

Abstract
Telomerase adds simple-sequence repeats to chromosome ends to offset the terminal sequence loss inherent in each cycle of genome replication. Inherited mutations in genes encoding subunits of the human telomerase holoenzyme give rise to disease phenotypes including hematopoietic failure and pulmonary fibrosis. Disease-associated variants of the human telomerase RNA are expressed in heterozygous combination with wild-type telomerase RNA. Here, we exploit a sensitized human primary cell assay system to investigate the biological function of disease-linked telomerase RNA variants and their impact on the function of coexpressed wild-type telomerase RNA. We find that telomerase RNA variants discovered in patients with dyskeratosis congenita or aplastic anemia show loss of function without any indication of dominant-negative impact on telomere maintenance by the coexpressed wild-type RNA. To reconcile this result with contradictory findings from reconstitution assays in vitro, we demonstrate that the lack of dominant-negative impact on telomere maintenance correlates with physiological assembly of active human telomerase holoenzyme ribonucleoproteins harboring monomers rather than higher-order multimers of telomerase RNA and telomerase reverse transcriptase. These findings support loss of function of telomerase RNA as a general mechanism of human disease.
DNA replication must be both accurate and complete in order to preserve genome integrity through many rounds of cell division. The linear nature of eukaryotic chromosomes complicates the requirements for genome replication, because the high-fidelity, primer-dependent polymerases that perform semiconservative DNA synthesis fail to duplicate chromosome termini. To compensate for incomplete end replication by DNA-templated DNA polymerases, eukaryotes evolved a specialized chromosome end maintenance mechanism. Most eukaryotic chromosome ends are capped by a tandem array of telomeric simple-sequence repeats with a 3′ single-stranded overhang (19). These telomeric repeats are necessary and sufficient to protect chromosome ends from inappropriate recombination, fusion, or degradation (39). In addition, tandem telomeric repeats allow chromosome end maintenance by telomerase.
Telomerase is a unique ribonucleoprotein (RNP) reverse transcriptase devoted to the task of telomeric repeat synthesis (4, 7). Telomerase recognizes authentic chromosome 3′ termini and extends them by new telomeric repeat synthesis. The telomerase RNA component (TER) contains the template for repeat synthesis, which is copied by the active site of telomerase reverse transcriptase (TERT). Assembly of TER and TERT using a heterologous cell extract such as rabbit reticulocyte lysate can reconstitute template-dependent DNA synthesis (2, 43). In vivo assembly of endogenous telomerase holoenzymes requires additional proteins that fold TER into a stable and active RNP conformation, assemble TER RNP with TERT, and regulate the association of catalytically active enzyme with its telomere substrates (9).
Single-celled organisms with constitutively active telomerase balance replication-linked loss of telomeric repeats with new repeat synthesis to achieve a telomere-length homeostasis. Numerous studies have investigated how telomere-interacting proteins can effectively count the number of repeats to establish an equilibrium set point for telomere length maintenance (22). In multicellular organisms, including humans, the extent to which telomere length depends on a telomere-based repeat-counting mechanism is unclear. Human somatic cells generally repress TERT expression (12). Even under conditions that strongly induce endogenous telomerase catalytic activation, only transient telomere elongation occurs before cells return to unabated telomere erosion (5). It seems likely that in the human organism, telomerase subunit expression levels and their regulated assembly to form active RNP are the predominant factors that determine telomere length (10). Even in human tumor cell lines, which upregulate TER and TERT relative to normal physiological levels, telomere length remains limited by expression of TER and/or TERT (13, 17).
