

Abstract
Evidence has been presented both for and against obligate retrograde movement of resident Golgi proteins through the endoplasmic reticulum (ER) during nocodazole-induced Golgi ministack formation. Here, we studied the nocodazole-induced formation of ministacks using phospholipase A2 (PLA2) antagonists, which have been shown previously to inhibit brefeldin A–stimulated Golgi-to-ER retrograde transport. Examination of clone 9 rat hepatocytes by immunofluorescence and immunoelectron microscopy revealed that a subset of PLA2 antagonists prevented nocodazole-induced ministack formation by inhibiting two different trafficking pathways for resident Golgi enzymes; at 25 μM, retrograde Golgi-to-ER transport was inhibited, whereas at 5 μM, Golgi-to-ER trafficking was permitted, but resident Golgi enzymes accumulated in the ER. Moreover, resident Golgi enzymes gradually redistributed from the juxtanuclear Golgi or Golgi ministacks to the ER in cells treated with these PLA2 antagonists alone. Not only was ER-to-Golgi transport of resident Golgi enzymes inhibited in cells treated with these PLA2 antagonists, but transport of the vesicular stomatitis virus G protein out of the ER was also prevented. These results support a model of obligate retrograde recycling of Golgi resident enzymes during nocodazole-induced ministack formation and provide additional evidence that resident Golgi enzymes slowly and constitutively cycle between the Golgi and ER.
INTRODUCTION
Microtubules are required to maintain the normal interconnected morphology of the Golgi complex at the microtubule-organizing center (MTOC) of unpolarized mammalian cells and to facilitate membrane traffic to and from the Golgi (for reviews, see Cole and Lippincott-Schwartz, 1995; Bloom and Goldstein, 1998; Lippincott-Schwartz, 1998). Many studies have shown that depolymerization of microtubules by treatment of cells with nocodazole or colchicine results in the formation of Golgi ministacks that are dispersed throughout the cell periphery (Pavelka and Ellinger, 1983; Rogalski and Singer, 1984; Thyberg and Moskalewski, 1985) and adjacent to endoplasmic reticulum (ER)-exit sites (Cole et al., 1996). Originally, it was believed that microtubule depolymerization led to the fragmentation of intact Golgi ribbons into smaller, disconnected ministacks that simply diffused throughout the cytoplasm (Rogalski and Singer, 1984). More recently, however, other studies have begun to suggest a very different model of ministack formation that may have more profound implications for our understanding of membrane trafficking to and from the Golgi complex. This model proposes that the membrane proteins of the Golgi complex constitutively and repeatedly cycle back through the ER and that nocodazole treatment reveals this pathway by inhibiting only the anterograde transport of proteins from ER-exit sites to the juxtanuclear Golgi and not the retrograde Golgi-to-ER movement. This model is based on evidence showing that the speed at which a Golgi protein cycles between the Golgi and ER correlated with its appearance at nocodazole-induced ministacks (Cole et al., 1996). Also, time-lapse imaging of nocodazole-treated cells expressing N-acetylgalactosaminyltransferase-II fused to the green fluorescent protein demonstrated that dispersed fluorescent ministacks did not form by fragmentation of the central Golgi ribbon with subsequent diffusion throughout the cytoplasm; rather dispersed fluorescent ministacks appeared to form de novo, growing in fluorescence intensity (Storrie et al., 1998). Constitutive Golgi-to-ER recycling was also observed in the absence of nocodazole, as shown in studies using chimeric resident Golgi proteins fused with the thermosensitive domain of the temperature-sensitive vesicular stomatitis virus G (VSVGts045) glycoprotein. The fusion proteins slowly redistributed from the Golgi to the ER, after a shift to the restrictive temperature because the VSVGts045 domain misfolded upon reaching the ER (Knipe et al., 1977), thus trapping the fusion protein in this organelle (Cole et al., 1998). Explaining the behavior of the Golgi in nocodazole-treated cells by the constitutive cycling of resident Golgi proteins through the ER can be termed the “recycling” model. Thus, the recycling model suggests that nocodazole treatment reveals a fundamental, constitutive cycling pathway between the Golgi complex and the ER for all resident Golgi proteins (Cole et al., 1996). Additional evidence that resident Golgi proteins constitutively recycle was obtained by visualizing Golgi dynamics in yeast (Wooding and Pelham, 1998). Moreover, retrograde movement of rapidly recycling proteins such as Golgi t-SNARES is well documented (Allan and Balch, 1999).
Recently, several experiments have been performed to test more directly the validity of the “fragmentation” and recycling mechanisms of Golgi ministack formation in the absence of microtubules. For example, the recycling model predicts that inhibition of export from the ER should impede Golgi ministack formation. In cells microinjected and incubated from 15 min to 3 h with a dominant-negative mutant of the Sar1 protein, a GTPase that is required for coatomer protein (COP) II–mediated vesicle transport out of the ER (Barlowe et al., 1994; Kuge et al., 1994; Aridor et al., 1995), Shima et al. (1998) found no effect on nocodazole-stimulated ministack formation. These results suggest that retrograde recycling of resident Golgi proteins through and out of the ER is not obligatory for ministack formation. However, using a different experimental procedure, Storrie et al. (1998) found that expression of the dominant-negative Sar1 protein for a longer period of time (3–10 h) caused the redistribution of resident proteins from both normal Golgi stacks and nocodazole-induced ministacks to the ER, results implicating retrograde traffic through the ER in ministack formation. Thus, these results have not yet resolved the issue, and other specific inhibitors or dominant-negative mutants that specifically disrupt Golgi-to-ER retrograde trafficking would be very helpful in determining which of the two models of nocodazole-induced Golgi ministack formation more accurately describes this pathway. Our recent studies of the retrograde trafficking of resident Golgi proteins to the ER may provide such tools (de Figueiredo et al., 1998).
We have begun to investigate the molecular mechanisms involved in the stimulation of tubule-mediated Golgi-to-ER retrograde trafficking by brefeldin A (BFA), a process that is facilitated by but not absolutely dependent on microtubules (Lippincott-Schwartz et al., 1990). On the basis of previous in vitro studies that suggested that tubule formation involved the direct action of a cytosolic enzyme activity (Banta et al., 1995), we found that a broad spectrum of chemical antagonists of cytosolic phospholipase A2 (PLA2) enzymes inhibited both BFA-stimulated retrograde traffic of resident Golgi proteins to the ER and tubulation of Golgi membranes (de Figueiredo et al., 1998). In other studies, we found that PLA2 antagonists also inhibited the retrograde trafficking of chimeric proteins, consisting of the thermosensitive domain of VSVGts045 fused to resident Golgi proteins (de Figueiredo and Brown, personal communication), that recycle from the Golgi complex and accumulate in the ER upon shift to the restrictive temperature (Cole et al., 1998). PLA2 enzymes are a large family of enzymes that hydrolyze glycerophospholipids primarily at the sn-2 position, yielding a lysophospholipid and a free fatty acid. These hydrolytic enzymes are thought to be involved not only in signal transduction via the release of arachidonic acid but also in the direct remodeling of membranes that could influence membrane-trafficking events (de Figueiredo et al., 1998).
