth factor receptor protein-tyrosine kinase as probes of structure and function. Embo J. 1985;4(11):2869–77. doi: 10.1002/j.1460-2075.1985.tb04016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, et al. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol Cell. 2002;10(2):283–94. doi: 10.1016/s1097-2765(02)00585-3. [DOI] [PubMed] [Google Scholar]
- Lame MW, Jones AD, et al. Protein targets of monocrotaline pyrrole in pulmonary artery endothelial cells. J Biol Chem. 2000;275(37):29091–9. doi: 10.1074/jbc.M001372200. [DOI] [PubMed] [Google Scholar]
- Langenfeld EM, Langenfeld J. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol Cancer Res. 2004;2(3):141–9. [PubMed] [Google Scholar]
- Lebrin F, Deckers M, et al. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65(3):599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Lehr HA, Mankoff DA, et al. Application of photoshop-based image analysis to quantification of hormone receptor expression in breast cancer. J Histochem Cytochem. 1997;45(11):1559–65. doi: 10.1177/002215549704501112. [DOI] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68(1):92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Huang J, et al. Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110(11):1499–506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- Mattocks AR, Jukes R, et al. Simple procedures for preparing putative toxic metabolites of pyrrolizidine alkaloids. Toxicon. 1989;27(5):561–7. doi: 10.1016/0041-0101(89)90117-7. [DOI] [PubMed] [Google Scholar]
- Mehra A, Attisano L, et al. Characterization of SMAD Phosphorylation and SMAD-Receptor Interaction. In: Howe PH, editor. Methods in Molecular Biology. Vol. 142. Totowa, NJ: Humana Press; 2000. pp. 67–78. [DOI] [PubMed] [Google Scholar]
- Mehra A, Wrana JL. TGF-beta and the Smad signal transduction pathway. Biochem Cell Biol. 2002;80(5):605–22. doi: 10.1139/o02-161. [DOI] [PubMed] [Google Scholar]
- Mineo C, Gill GN, et al. Regulated migration of epidermal growth factor receptor from caveolae. J Biol Chem. 1999;274(43):30636–43. doi: 10.1074/jbc.274.43.30636. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, et al. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol. 2003;285(6):L1179–83. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- Miyazono K. Signal transduction by bone morphogenetic protein receptors: functional roles of Smad proteins. Bone. 1999;25(1):91–3. doi: 10.1016/s8756-3282(99)00113-1. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Lin HY, et al. The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J Biol Chem. 1993;268(30):22215–8. [PubMed] [Google Scholar]
- Mukhopadhyay S, Shah M, et al. Monocrotaline pyrrole-induced megalocytosis of lung and breast epithelial cells: Disruption of plasma membrane and Golgi dynamics and an enhanced unfolded protein response. Toxicol Appl Pharmacol. 2005 doi: 10.1016/j.taap.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Shah M, et al. Monocrotaline pyrrole-induced megalocytosis of lung and breast epithelial cells: Disruption of plasma membrane and Golgi dynamics and an enhanced unfolded protein response. Toxicol Appl Pharmacol. 2006;211(3):209–20. doi: 10.1016/j.taap.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Nakao A, Imamura T, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. Embo J. 1997;16(17):5353–62. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JH, Wheeler L, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345(5):319–24. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- Nicolas FJ, De Bosscher K, et al. Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci. 2004;117(Pt 18):4113–25. doi: 10.1242/jcs.01289. [DOI] [PubMed] [Google Scholar]
- Nohe A, Hassel S, et al. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002;277(7):5330–8. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- Nohe A, Keating E, et al. Signal transduction of bone morphogenetic protein receptors. Cell Signal. 2004;16(3):291–9. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Nohe A, Keating E, et al. Dynamics and interaction of caveolin-1 isoforms with BMP-receptors. J Cell Sci. 2005;118(Pt 3):643–50. doi: 10.1242/jcs.01402. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, et al. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273(10):5419–22. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Panopoulou E, Gillooly DJ, et al. Early endosomal regulation of Smad-dependent signaling in endothelial cells. J Biol Chem. 2002;277(20):18046–52. doi: 10.1074/jbc.M107983200. [DOI] [PubMed] [Google Scholar]