Studies of human disease, along with related studies using model organisms, support the hypothesis that telomerase function is limited by steady-state accumulation of TER (20, 50). Patients with X-linked or autosomal dominant (AD) dyskeratosis congenita (DC) inherit altered sequence of the TER-binding protein dyskerin (X-linked DC) or heterozygous mutation of the gene encoding TER (AD DC). X-linked DC patient cells express one of a large variety of single-amino-acid substitutions of dyskerin and as a consequence have TER levels reduced to 20 to 40% of normal (33, 48, 49). Cells from an AD DC patient with a heterozygous mutation that prevents TER accumulation have ∼50% of the normal level of TER, which is produced entirely from the wild-type allele (45). Even these modest reductions in TER accumulation impose severe disease phenotypes and early mortality, typically due to bone marrow failure (18, 40). Also, constitutive expression of TERT in primary cells from patients with X-linked or AD DC fails to give an extent of telomere elongation comparable to that obtained in normal primary cells (45, 49). Thus, as little as 50% reduction in the steady-state accumulation of human TER compromises telomerase function at telomeres.
In some patients with AD DC or aplastic anemia (AA), the TER sequence change disrupts TER function without apparent impact on TER steady-state accumulation. TER variants in this second class are not altered in the sequence motifs known to be essential for precursor processing and assembly into biologically stable RNP (18, 40). Transient transfection assays support the prediction that these TER variants accumulate in vivo as RNP with reduced catalytic activity (16, 40). To mimic the disease context in which a TER variant is expressed in heterozygous combination with wild-type TER, activity assays have coexpressed or combined wild-type and variant TERs. In most cases, these studies report that a TER variant does not inhibit the coexpressed wild-type TER (16, 40). In other cases, in particular for disease-associated TER variants with sequence changes in the template, the results suggest that the TER variant has a strong dominant-negative impact on wild-type TER activity (51). In vitro reconstitution studies performed prior to the discovery of disease-associated TER variants also used mixed template-altered and wild-type TERs with the conclusion that there is a strong dominant-negative impact of mutant TER. Functional interference was attributed to the presence of multiple TER molecules in a single catalytically active human telomerase RNP particle (2, 44).
Critical evaluation of the disease-relevant impact of TER variants on coexpressed wild-type TER function should utilize human primary cells as the assay system, in a setting where TER is limiting for telomerase catalytic activity and telomere elongation. Here, we exploit X-linked DC patient primary fibroblasts expressing TERT as this system. We find that AD DC and AA TER alleles show loss of function without any evidence for a dominant-negative impact on telomere maintenance by wild-type TER. We correlate this lack of dominant-negative impact with the demonstration that TER and TERT assemble as monomers in active human telomerase holoenzyme.
MATERIALS AND METHODS
Cell culture, retroviral integration, and transfection.
Primary dermal fibroblast cultures of GM01774 (X-linked DC patient cells) and GM01787 (family-matched normal control cells) with an integrated TERT expression vector were grown and infected with retrovirus as previously described (49). The TER retroviral expression vector uses the U3-TER-500 cassette (16) cloned into the NheI site of pBABEpuro to generate pBABE-U3-TER (49). Chemical selection for integration began 48 h after infection using 2.5 μg/ml puromycin. Surviving cells were pooled and grown as polyclonal cultures, with population doublings counted from the first postselection split. 293T and VA13+TERT cells were grown and used for transient transfection by the calcium phosphate method as previously described (32).
TER, TERT, telomerase activity, and telomere length.
RNA samples were prepared using TRIzol. Northern blot hybridization for TER, the endogenous cross-reacting RNA loading control, and/or the recovery control for precipitation was performed using end-labeled 2′-O-methyl RNA oligonucleotide probe as described previously (16). Immunoblots for TERT used affinity-purified rabbit polyclonal human TERT peptide antibody (33). Telomerase activity assays used whole-cell extracts made by freeze-thaw lysis and telomeric repeat amplification protocol (TRAP) conditions described previously (32), including a competitive internal amplification control longer than the longest TRAP products to control for suppression of amplification of the repeat ladder. Telomere length was assayed using genomic DNA digested with HinfI and RsaI. Following electrophoresis, in-gel hybridization was performed using the end-labeled oligonucleotide (T2AG3)3 (34).
RNA-based affinity purification.