In this work we examined the mechanism of nocodazole-mediated Golgi ministack formation, with the hypothesis that if retrograde transport from the Golgi to the ER is necessary for this process, then PLA2 antagonists should inhibit formation of dispersed Golgi ministacks. We report that a variety of PLA2 antagonists inhibited the nocodazole-induced disappearance of the juxtanuclear Golgi and the subsequent appearance of Golgi ministacks, results consistent with the retrograde-recycling model of ministack formation. Surprisingly, in the presence of low concentrations of certain PLA2 antagonists [e.g., N-(p-amylcinnamoyl)anthranilic acid (ACA) and 2-(p-amylcinnamoyl)amino-4-chlorobenzoic acid (ONO-RS-082)], ∀-mannosidase II (ManII) accumulated in the ER of nocodazole-treated cells, providing additional evidence of an ER intermediate in ministack formation as well as suggesting that certain PLA2 antagonists may inhibit ER-to-Golgi anterograde transport. Importantly, these data suggest a novel role for a PLA2 activity(s) in anterograde ER-to-Golgi transport. Also, we provide evidence that ManII normally cycles between the Golgi and ER in cells containing a juxtanuclear Golgi or nocodazole-induced ministacks. These results are consistent with previous studies that suggested resident Golgi enzymes constitutively cycle slowly between the Golgi and the ER (Cole et al., 1996) and suggest that this recycling to the ER is the mechanism responsible for the formation of Golgi ministacks in nocodazole-treated cells.
MATERIALS AND METHODS
Reagents and Antibodies
The PLA2 antagonists ACA, arachidonyl trifluoromethyl ketone (AACOCF3), E-6-(bromomethylene)tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one or bromoenol lactone (BEL), ONO-RS-082, and palmityl trifluoromethyl ketone (PACOCF3) were purchased from Biomol Research Laboratories (Plymouth Meeting, PA). Stock solutions of 40 mM ACA in EtOH at −20°C, 20 mM AACOCF3 in DMSO at −80°C, and 10 mg/ml PACOCF3 in DMSO at −80°C were prepared and stored. Concentrated stock solutions (at least 500×) of BEL (in DMSO) and ONO-RS-082 (in EtOH) were made fresh before experiments were performed. Nocodazole, BFA, and cycloheximide (Sigma Chemical, St. Louis, MO) were stored as stock solutions of 6 mg/ml in DMSO at −20°C, 10 mg/ml in EtOH at −20°C, and 2 mg/ml in H2O at 4°C, respectively. VSVGts045 was kindly provided by Dr. Vivek Malhotra (University of California, San Diego, San Diego, CA). The following antibodies were generously supplied to us: the polyclonal antibody against α-mannosidase II (Dr. Marilyn G. Farquhar, University of California, San Diego, and Dr. Kelly Moreman, University of Georgia, Athens, GA), the monoclonal anti-β-COP antibody M3A5 (Dr. W. Balch, Scripps Research Institute, La Jolla, CA), and the monoclonal antibody P5D4 against the VSVG protein (Dr. Vivek Malhotra, University of California, San Diego). Monoclonal anti-α-tubulin antibody was purchased from Amersham (Arlington Heights, IL). The monoclonal anti-protein disulfide isomerase (PDI) antibody was purchased from Affinity Bioreagents (Golden, CO), and all secondary fluorescent antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cell Culture and Treatments to Investigate Membrane-trafficking Pathways
Clone 9 rat hepatocytes were grown on glass coverslips in modified Eagle’s minimal essential medium (MEM) with 10% fetal calf serum (FCS) and 50 U/ml penicillin + 50 μg/ml streptomycin from Life Technologies (Grand Island, NY) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
All inhibitors and drugs were diluted at least 1:500 in serum-free MEM with appropriate solvent controls being conducted. In assays examining nocodazole-induced ministack formation, cells were washed twice in serum-free MEM, incubated at 4°C with or without PLA2 antagonists in MEM for 20 min, and subsequently shifted to 37°C in MEM containing nocodazole (6 μg/ml), with or without PLA2 antagonists. In nocodazole washout experiments, cells were washed twice in serum-free MEM and incubated at 37°C with nocodazole (6 μg/ml) for 2 h to form Golgi ministacks. To follow the recovery of the Golgi complex, the cells were washed twice in serum-free MEM (to remove nocodazole) and allowed to recover in serum-free MEM for various times before fixing and processing for immunofluorescence microscopy. To follow the effect of ONO-RS-082 on the recovery of the Golgi complex from ministacks, cells were incubated in 10 μM ONO-RS-082 for 10 min in the continued presence of nocodazole, washed twice in serum-free MEM (to remove nocodazole), and incubated in 10 μM ONO-RS-082 alone for various times before fixing and processing for immunofluorescence microscopy.
To ensure that the change in distribution of membrane markers, e.g., ManII, was not caused by new protein synthesis, trafficking experiments were done in the presence of 2 μg/ml cycloheximide (see Figures 1–6 and 8–10), as we have used previously on clone 9 cells (Brown et al., 1984), or 50 μg/ml cycloheximide (see Figure 7) to inhibit protein synthesis.
Figure 1.

PLA2 antagonists inhibit the nocodazole-induced formation of Golgi ministacks. Clone 9 rat hepatocytes were left untreated (A and B) or incubated at 4°C for 20 min before shifting to 37°C in 6 μg/ml nocodazole (Noc) for 2 h (C and D). To examine the effect of PLA2 antagonists on ministack formation, we incubated the cells at 4°C for 20 min with 5 μM BEL (E and F), 25 μM AACOCF3 (G and H), or 25 μM ONO-RS-082 (ONO; I and J) and then shifted the cells to 37°C in the continued presence of the same PLA2 antagonist plus 6 μg/ml nocodazole for 2 h (E–H) or 1 h (I and J). All conditions included 2 μg/ml cycloheximide. Cells were fixed and processed for immunofluorescence microscopy (see MATERIALS AND METHODS) using a polyclonal antibody against the medial Golgi enzyme ManII (A, C, E, G, and I) and a monoclonal antibody against α-tubulin (B, D, F, H, and J).