- Penheiter SG, Mitchell H, et al. Internalization-dependent and -independent requirements for transforming growth factor beta receptor signaling via the Smad pathway. Mol Cell Biol. 2002;22(13):4750–9. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierreux CE, Nicolas FJ, et al. Transforming growth factor beta-independent shuttling of Smad4 between the cytoplasm and nucleus. Mol Cell Biol. 2000;20(23):9041–54. doi: 10.1128/mcb.20.23.9041-9054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos M, Lame MW, et al. The BMP type II receptor is located in lipid rafts, including caveolae, of pulmonary endothelium in vivo and in vitro. Vascul Pharmacol. 2006;44(1):50–9. doi: 10.1016/j.vph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276(41):38121–38. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Razani B, Zhang XL, et al. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276(9):6727–38. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pascual F, Redondo-Horcajo M, et al. Functional cooperation between Smad proteins and activator protein-1 regulates transforming growth factor-beta-mediated induction of endothelin-1 expression. Circ Res. 2003;92(12):1288–95. doi: 10.1161/01.RES.0000078491.79697.7F. [DOI] [PubMed] [Google Scholar]
- Rosendahl A, Checchin D, et al. Activation of the TGF-beta/activin-Smad2 pathway during allergic airway inflammation. Am J Respir Cell Mol Biol. 2001;25(1):60–8. doi: 10.1165/ajrcmb.25.1.4396. [DOI] [PubMed] [Google Scholar]
- Rosendahl A, Pardali E, et al. Activation of bone morphogenetic protein/Smad signaling in bronchial epithelial cells during airway inflammation. Am J Respir Cell Mol Biol. 2002;27(2):160–9. doi: 10.1165/ajrcmb.27.2.4779. [DOI] [PubMed] [Google Scholar]
- Rosenzweig BL, Imamura T, et al. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci U S A. 1995;92(17):7632–6. doi: 10.1073/pnas.92.17.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Masuda Y, et al. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J Biol Chem. 2001;276(21):17871–7. doi: 10.1074/jbc.M008422200. [DOI] [PubMed] [Google Scholar]
- Schlegel A, Lisanti MP. The caveolin triad: caveolae biogenesis, cholesterol trafficking, and signal transduction. Cytokine Growth Factor Rev. 2001;12(1):41–51. doi: 10.1016/s1359-6101(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Shah M, Patel K, et al. Monocrotaline pyrrole-induced endothelial cell megalocytosis involves a Golgi blockade mechanism. Am J Physiol Cell Physiol. 2005;288(4):C850–62. doi: 10.1152/ajpcell.00327.2004. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Bernstein ML, et al. Site-specific responses to monocrotaline-induced vascular injury: evidence for two distinct mechanisms of remodeling. Am J Respir Cell Mol Biol. 1996;15(3):390–7. doi: 10.1165/ajrcmb.15.3.8810644. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Schuster DP, et al. The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest. 1996;98(2):434–42. doi: 10.1172/JCI118809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HC, Lame MW, et al. Cell cycle alterations associated with covalent binding of monocrotaline pyrrole to pulmonary artery endothelial cell DNA. Toxicol Appl Pharmacol. 1996;141(1):319–29. doi: 10.1006/taap.1996.0289. [DOI] [PubMed] [Google Scholar]
- Valdimarsdottir G, Goumans MJ, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106(17):2263–70. doi: 10.1161/01.cir.0000033830.36431.46. [DOI] [PubMed] [Google Scholar]
- van den Driesche S, Mummery CL, et al. Hereditary hemorrhagic telangiectasia: an update on transforming growth factor beta signaling in vasculogenesis and angiogenesis. Cardiovasc Res. 2003;58(1):20–31. doi: 10.1016/s0008-6363(02)00852-0. [DOI] [PubMed] [Google Scholar]
- Venable JH, Coggeshall R. A Simplified Lead Citrate Stain for Use in Electron Microscopy. J Cell Biol. 1965;25:407–8. doi: 10.1083/jcb.25.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Tuder RM. Cellular and molecular mechanisms in the pathogenesis of severe pulmonary hypertension. Eur Respir J. 1995;8(12):2129–38. doi: 10.1183/09031936.95.08122129. [DOI] [PubMed] [Google Scholar]
- Wicks SJ, Lui S, et al. Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol Cell Biol. 2000;20(21):8103–11. doi: 10.1128/mcb.20.21.8103-8111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DW, Lame MW, et al. Monocrotaline pyrrole interacts with actin and increases thrombin-mediated permeability in pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1998;152(1):138–44. doi: 10.1006/taap.1998.8488. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev. 2000;11(1–2):5–13. doi: 10.1016/s1359-6101(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Watson N, et al. Nucleocytoplasmic shuttling of Smad1 conferred by its nuclear localization and nuclear export signals. J Biol Chem. 2001;276(42):39404–10. doi: 10.1074/jbc.M103117200. [DOI] [PubMed] [Google Scholar]
- Yang X, Long L, et al. Dysfunctional Smad Signaling Contributes to Abnormal Smooth Muscle Cell Proliferation in Familial Pulmonary Arterial Hypertension. Circ Res. 2005 doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- Yang X, Long L, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96(10):1053–63. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- Yeager ME, Halley GR, et al. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88(1):E2–E11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Geverd DA. Regulation of Smad3 expression in bleomycin-induced pulmonary fibrosis: a negative feedback loop of TGF-beta signaling. Biochem Biophys Res Commun. 2002;294(2):319–23. doi: 10.1016/S0006-291X(02)00471-0. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Liu Y, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99(17):11375–80. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]