RNA hairpin sequence, recombinant coat protein, and general purification procedures have been described previously (21). For tagged TER affinity purification, whole-cell extracts were diluted to 1 mg/ml in binding buffer (20 mM HEPES buffer at pH 7.9, 150 mM NaCl, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 0.1% NP-40, 1 mM dithiothreitol, phenylmethylsulfonyl fluoride) and clarified by centrifugation to produce input. Lysate was supplemented with 3.5 μg/ml tagged Pseudomonas phage 7 (PP7) coat protein and rotated end over end for 60 min at 4°C before addition of 20 μl/ml rabbit immunoglobulin G (IgG) agarose (Sigma) and 90 min of additional rotation. For single-step purifications, bound samples were washed three times in wash buffer, which is binding buffer with 0.2% Triton X-100 and 0.2% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} for 15 min at room temperature followed by RNA extraction with TRIzol. If a subsequent affinity purification step for coexpressed FLAG3-TERT was to be performed, first-step-bound samples were washed three times with wash buffer for 5 min at 4°C and then washed twice with binding buffer for 5 min at 4°C. Elution was performed by cleavage between the coat protein and its tag using ∼30 ng/μl S219V tobacco etch virus (TEV) protease (23) in one packed resin volume of elution buffer (binding buffer with 0.2 U/μl RNaseIN [Promega], 100 ng/μl molecular-grade bovine serum albumin, 100 ng/μl tRNA, 100 ng/μl casein), with end-over-end rotation for 1 h at 4°C. Resin was recovered by centrifugation and rinsed with another packed resin volume of elution buffer. Elution supernatants were cleared of contaminating beads using Micro Bio spin columns (Bio-Rad) and transferred to an ultralow-retention tube. Second-step purification used 5 μl FLAG M2 antibody resin (Sigma) rotated end over end to allow binding for 45 min at 4°C. Final bound samples were washed three times in binding buffer for 5 min at 4°C followed by RNA extraction with TRIzol.
Protein-based affinity purification.
For tagged TERT affinity purification, whole-cell extracts were diluted to 0.7 mg/ml in binding buffer and clarified by centrifugation to produce input. Rabbit IgG agarose or FLAG M2 antibody resin washed in binding buffer was added using 5 μl packed beads per 350 μl extract and rotated end over end for 2 h at 4°C. Bound samples were washed twice in binding buffer for 5 min at room temperature then transferred to ultralow-retention tubes for a final wash in elution buffer, which is binding buffer with 0.1% Triton X-100 and 0.2% CHAPS. Elution was performed using TEV protease (described above) or 10 to 15 ng/μl FLAG peptide in 50 μl elution buffer (wash buffer with 100 ng/μl molecular-grade bovine serum albumin and 100 ng/μl tRNA) by rotation end over end for 15 min at room temperature. Resin was recovered by centrifugation and rinsed with another 50 μl elution buffer. Both elution supernatants were cleared of contaminating beads using Micro Bio spin columns and transferred to an ultralow-retention tube for second-step purification using the alternate resin. Binding was performed by end-over-end rotation for 15 to 20 min at room temperature followed by washing and elution as described above, except for use of a reduced 10-μl elution volume and 10-min incubation. Because TEV protease in the IgG agarose elutions inhibits the TRAP assay (data not shown), the His-tagged protease was removed from eluted samples by a 10-min end-over-end rotation with 5 μl packed Ni-nitrilotriacetic acid agarose followed by supernatant recovery.
RESULTS
A primary cell system for testing the biological activity of human TER variants.
An experimental system for assaying the telomere synthesis function of TER variants in human primary cells has been lacking. Primary cells constitutively express and accumulate mature TER (15). Mature TER overaccumulation in primary fibroblasts, at least in cell cultures that also express TERT, appears strongly growth inhibitory (49). In addition, reducing the level of endogenous TER in primary cells is technically difficult. Experimental RNA interference is challenged by the highly folded structure and nuclear localization of TER and by its long half-life of many days or weeks in vivo (53).