Figure 6.

Redistribution of ManII in cells treated with ONO-RS-082 or ACA alone. Clone 9 cells were treated with 5 μM BEL (A), 5 μM ONO-RS-082 (B–D), or 25 μM ACA (E and F) for 2 h before fixing and processing for immunofluorescence using a polyclonal antibody against ManII (A–C and E) and a monoclonal antibody against PDI (D and F). All conditions included 2 μg/ml cycloheximide.
Figure 8.

ManII, originating from nocodazole-induced ministacks, accumulates in the ER in cells incubated with ONO-RS-082 or ACA but not AACOCF3. Cells were incubated with 6 μg/ml nocodazole for 2 h to form Golgi ministacks and then incubated with nocodazole alone (A), 25 μM ACA and nocodazole (B), 5 μM ONO-RS-082 and nocodazole (C), or 25 μM AACOCF3 and nocodazole (D) for an additional 2 h. All conditions included 2 μg/ml cycloheximide. Cells were fixed and processed for immunofluorescence using a polyclonal antibody against ManII.
Figure 10.

ONO-RS-082 prevents the complete reformation of the Golgi complex after nocodazole washout. Golgi ministacks were formed by incubating cells at 37°C in 6 μg/ml nocodazole for 2 h. Cells were then washed twice in nocodazole-free media and allowed to recover in either media alone (A–F) or 10 μM ONO-RS-082 (G–L) for various times. All conditions included 2 μg/ml cycloheximide. Cells were fixed and processed for double-label immunofluorescence microscopy using a polyclonal antibody against ManII (A, C, E, G, I, and K) and a monoclonal antibody against α-tubulin (B, D, F, H, J, and L). Short ManII-positive tubules are indicated by arrows (A and C).
Figure 7.
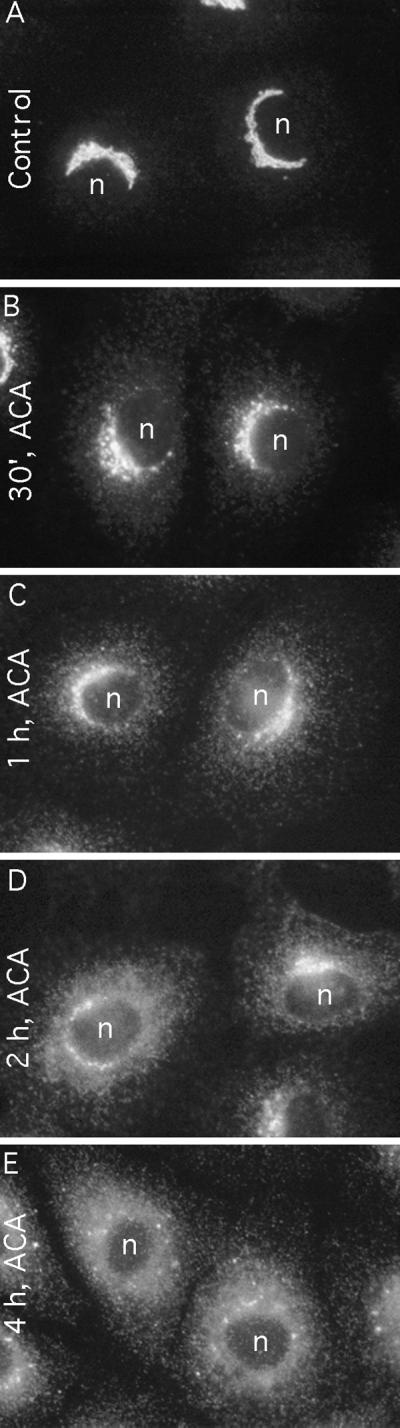
Kinetic analysis of ACA-induced redistribution of ManII to the ER. Cells were incubated in the presence or absence of ACA (25 μM) for up to 4 h as indicated on the micrographs and then were fixed and stained for ManII immunofluorescence. In all cases, cells were pretreated with 50 μg/ml cycloheximide for 15 min and then incubated in its continuous presence after addition of ACA. n, nucleus.
VSVGts045 Membrane-trafficking Assay
Clone 9 cells were infected with VSVGts045 by washing the cells three times in serum-free MEM followed by incubation at 40°C with VSVGts045 in serum-free MEM for 45 min. An equal volume of MEM + 10% FCS was added, and cells were incubated for 30 min at 40°C. Cells were washed twice in MEM + 10% FCS and kept at 40°C for 3 h to accumulate the VSVG protein in the ER. Cells were incubated in the absence or presence of PLA2 antagonists for 30 min at 40°C and shifted to the permissive temperature of 32°C in the continued presence or absence of PLA2 antagonists for 30 min to allow transport of the VSVG protein from the ER to the Golgi. Cells were fixed and processed for immunofluorescence microscopy as described below, VSVG was visualized using the monoclonal antibody P5D4, and the Golgi was visualized using a polyclonal antibody against ManII.
To quantify the transport of the VSVG protein from the ER to the Golgi, we used the immunofluorescence microscopy assay described above. Successful transport of the VSVG protein to the Golgi was defined as colocalization of the VSVG protein and ManII. Each value represents the average of two experiments with 100 cells counted in each experiment.
Immunofluorescence and Immunoelectron Microscopy
Immunofluorescence microscopy was performed as described previously (Wood et al., 1991). Briefly, cells were fixed in 3.7% formalin in phosphate-buffered saline (PBS), pH 7.4, for 10 min at room temperature, washed three times for 5 min each in PBS, and permeabilized for 5 min in 0.1% Triton X-100 in PBS. Cells were incubated with the primary antibody for 1 h at room temperature, washed three times for 5 min each in PBS, incubated with the secondary antibody for 1 h at room temperature, and washed three times for 5 min each in PBS before being mounted on slides. Images were collected on a Zeiss Axiovert 100TV fluorescent microscope using a digital charge-coupled device camera (Princeton Instruments, Trenton, NJ) controlled by Metamorph software (Universal Imaging, West Chester, PA). Figures were assembled using Adobe Photoshop (Adobe Systems, San Jose, CA).
To visualize the Golgi complex by immunoperoxidase electron microscopy, cells were fixed with periodate-lysine-paraformaldehyde fixative (McLean and Nakane, 1974), permeabilized, and incubated with a polyclonal antibody against ManII. The cells were then incubated with sheep anti-rabbit-HRP conjugates and processed for diaminobenzidine cytochemistry as described previously (Brown and Farquhar, 1989).