Primary fibroblasts from X-linked DC patients forced to constitutively express TERT from an integrated retrovirus (designated as X-linked DC+TERT cells) have ∼25% of the level of TER and much shorter telomeres than normal primary fibroblasts expressing TERT (designated as WT+TERT cells). The low level of endogenous TER in X-linked DC+TERT cells is sufficient for modest elongation of critically short telomeres that would otherwise impose senescence, but bulk telomere length continues to shorten (49). X-linked DC+TERT cells that also express extra TER precursor (designated as X-linked DC+TERT+TER cells) show increased mature TER accumulation, correspondingly increased telomerase catalytic activity in cell extract, and telomere elongation (49). Importantly, the total level of mature TER in X-linked DC+TERT+TER cells is close to that in WT+TERT cells, with the consequence that no growth inhibition is observed. Because recombinant TER accumulates in ∼5-fold excess of endogenous TER in X-linked DC+TERT+TER cells, this system provides a sensitized context for evaluation of variant TER function and its potential dominant-negative impact on the catalytic activity and telomere maintenance function of wild-type TER.
We examined the biological activities of several clinically characterized human TER variants (Fig. 1) using the previously characterized wild-type TER as a positive control and empty TER expression vector as a negative control. To express biologically functional mature TER in vivo, we used a vector context in which mature TER sequence is preceded by the U3 small nucleolar RNA polymerase II promoter and followed by a downstream region lacking a polyadenylation signal (16). To insulate this expression cassette from overlapping mRNA transcription, it is placed within the long terminal repeat of an integrated retrovirus (49). Other expression systems that position TER sequence between an mRNA promoter and polyadenylation signal will yield a mixed-transcript population with aberrantly 3′-extended TER (16, 27, 31), which can reconstitute RNP active in vitro that is likely to be mislocalized in vivo.
FIG. 1.
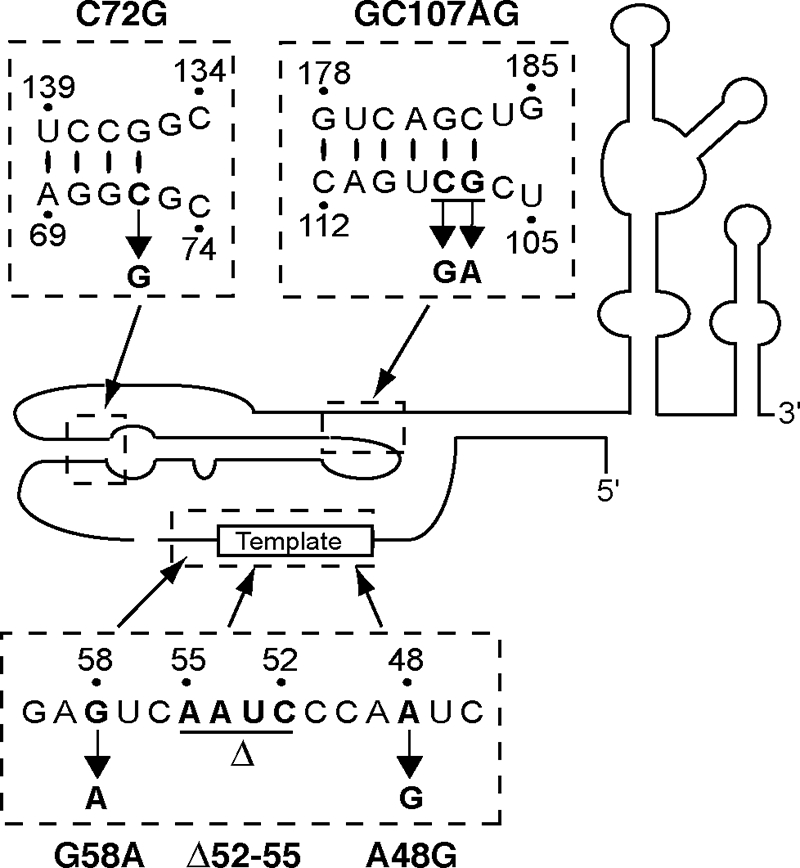
Structure of human TER and TER variants. The secondary structure of mature TER is schematized. Dashed boxes illustrate primary sequence changes studied in this work.
Stable expression of TER variants with sequence changes in the pseudoknot region.
Many studies have probed the significance of human TER pseudoknot structure using mutagenesis (37). In vitro assays of catalytic activity demonstrate severe inhibition resulting from mutations in the pseudoknot stems and conserved residues of the connecting loops. The rationale for the impact of many different sequence substitutions became evident with nuclear magnetic resonance structure determination of a model of the human TER pseudoknot, which revealed a triple-helical base pairing of one highly conserved loop and stem (36). An AD DC mutation disrupts pseudoknot structure by replacing GC at residues 107 and 108 with AG (GC107AG), unpairing the end of a helix beyond the region of base triple formation (Fig. 1). When reconstituted with TERT using a transient transfection vector that produces mature TER, the GC107AG variant showed very severely reduced telomerase activation (16). Another catalytically impaired TER variant was identified in a patient with AA, in which C at residue 72 is replaced by G (C72G). This substitution disrupts a predicted base pair within the longer pseudoknot stem (Fig. 1). When reconstituted with TERT using a transient transfection vector that produces mature TER, the C72G TER variant also showed severely reduced telomerase activity; however, a low level of activity above that of the GC107AG variant remained detectable (16). Neither of these pseudoknot region TER variants shows a dominant-negative impact on reconstitution of catalytic activity by wild-type TER in any of the various assay formats employed (17, 39).
We used X-linked DC+TERT cells to stably express the GC107AG and C72G TER variants in parallel with wild-type TER and empty vector controls. Cell cultures were sampled at increasing population doubling level (PDL) following selection for vector integration to examine TER accumulation, telomerase activation, and telomere length. Introduction of the empty vector negative control did not increase the steady-state level of mature TER monitored by Northern blotting, whereas introduction of recombinant wild-type, GC107AG, or C72G TER did increase the overall level of TER (Fig. 2A). Slightly lower steady-state accumulation levels were observed for the recombinant TER variants compared to the wild type, but all of the recombinant TERs accumulated in molar excess of endogenous wild-type TER. Next, cell extracts harboring TER RNPs were assayed for telomerase catalytic activity with the PCR-amplified TRAP assay, comparing products across a titration of cell extract normalized for total protein. Over many TRAP assay repetitions, the expression of recombinant wild-type TER but not of either recombinant TER variant led to an obvious increase in telomerase catalytic activation (Fig. 2B [additional data not shown]). This is consistent with the low level of telomerase catalytic activation expected for the pseudoknot region TER variants (15), which should be substantially less than the level of telomerase catalytic activation accomplished by the endogenously encoded wild-type TER.
FIG. 2.

Stable expression and functional assay of TER variants in the pseudoknot region. (A) Northern blot of mature TER and a loading control (LC) detected in total RNA from cell extract. “Relative TER” indicates the hybridization signal for TER after normalization to the LC and expressed relative to recombinant wild-type (WT) TER. Note that mature TER migrates as a doublet due to partial folding during electrophoresis. (B) TRAP assay with 2.5-fold serial dilutions of extract. The internal control for PCR amplification was weaker than the telomerase products in this assay and is not shown. (C) Telomeric restriction fragments detected with a telomeric repeat probe.
We assayed telomere length as the biological indicator of telomerase function. We monitored telomere length by in-gel hybridization of telomeric restriction fragments using a radiolabeled telomeric repeat probe. With increasing PDL after selection, X-linked DC+TERT cells with recombinant wild-type TER gained telomere length as expected, while telomeres in cells with the empty vector did not (Fig. 2C). Telomeres in cells expressing C72G TER elongated slightly and then were stably maintained. In contrast, telomeres in cells expressing GC107AG TER were indistinguishable from those of the empty vector control (Fig. 2C). These findings suggest that C72G and GC107AG TER alleles demonstrate partial and near-complete or complete loss of function for telomere elongation, respectively. The same results were observed for independent trials of retroviral infection, selection, and culture growth (data not shown). Importantly, even though X-linked DC+TERT+TER cells expressing pseudoknot region TER variants accumulate more recombinant than endogenous TER, recombinant TER did not detectably inhibit endogenous wild-type TER function: cell cultures expressing the GC107AG or C72G TER variants continued to grow rather than enter senescence and did not show accelerated telomere shortening. The combined results above imply that variant TER expression in vivo does not impose a substantial dominant-negative impact on the catalytic or biological activity of coexpressed wild-type TER.