RESULTS
PLA2 Antagonists Inhibit Nocodazole-induced Ministack Formation
We reasoned that if nocodazole-induced Golgi ministack formation requires obligatory recycling of Golgi membranes to the ER, then PLA2 antagonists, which inhibit retrograde traffic from the Golgi to the ER (de Figueiredo et al., 1998), should also inhibit this pathway. To examine the effect of PLA2 antagonists on nocodazole-induced Golgi ministack formation, clone 9 rat hepatocytes were incubated at 4°C for 20 min and then transferred to 37°C in nocodazole (6 μg/ml) for 2 h to depolymerize cold-sensitive microtubules (Turner and Tartakoff, 1989; Cole et al., 1996), in the absence or presence of PLA2 antagonists. Cells were then fixed and processed for double-label indirect-immunofluorescence microscopy using a polyclonal antibody against the medial Golgi enzyme ManII and a monoclonal antibody against α-tubulin.
In control cells the juxtanuclear Golgi ribbon (Figure 1A) sits near the MTOC (Figure 1B). However, when treated with nocodazole for 2 h, microtubules were depolymerized (Figure 1D), and as expected, the Golgi complex was seen as ministacks dispersed throughout the cytoplasm (Figure 1C). Pretreatment with 5 μM BEL, an irreversible inhibitor that covalently modifies the active site of PLA2, prevented the formation of nocodazole-induced dispersed Golgi ministacks, leaving the Golgi as a typical juxtanuclear ribbon (Figure 1E), even though microtubules were depolymerized (Figure 1F). The PLA2 antagonists AACOCF3 (Figure 1, G and H) and PACOCF3 (our unpublished data), which are substrate analogues, also inhibited ministack formation. Similarly, 25 μM of the reversible PLA2 inhibitor ONO-RS-082 prevented ministack formation (Figure 1, I and J); however, in this case, cells were only incubated for 1 h in nocodazole and ONO-RS-082, because longer times in 25 μM ONO-RS-082 were toxic to the cells. Under the conditions used, 1 h in nocodazole was sufficient to form ministacks (our unpublished data). These results suggested that nocodazole-induced ministack formation was not simply caused by the loss of microtubules that no longer tethered the Golgi but instead was a PLA2-dependent event.
The disconnected morphology of the juxtanuclear Golgi complex in cells treated with PLA2 antagonists and nocodazole (Figure 1, E, G, and I) is similar to that seen in cells treated with PLA2 antagonists alone (de Figueiredo et al., 1998). We have reported recently that this morphological change is caused by the inhibition of membrane tubules that form bridges between spatially separate stacks (de Figueiredo et al., 1999).
To examine more closely the structure of the Golgi complex in cells treated with nocodazole and PLA2 antagonists, we performed immunoperoxidase electron microscopy using a polyclonal antibody against ManII. In cells treated with 5 μM BEL before addition of nocodazole, the stacked morphology of the Golgi complex was unchanged compared with the Golgi in control cells (compare Figure 2, C and A) or in cells treated with nocodazole alone (compare Figure 2, C and B). The Golgi stacks were generally smaller in cells treated with nocodazole or nocodazole and BEL compared with that in control cells. Thus, there appeared to be no obvious disruption of the stacked architecture of Golgi cisternae in cells where the formation of ministacks was inhibited by BEL.
Figure 2.

Immunoperoxidase EM analysis of cells treated with BEL and nocodazole. Cells were either untreated (A), treated with nocodazole (6 μg/ml) for 2 h (B), or pretreated with 5 μM BEL followed by nocodazole (6 μg/ml) in the continued presence of BEL for 2 h (C). Cells were then fixed and incubated with rabbit anti-ManII antibodies, followed by sheep anti-rabbit-HRP conjugates, and processed for diaminobenzidine cytochemistry as described previously (Brown and Farquhar, 1989) (see MATERIALS AND METHODS).
Evidence of an ER Intermediate in Nocodazole-induced Ministack Formation
Although a variety of PLA2 antagonists inhibited nocodazole-induced ministack formation, we noticed that the distribution of ManII, in addition to remaining in juxtanuclear Golgi complexes, became somewhat more diffuse with certain antagonists (Figure 1G, nocodazole + AACOCF3). These observations suggested that some ManII may have recycled to the ER. Support for this idea came when we found a dose-dependent, qualitative difference in ManII staining in cells treated with nocodazole and ONO-RS-082. Whereas 25 μM ONO-RS-082 prevented nocodazole-induced changes in the Golgi complex (Figure 1I), 5 μM ONO-RS-082 resulted in the gradual accumulation of ManII in a diffuse and nuclear envelope staining pattern after 1 h (Figure 3A) and 2 h (Figure 3C), suggesting that ManII was in the ER. Also, a structural analogue of ONO-RS-082, ACA, trapped ManII in an ER-like staining pattern in cells in which ministack formation was inhibited (Figure 3, E and F). ManII was found in the nuclear envelope in cells treated with ONO-RS-082 or ACA and nocodazole (Figure 3, C and E, arrows), indicative of its presence in the ER.
Figure 3.

ManII accumulates in an ER intermediate before ministack formation. Clone 9 cells were incubated in 5 μM ONO-RS-082 (A–D) or 25 μM ACA (E and F) at 4°C for 20 min and shifted to 37°C in the continued presence of antagonist plus 6 μg/ml nocodazole for 1 h (A and B) or 2 h (C–F). All conditions included 2 μg/ml cycloheximide. Cells were fixed and processed for double-label immunofluorescence using a polyclonal antibody against ManII (A, C, and E) and a monoclonal antibody against α-tubulin (B, D, and F). The accumulation of ManII in a ring around the nucleus (C and E, arrows) is indicative of its localization in the nuclear envelope and ER.
The dose-dependent inhibition of nocodazole-induced ministack formation by ONO-RS-082 was quantified using an immunofluorescence assay to visualize ManII (Figure 4). Clone 9 cells were pretreated with varying concentrations of ONO-RS-082 at 4°C before shifting to 37°C in the presence of 6 μg/ml nocodazole and antagonist for 2 h. Dispersed Golgi ministack formation was potently inhibited by ONO-RS-082 with an IC50 of ∼3 μM (Figure 4, open squares). In cells treated with low concentrations of ONO-RS-082, ManII staining appeared to be ER-like (Figure 4, open triangles), with the maximum percentage of cells containing ManII in the ER at 5 μM. Increasing the concentration of ONO-RS-082 prevented ManII from leaving the juxtanuclear region (Figure 4, open circles). Additional evidence that ManII accumulated in the ER of cells treated with ONO-RS-082 and nocodazole was provided by double-label immunofluorescence colocalization of ManII and protein disulfide isomerase (PDI) (Figure 5, C and D), a resident ER protein (Noiva and Lennarz, 1992; Sitia and Meldolesi, 1992). These data suggested that movement of resident transmembrane Golgi enzymes into ministacks in nocodazole-treated cells required two ONO-RS-082-sensitive steps: anterograde ER-to-Golgi transport and less sensitive retrograde Golgi-to-ER transport.
Figure 4.

Dose response of ONO-RS-082 inhibition of nocodazole-induced ministack formation. Cells were pretreated with various concentrations of ONO-RS-082 followed by nocodazole for 2 h before fixing and processing for immunofluorescence microscopy, using a polyclonal antibody against ManII to visualize the Golgi. The percentage of cells with ManII in the juxtanuclear region, ER (as determined by nuclear envelope staining), or ministacks was quantified, with each point representing the average of two experiments with 200 cells counted in each experiment.
Figure 5.

Colocalization of ManII with the resident ER enzyme PDI in cells treated with 5 μM ONO-RS-082 and nocodazole. Clone 9 cells were untreated (A and B) or treated with 5 μM ONO-RS-082 (C and D) at 4°C for 20 min before shifting to 37°C in the continued presence of antagonist plus 6 μg/ml nocodazole for 2 h. All conditions included 2 μg/ml cycloheximide. Cells were processed for double-label immunofluorescence microscopy using a polyclonal antibody against ManII (A and C) and a monoclonal antibody against PDI (B and D).
Evidence of Constitutive Recycling of Resident Golgi Enzymes through the ER
Our data suggest that ONO-RS-082 and ACA inhibit the anterograde movement of ManII from the ER to nascent ministacks in the absence of microtubules. If nocodazole treatment is revealing the constitutive cycling of ManII between the Golgi complex and ER, then we would predict that ManII should accumulate in the ER of cells treated with ONO-RS-082 or ACA alone for 2 h. Indeed, when cells were treated with 5 μM ONO-RS-082 alone, ManII gradually over 2 h accumulated in the nuclear envelope and ER, whereas staining was significantly reduced, but not totally absent, in the juxtanuclear Golgi region (Figure 6B). The diffuse ManII staining colocalized with PDI in cells treated with ONO-RS-082 alone (Figure 6, C and D) or ACA alone (Figure 6, E and F). This accumulation in the ER was reversible because ManII returned to the juxtanuclear region after ONO-RS-082 was removed from cells (our unpublished data). Interestingly, ManII did not accumulate in the ER in cells treated with 5 μM BEL for 2 h but instead remained in the juxtanuclear region (Figure 6A), suggesting that retrograde Golgi-to-ER cycling is more sensitive to BEL than to ONO-RS-082.
A more complete kinetic analysis of cells treated with ACA alone revealed the slow accumulation of Golgi-derived ManII in the ER (Figure 7). By 30 min after addition, ManII was still prominently in the Golgi complex, but faint, diffuse staining could be detected (Figure 7A). Between 30 min and 4 h after addition of ACA, the juxtanuclear Golgi ribbon appeared to fragment somewhat and diminish in staining intensity, whereas the diffuse ER-like staining increased until, by 4 h, no Golgi staining was detected (Figure 7B–E). The accumulation of ManII in the ER represented protein recycled from the Golgi, and not newly synthesized protein, because cycloheximide was present throughout the experiment. Similar results were obtained with ONO-RS-082; however, even after 4 h of treatment, a small amount of Golgi staining could be detected (our unpublished data).
Because the absence of microtubules has little effect on retrograde cycling of resident Golgi proteins from nocodazole-induced Golgi ministacks to the ER (Cole et al., 1996; Storrie et al., 1998), we predicted that ManII originating from nocodazole-induced ministacks should accumulate in the ER in the presence of low concentrations of ACA or ONO-RS-082. To test this hypothesis we first treated cells with nocodazole for 2 h to form dispersed Golgi ministacks and then added PLA2 antagonists in the continued presence of nocodazole for an additional 2 h. As seen by indirect immunofluorescence microscopy, a significant portion of ManII shifted from the ministacks (Figure 8A) to the ER in cells treated with 25 μM ACA (Figure 8B) or 5 μM ONO-RS-082 (Figure 8C) for 2 h. These results provided additional evidence that resident Golgi enzymes continuously cycle between nocodazole-induced Golgi ministacks and the ER and that ACA and ONO-RS-082 inhibit the transport of ManII from the ER to Golgi ministacks. In Golgi ministack-containing cells treated with 25 μM AACOCF3 for 2 h, ManII did not significantly accumulate in the ER but instead remained mainly in peripheral Golgi ministacks (Figure 8D). Together, these data are consistent with a model in which resident Golgi enzymes continuously cycle between the Golgi and the ER and suggest that there are two PLA2-dependent steps: a retrograde Golgi-to-ER step that is more sensitive to BEL, AACOCF3, and PACOCF3 and an anterograde ER-export step that is more sensitive to ACA and ONO-RS-082.
Transport of the VSVG Protein in the Presence of PLA2 Antagonists
Because the ER-to-Golgi transport of a cycling, resident Golgi protein was affected by ONO-RS-082, we wished to examine the effects of PLA2 antagonists on the ER-to-Golgi transport of a newly synthesized transmembrane protein that is transported via the secretory pathway. To do this we used the VSVts045 temperature-sensitive mutant of VSVG protein to examine the synchronous movement of VSVG from the ER to the Golgi in the presence or absence of PLA2 antagonists by indirect double-label immunofluorescence microscopy. Clone 9 cells were infected with VSVts045 and kept at the restrictive temperature of 40°C for 3 h to accumulate VSVG in the ER. In cells kept at 40°C, essentially all of the VSVG protein remained in the ER, as seen by the diffuse staining using a monoclonal anti-VSVG protein antibody (Figure 9A); only ∼1% of the cells contained VSVG in the Golgi (Figure 9J). However, after a shift to the permissive temperature of 32°C for 30 min, VSVG was efficiently transported to the Golgi (Figure 9J, 96% of the cells contained VSVG in the Golgi), as evidenced by the colocalization of VSVG with ManII (Figure 9, C and D). In cells that were pretreated with 5 μM ONO-RS-082 for 30 min at 40°C before shifting to 32°C in the continued presence of ONO-RS-082 for 30 min, transport of the VSVG protein to the Golgi was inhibited (Figure 9J, 12% of the cells contained VSVG in the Golgi), because VSVG and ManII staining did not colocalize (Figure 9, E and F). Conversely, treatment with 5 μM BEL at 40°C before shifting to 32°C in the continued presence of BEL did not prevent transport of VSVG to the Golgi (Figure 9, G and H). These results are consistent with the above studies showing that certain PLA2 antagonists caused accumulation of ManII in the ER during nocodazole treatment. Moreover, they suggest that there is a PLA2-dependent transport step between the ER and the Golgi for both Golgi proteins, i.e., ManII, and those destined for the cell surface, i.e., VSVG.