Stable expression of TER variants with sequence changes in the template region.
Mutations in the TER template can alter the sequence of repeat synthesis, and even a few altered telomeric repeats mixed with wild-type repeats can be deleterious for telomere function. Therefore, it is surprising that two relatively recently identified DC TER variants (42) have sequence changes in the template itself (Fig. 1). Transient reconstitution assays of TER variants with replacement of residue A48 with G (A48G) or deletion of residues 52 to 55 (Δ52-55) yielded no activity in TRAP assays using the canonical repeat amplification primers, but these variants showed a profound dominant-negative impact on the activity of wild-type TER reconstituted in combination with the variant (51). We used X-linked DC+TERT cells to examine the biological function of these two TER variants, as well as a template-flanking replacement of residue G58 with A (G58A). The G58A variant was originally identified in association with AA but later reclassified as a polymorphism (41, 46, 52). When reconstituted with TERT by transient transfection, G58A TER supports near-normal or slightly reduced telomerase activation, depending on expression context and assay conditions (16, 30).
Retroviruses expressing the A48G, Δ52-55, and G58A TER variants were introduced into X-linked DC+TERT cells in parallel with the wild-type TER and empty vector controls. Following release from selection, cell cultures at increasing PDL were sampled for TER accumulation, telomerase activation, and telomere length. Expression of recombinant wild-type TER, the G58A TER polymorphism, and the Δ52-55 TER variant increased mature TER accumulation to a substantial excess over the level of endogenously encoded wild-type TER (Fig. 3A). Curiously, however, recombinant A48G TER expression yielded little if any increase in steady-state accumulation of mature TER (Fig. 3A). This unexpected observation was reproducible in independent trials of retroviral infection, selection, and culture growth (data not shown). The A48G sequence change does not affect hybridization of the oligonucleotide used for TER detection by Northern blotting. Also, cells selected for integration of the A48G expression vector recovered from selection comparably to the parallel control cultures and did not show any evidence of growth inhibition over subsequent population doublings, suggesting that A48G TER precursor expression was not toxic. Furthermore, the same retroviral vector encoding A48G TER produced mature TER by transient transfection at a level slightly reduced from that of the wild type but still robustly detectable (data not shown), and previous transient transfection assays using a different A48G TER expression vector found its accumulation equal to that of wild-type TER (51). Together these results suggest that the steady-state accumulation of mature TER stably expressed in primary cells depends on variables beyond those that influence short-term accumulation of mature TER following transient transfection of transformed cells.
FIG. 3.

Stable expression and functional assay of TER variants in the template region. (A) Northern blot of mature TER and a loading control (LC) detected in total RNA from cell extract. “Relative TER” indicates hybridization signal for TER after normalization to the LC and expressed relative to recombinant wild-type (WT) TER. (B) TRAP assay with 2.5-fold serial dilutions of extract. The internal control for PCR amplification is shown. (C) Telomeric restriction fragments detected with a telomeric repeat probe.
Extracts from X-linked DC+TERT+TER cell cultures were assayed to determine the level of telomerase catalytic activation (Fig. 3B). Telomerase activity in extract of cells expressing wild-type recombinant TER or the G58A polymorphism was increased relative to telomerase activity in extract from vector control cells, whereas minimal if any increase in telomerase activity was observed for cell cultures with Δ52-55 TER or the accumulation-compromised A48G TER. Importantly, no dominant-negative impact of Δ52-55 TER expression on the catalytic activity of coexpressed endogenous wild-type TER was detected, despite the excess of Δ52-55 TER relative to wild-type TER. This finding contrasts with the conclusion from a previous study that used transient transfection or RRL to coexpress wild-type and A48G or Δ52-55 TERs, which reported dominant-negative inhibition of activity reconstituted by wild-type TER to below 1% of its normal level (51). Functional interference by TER variants than compete for assembly with limiting TERT could be at most 50%, if both RNAs are present in equal amounts. This is much less severe inhibition than that reported as the dominant-negative impact of mutant template TERs in previous reconstitution assays (44, 51).