Figure 9.

ONO-RS-082, but not BEL, inhibits ER-to-Golgi transport of the VSVG protein. Clone 9 cells were infected with VSVts045 at 40°C and kept at the restrictive temperature (40°C) for 3 h to accumulate the VSVG protein in the ER (see MATERIALS AND METHODS). Transport of the VSVG protein was followed by immunofluorescence microscopy using a monoclonal antibody against the VSVG protein (A, C, E, and G) and a polyclonal antibody against ManII (B, D, F, and H). Cells were kept at 40°C (A and B) or shifted to the permissive temperature 32°C for 30 min in the absence (C and D) or presence of ONO-RS-082 (E and F) or BEL (G and H). In J, the percentage of cells with the VSVG protein in the Golgi at 32°C in the presence of ONO-RS-082 and BEL was determined. Colocalization of the VSVG protein and ManII was counted as transport of the VSVG protein to the Golgi. Each column represents the average of two experiments with 100 cells counted in each experiment.
PLA2 Antagonists Inhibit Golgi Complex Reformation from Ministacks
Previous work in our lab has shown that a PLA2 activity(s) is necessary to reform a continuous interconnected Golgi complex during recovery from pharmacological disruption by BFA and ilimaquinone (de Figueiredo et al., 1999), and we wished to examine the effect of PLA2 antagonists on the reformation of the Golgi after removal of nocodazole. To do this, ministacks were formed by treating cells with nocodazole alone for 2 h; then cells were washed free of nocodazole and allowed to recover in the absence or presence of 5 μM ONO-RS-082 for various periods of time. Normal reformation of the Golgi and repolymerization of microtubules occurred rapidly, because ManII-positive elements began to cluster and move to the juxtanuclear region within 15 min after nocodazole removal (Figure 10, A and B). Golgi recovery in the absence of PLA2 antagonists was complete by 60 min (Figure 10, E and F) and appeared to involve the formation of ManII-positive tubules extending from Golgi elements (Figure 10, A and C, arrows). In the presence of 5 μM ONO-RS-082 only partial reassembly of the Golgi occurred after removal of nocodazole (Figure 10G–L). Under these conditions Golgi elements were able to move to the juxtanuclear region, but they did not coalesce into an intact Golgi ribbon (Figure 10G–L), resulting in a fragmented juxtanuclear structure similar to that seen after 1 h in ONO-RS-082 alone (de Figueiredo et al., 1998). Repolymerization of microtubules was not inhibited (Figure 10, H, J, and L). Thus, it appears that ONO-RS-082 did not interfere with the movement of Golgi elements toward the minus end of microtubules but rather inhibited the final coalescence of Golgi elements into a connected and continuous Golgi complex.
DISCUSSION
The dynamic and continuous cycling of resident Golgi proteins through the ER has been proposed as the mechanism to explain the formation of Golgi ministacks in the absence of microtubules (Cole et al., 1996). BFA-stimulated retrograde redistribution of Golgi proteins to the ER has been shown previously to be inhibited by PLA2 antagonists (de Figueiredo et al., 1998). On the basis of these results, we predicted that if retrograde recycling of Golgi proteins through the ER was required for nocodazole-induced ministack formation, then PLA2 antagonists might inhibit ministack formation. Indeed, in the present work, we showed that low concentrations of PLA2 antagonists inhibited nocodazole-induced ministack formation, suggesting that a PLA2 activity(s) was involved in this process. We infer from our results that PLA2 antagonists targeted specific cytosolic PLA2(s) and did not nonspecifically poison cells because of the following: 1) the inhibition of ministack formation was both rapid and reversible, and under all experimental conditions cells remained viable as determined by trypan blue exclusion; 2) a wide range of inhibitors of cytoplasmic PLA2 activity was tested (Ca2+-dependent and independent, covalent active site–binding and substrate analogues) that act by different mechanisms, and all inhibited ministack formation; and 3) the low micromolar concentrations used to inhibit ministack formation were comparable with the concentrations that inhibited both BFA-stimulated Golgi-to-ER trafficking (de Figueiredo et al., 1998) and known intracellular cytosolic PLA2 activities (Hazen et al., 1991; Gelb et al., 1994; Ackermann et al., 1995).
Inhibition of nocodazole-induced ministack formation by BEL, PACOCF3, and AACOCF3 resulted in the formation of large, disconnected Golgi fragments that remained in the juxtanuclear region. This change in Golgi morphology was identical to that reported when cells were treated with PLA2 antagonists alone (de Figueiredo et al., 1998) and was shown recently to be caused by the inhibition of the dynamic formation of membrane tubules that serve to connect spatially separate Golgi stacks into a single, interconnected organelle (de Figueiredo et al., 1999). Thus, our results are not consistent with nocodazole-induced formation of ministacks by fragmentation and passive diffusion, because the Golgi complex remained in the juxtanuclear region in the absence of microtubules. More relevant, there was a PLA2-dependent step early in the redistribution of Golgi enzymes to form ministacks induced by nocodazole. These results agree with previous studies that proposed that passive diffusion of Golgi elements was insufficient to explain ministack formation because it was an energy-dependent, N-ethyl maleimide- sensitive process (Turner and Tartakoff, 1989; Cole et al., 1996).
Our conclusion that nocodazole-induced ministack formation requires the recycling of resident Golgi enzymes through the ER was based on the unexpected finding that a subset of PLA2 antagonists exhibited a concentration-dependent, differential block at two separate stages of the recycling pathway: 25 μM ONO-RS-082 caused ManII to remain primarily in juxtanuclear complexes, whereas 5 μM ONO-RS-082 caused ManII to gradually accumulate in the ER. Importantly, ManII that accumulated in the ER must have originated from the juxtanuclear region because no ministacks were observed before its appearance in the ER. These data are entirely consistent with the finding that expression of a dominant-negative mutant of Sar1p inhibited ministack formation and accumulated Golgi markers in the ER (Storrie et al., 1998). The fact that Golgi markers accumulated in the ER before ministack formation in the presence of PLA2 antagonists and the fact that the PLA2 antagonists that inhibited retrograde Golgi-to-ER trafficking also inhibited the disappearance of the Golgi complex provide strong evidence of the recycling model of ministack formation.
The underlying mechanism of the recycling model of ministack formation was suggested by Lippincott-Schwartz and colleagues to be the slow constitutive cycling of resident Golgi proteins through the ER, possibly representing a quality control pathway to degrade proteins targeted for destruction (Cole et al., 1998). A model of continuous recycling was used to explain the gradual Golgi disappearance and ER accumulation of chimeric VSVGts045-resident Golgi proteins shifted to the restrictive temperature in living cells (Cole et al., 1998), as well as the accumulation of Golgi markers in the ER of nocodazole-treated cells expressing a dominant-negative mutant of Sar1p (Storrie et al., 1998). Our data showing that ManII accumulated in the ER in the presence of ACA or 5 μM ONO-RS-082 under three different conditions—1) in the presence of the antagonists alone, 2) when the antagonists are added before nocodazole-induced ministack formation, and 3) when the antagonists are added after ministack formation—also support the model of constitutive cycling of resident Golgi proteins through the ER. Thus, recent studies describing the dynamics of Golgi proteins suggest that Golgi proteins cannot be simply classified as resident or recycling but should rather be distinguished on the basis of their rates of cycling between the Golgi complex and the ER.
Previous work from our lab showed that 1 μM ONO-RS-082 did not affect ER-to-Golgi transport of the VSVG protein (de Figueiredo et al., 1999), but in the present study we found that 5 μM ONO-RS-082 inhibited ER-to-Golgi transport. Interestingly, an intracellular PLA2 enzyme has been implicated in ER-to-Golgi transport of apoproteins in vitro (Slomiany et al., 1992), and a PLA2 antagonist has been reported to block secretion of prolactin in vivo (Tagaya et al., 1993). The exact molecular target involved in ER-to-Golgi transport that is affected by ONO-RS-082 and ACA is unknown, but it is tempting to speculate that they may be inhibiting the formation of tubules or pleomorphic structures containing VSVG–green fluorescent protein that have been observed moving from the ER to the Golgi in living cells (Presley et al., 1997). The fact that antagonists that were more specific for Ca2+-independent (BEL, AACOCF3, and PACOCF3) than Ca2+-dependent PLA2s (Hazen et al., 1991; Ackermann et al., 1995) had no effect on anterograde transport suggests that multiple PLA2s act at different transport steps or various transport steps have a differential dependence on PLA2 activity. Caution must, of course, be exercised when interpreting data based on inhibitors because they may have indirect effects. And, irrespective of any conclusions about the potential role for a cytoplasmic PLA2(s) in mediating intracellular trafficking, the PLA2 antagonists were primarily valuable in these studies for revealing the accumulation of recycling Golgi proteins in the ER during nocodazole-induced ministack formation.
Reformation of an interconnected Golgi complex after mitotic and pharmacological breakdown may involve tubular connections between coalescing stacks (Lucocq et al., 1989; Rabouille et al., 1995a,b; de Figueiredo et al., 1999; Polishchuk et al., 1999). Also, tubular extensions connecting adjacent Golgi cisternae have been observed by freeze-etch electron microscopy (EM) (Weidman et al., 1993) and thin and thick section EM (Novikoff et al., 1971; Rambourg et al., 1979; Rambourg and Clermont, 1990). Recent work from our lab has implicated PLA2-dependent tubular membrane extensions between adjacent Golgi stacks in the coalescence of an interconnected organelle after the removal of BFA and ilimaquinone (de Figueiredo et al., 1999). Thus, our finding that PLA2 antagonists inhibited a late step in Golgi reassembly after nocodazole washout suggests a common, PLA2-dependent pathway for Golgi reformation whether the Golgi membranes originated from the ER or preexisting ministacks scattered throughout the cytoplasm. The specific protein(s) involved in Golgi coalescence in the juxtanuclear region, whose activity is inhibited by PLA2 antagonists, remains to be identified.
In summary, our results confirm and extend previous studies that concluded that resident Golgi enzymes constitutively recycle through the ER. Moreover, our results support the idea that nocodazole-induced Golgi ministack formation results from disrupting this Golgi–ER cycle by inhibiting the microtubule-dependent centripetal movement of nascent Golgi elements from dispersed ER-exit sites to the juxtanuclear Golgi region, as evidenced by the ER accumulation of Golgi proteins in the presence of certain PLA2 antagonists. The use of PLA2 antagonists in future studies should facilitate the dissection of the complex coordination of anterograde and retrograde trafficking of the Golgi–ER system.
ACKNOWLEDGMENTS
We are grateful to Drs. Marilyn Farquhar and Kelly Moreman for the gift of anti-ManII antibodies, Dr. Vivek Malhotra for supplying VSVts045 and the anti-VSVG protein antibodies, and Dr. Bill Balch for the anti-β-COP antibodies. We thank Marian Strang for her assistance with the electron microscopy experiments. Also, we thank Dr. Esther Racoosin for helpful scientific discussions and critical review of the manuscript and Dr. Anthony Bretscher for the generous use of his fluorescence microscope. This work was supported by National Institutes of Health grant DK-51596 (to W.J.B.).