Consistent with the increase in telomerase catalytic activation detected in vitro, cells expressing recombinant wild-type or G58A TER gained telomere length with increasing PDL after selection (Fig. 3C). In contrast, cells expressing empty vector, the poorly accumulating A48G TER, or Δ52-55 TER possessed very short telomeres that did not undergo bulk elongation (Fig. 3C). Accumulation of Δ52-55 TER did not inhibit the function of coexpressed wild-type TER, because cells expressing Δ52-55 TER did not show accelerated telomere shortening compared to cells expressing the empty vector control and continued to grow without senescence. The same results were observed for independent trials of retroviral infection, selection, and culture growth (data not shown). We conclude that the disease-associated human TER variants examined here do not have any apparent dominant-negative impact on coexpressed wild-type TER function in vivo, even when the TER variant has a sequence change in the template region.
Discrimination of TER monomer versus multimer in RNPs reconstituted in vivo.
The absence of a dominant-negative impact from expression of disease-associated TER variants suggests functional independence of TER monomers in vivo. This functional independence could derive from physical independence of TER monomers, which would occur if a single TER assembles into each active telomerase RNP. TER and TERT multimerization in endogenously assembled human telomerase holoenzyme has been inferred from gel filtration results, by comparison of telomerase holoenzyme fractionation with that of protein standards (8), but no direct assay has tested this conclusion. In vitro reconstitution assays originated the conclusion that TER multimerizes in the active RNP and that subunits show dominant-negative interdependence for activity (44), but more recent single-particle biophysical studies suggest that human telomerase can be reconstituted in vitro as an RNP with monomers of TER and TERT (1). To directly test whether TERs coassemble into a shared particle of telomerase RNP in vivo, we developed an RNA-based purification method for isolation of endogenously assembled RNPs (21). Here we use the first step of this method, which exploits the natural affinity and specificity of PP7 coat protein for its RNA hairpin binding site (28). Unlike the related phage MS2 coat protein and RNA hairpin (26), the PP7 components retain high affinity and specificity under the desired buffer conditions of physiological ionic strength (20).
Human TER accumulates in vivo and assembles into active RNP when tagged by extension of its 5′ end (31). We compared the accumulation and activity of TER tagged at its 5′ end with one, three, or six copies of the 25-nucleotide hairpin binding site for PP7 coat protein using human VA13 cells, which lack endogenous TER or TERT. VA13 cells stably expressing TERT from an integrated retrovirus were transiently transfected to express a tagged TER, untagged wild-type TER, or empty vector control. Northern blots of cell extracts reveal that the tagged TERs did accumulate (Fig. 4A). A tag with three hairpins fully resolved tagged TER mobility from that of untagged TER while still allowing robust transient accumulation. TRAP assays of cell extract demonstrate that each of the tagged TERs assembled with TERT to form catalytically active RNP (Fig. 4B). Accumulation of 5′-tagged TER stably expressed from an integrated retrovirus in long-term cell cultures was not as robust as wild-type TER accumulation (data not shown). Thus, as described above for the A48G TER variant, tagged TER overexpression by short-term transient transfection overcomes a limit on accumulation that is more evident when recombinant TER is stably expressed from an integrated vector.
FIG. 4.

Tagged TER accumulation and assembly into active RNP. (A) Northern blot of total RNA and a loading control (LC) from extracts of VA13+TERT cells transfected to express untagged TER or TER with a 5′ tag of one, three, or six sequential PP7 hairpins. Note that PP7-tagged TERs, like untagged TER, migrate in multiple forms due at least in part to partial folding during electrophoresis. (B) TRAP assay with fivefold serial dilutions of extract. The internal control (IC) for PCR amplification is shown.