Abbreviations used:
- AACOCF3
arachidonyl trifluoromethyl ketone
- ACA
N-(p-amylcinnamoyl)anthranilic acid
- BEL
bromoenol lactone
- BFA
brefeldin A
- COP
coatomer protein
- EM
electron microscopy
- ER
endoplasmic reticulum
- FCS
fetal calf serum
- ManII
α-mannosidase II
- MEM
Eagle’s minimal essential medium
- MTOC
microtubule-organizing center
- ONO-RS-082
2-(p-amylcinnamoyl)amino-4-chlorobenzoic acid
- PACOCF3, palmityl trifluoromethyl ketone
PBS, phosphate-buffered saline
- PDI
protein disulfide isomerase
- PLA2
phospholipase A2
- VSVGts045
temperature-sensitive vesicular stomatitis virus G protein
REFERENCES
- Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- Allan BB, Balch WE. Protein sorting by directed maturation of Golgi compartments. Science. 1999;285:63–66. doi: 10.1126/science.285.5424.63. [DOI] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta M, Polizotto RS, Wood SA, de Figueiredo P, Brown WJ. Characterization of a cytosolic activity that induces the formation of Golgi membrane tubules in a cell-free reconstitution system. Biochemistry. 1995;34:13359–13366. doi: 10.1021/bi00041a012. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Goldstein LS. Cruising along microtubule highways: how membranes move through the secretory pathway. J Cell Biol. 1998;140:1277–1280. doi: 10.1083/jcb.140.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WJ, Constantinescu E, Farquhar MG. Redistribution of mannose-6-phosphate receptors induced by tunicamycin and chloroquine. J Cell Biol. 1984;99:320–326. doi: 10.1083/jcb.99.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WJ, Farquhar MG. Immunoperoxidase methods for the localization of antigens in cultured cells and tissue sections by electron microscopy. Methods Cell Biol. 1989;31:553–569. doi: 10.1016/s0091-679x(08)61626-x. [DOI] [PubMed] [Google Scholar]
- Cole NB, Ellenberg J, Song J, DiEuliis D, Lippincott-Schwartz J. Retrograde transport of Golgi-localized proteins to the ER. J Cell Biol. 1998;140:1–15. doi: 10.1083/jcb.140.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Lippincott-Schwartz J. Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol. 1995;7:55–64. doi: 10.1016/0955-0674(95)80045-x. [DOI] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo P, Drecktrah D, Katzenellenbogen JA, Strang M, Brown WJ. Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc Natl Acad Sci USA. 1998;95:8642–8647. doi: 10.1073/pnas.95.15.8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo P, Polizotto RS, Drecktrah D, Brown WJ. Membrane tubule-mediated reassembly and maintenance of the Golgi complex is disrupted by phospholipase A2 antagonists. Mol Biol Cell. 1999;10:1763–1782. doi: 10.1091/mbc.10.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb MH, Jain MK, Berg OG. Inhibition of phospholipase A2. FASEB J. 1994;8:917–924. doi: 10.1096/fasebj.8.12.8088457. [DOI] [PubMed] [Google Scholar]
- Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- Knipe D, Baltimore MD, Lodish HF. Maturation of viral proteins in cells infected with temperature-sensitive mutants of vesicular stomatitis virus. J Virol. 1977;21:1149–1158. doi: 10.1128/jvi.21.3.1149-1158.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge O, Dascher C, Orci L, Rowe T, Amherdt M, Plutner H, Ravazzola M, Tanigawa G, Rothman JE, Balch WE. Sar1 promotes vesicle budding form the endoplasmic reticulum but not Golgi compartments. J Cell Biol. 1994;125:51–65. doi: 10.1083/jcb.125.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J. Cytoskeletal proteins and Golgi dynamics. Curr Opin Cell Biol. 1998;10:52–59. doi: 10.1016/s0955-0674(98)80086-0. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Lucocq JM, Berger EG, Warren G. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J Cell Biol. 1989;109:463–474. doi: 10.1083/jcb.109.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixative for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Noiva R, Lennarz WJ. Protein disulfide isomerase. A multifunctional protein resident in the lumen of the endoplasmic reticulum. J Biol Chem. 1992;267:3553–3556. [PubMed] [Google Scholar]
- Novikoff PM, Novikoff AB, Quintana N, Hauw JJ. Golgi apparatus, GERL, and lysosomes of neurons in rat dorsal root ganglia, studied by thick section and thin section cytochemistry. J Cell Biol. 1971;50:859–886. doi: 10.1083/jcb.50.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka M, Ellinger A. Effect of colchicine on the Golgi complex of rat pancreatic acinar cells. J Cell Biol. 1983;97:737–748. doi: 10.1083/jcb.97.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk RS, Polishchuk EV, Mironov AA. Coalescence of Golgi fragments in microtubule-deprived living cells. Eur J Cell Biol. 1999;78:170–185. doi: 10.1016/S0171-9335(99)80096-X. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters JM, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995a;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Mistelli T, Watson R, Warren G. Reassembly of Golgi stacks form mitotic Golgi fragments in a cell-free system. J Cell Biol. 1995b;129:605–618. doi: 10.1083/jcb.129.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambourg A, Clermont Y. Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur J Cell Biol. 1990;51:189–200. [PubMed] [Google Scholar]
- Rambourg A, Clermont Y, Hermo L. Three-dimensional structure of the Golgi apparatus in Sertoli cells of the rat. Am J Anat. 1979;154:455–476. doi: 10.1002/aja.1001540402. [DOI] [PubMed] [Google Scholar]
- Rogalski AA, Singer SJ. Associations of elements of the Golgi apparatus with microtubules. J Cell Biol. 1984;99:1092–1100. doi: 10.1083/jcb.99.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Cabrera-Poch N, Pepperkok R, Warren G. An ordered inheritance strategy for the Golgi apparatus: visualization of mitotic disassembly reveals a role for the mitotic spindle. J Cell Biol. 1998;141:955–966. doi: 10.1083/jcb.141.4.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitia R, Meldolesi J. Endoplasmic reticulum: a dynamic patchwork of specialized subregions. Mol Biol Cell. 1992;3:1067–1072. doi: 10.1091/mbc.3.10.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomiany A, Grzelinska E, Kasinathan C, Yamaki K, Palecz D, Slomiany BL. Function of intracellular phospholipase A2 in vectoral transport of apoproteins from ER to Golgi. Int J Biochem. 1992;24:1397–1406. doi: 10.1016/0020-711x(92)90065-9. [DOI] [PubMed] [Google Scholar]
- Storrie B, White J, Rottger S, Stelzer EHK, Suganuma T, Nilsson T. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J Cell Biol. 1998;143:1505–1521. doi: 10.1083/jcb.143.6.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaya M, Henomatsu N, Yoshimori T, Yamamoto A, Tashiro Y, Fukui T. Correlation between phospholipase A2 activity and intra-Golgi protein transport reconstituted in a cell free system. FEBS Lett. 1993;324:201–204. doi: 10.1016/0014-5793(93)81393-e. [DOI] [PubMed] [Google Scholar]
- Thyberg J, Moskalewski S. Microtubules and the organization of the Golgi complex. Exp Cell Res. 1985;159:1–6. doi: 10.1016/s0014-4827(85)80032-x. [DOI] [PubMed] [Google Scholar]
- Turner JR, Tartakoff AM. The response of the Golgi complex to microtubule alterations: the roles of metabolic energy and membrane traffic in Golgi complex organization. J Cell Biol. 1989;109:2081–2088. doi: 10.1083/jcb.109.5.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidman P, Roth R, Heuser J. Golgi membrane dynamics imaged by freeze-etch electron microscopy: views of different membrane coatings involved in tubulation versus vesiculation. Cell. 1993;75:123–133. [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- Wooding S, Pelham HRB. The dynamics of Golgi protein traffic visualized in living yeast cells. Mol Biol Cell. 1998;9:2667–2680. doi: 10.1091/mbc.9.9.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]