To investigate the multimerization state of TER in human telomerase RNPs assembled in vivo, we used transient transfection of 293T cells to express TER tagged with three PP7 hairpins, untagged TER, or a weighted mixture of both (Fig. 5A, lanes 2 to 4). We then purified tagged TER from whole-cell extract using PP7 coat protein (Fig. 5A, lanes 5 to 7). Recombinant coat protein fused to tandem protein A domains (the ZZ tag) was added to cell extract, followed by recovery of coat protein and associated RNPs with IgG agarose. From cell extract containing only untagged TER, no TER was recovered by PP7 coat protein as expected. From extracts containing tagged TER, only tagged TER was recovered by affinity purification. Specific enrichment for tagged TER was observed even when much more untagged than tagged TER was present in the extract. This result suggests that coexpressed TERs do not assemble into a multimeric complex.
FIG. 5.

Affinity purification of telomerase RNPs by tagged TER. 293T cells were transfected to express combinations of untagged and/or tagged TER, with coexpression of FLAG3-tagged TERT in panel B as indicated. TERs were detected by Northern blotting along with a recovery control (RC) added to each sample before RNA extraction. Recombinant untagged TER was used as a marker on all blots and is shown in lane 1 of panel A. Relative loadings of input and bound (IP) samples were 5% and 100% in panel A or 2%, 10%, and 90% of sequential samples in panel B.
To ensure that the pool of telomerase RNP assembled with TERT was represented in our affinity purification of tagged TER, we added a second step of affinity purification to select for telomerase RNPs with TERT. 293T cells were cotransfected to express FLAG3-tagged TERT as well as hairpin-tagged and/or untagged TER (Fig. 5B, lanes 1 to 4). First, RNPs with tagged TER were recovered on IgG agarose using PP7 coat protein and eluted by cleavage between the coat protein and ZZ tag with TEV protease (Fig. 5B, lanes 5 to 8). Second, eluted RNPs that also harbor tagged TERT were recovered by binding to FLAG antibody resin (Fig. 5, lanes 9 to 12). When TERs were not coexpressed with FLAG3-TERT, complexes containing tagged TER from the first step of affinity purification remained in the unbound fraction of the second-step purification, providing a control for nonspecific background. On the other hand, when TERs were coexpressed with FLAG3-TERT, complexes containing tagged TER were indeed recovered with the FLAG antibody resin (Fig. 5B, lanes 10 and 11). These results support the conclusion that telomerase RNPs assembled in vivo, including telomerase RNPs with bound TERT, are monomeric for TER. Because only the extract-soluble pool of telomerase RNP is accessible for biochemical assays, it remains possible that some RNP is assembled into a particle of higher-order subunit multimerization that is either not soluble or is disrupted by cell lysis.
Discrimination of TERT monomer versus multimer in enzyme reconstituted in vivo.
As a complementary approach, we used catalytic activity assays to address whether active telomerase complexes assembled in vivo are monomeric or multimeric for TERT. We used transient transfection of 293T cells to express either ZZ-TERT, FLAG3-TERT (FL), TERT with a combined ZZ-FLAG3 tag (ZZFL), or a mixture of ZZ-TERT and FL-TERT (ZZ+FL). If two or more TERT proteins coassemble into an active RNP particle, coexpression of ZZ-TERT and FL-TERT should allow recovery of telomerase activity after tandem steps of IgG agarose and FLAG antibody chromatography. On the other hand, if these TERTs assemble into separate RNPs, each monomeric for TERT, either single step of affinity purification will enrich active RNP but tandem steps of purification will not. The ZZFL-TERT serves as a positive control for active RNP recovery following tandem steps of affinity purification, because both tags are present on the same polypeptide.
Plasmid concentrations were optimized to yield closely comparable levels of telomerase catalytic activation (Fig. 6A) and TERT protein (Fig. 6B) across transfected cell extracts. TERT protein levels were assayed by immunoblotting with rabbit polyclonal anti-TERT IgG, which recognizes a peptide within TERT using its variable region and also binds to the ZZ tag through its constant region. Therefore, to accurately compare TERT levels, extracts were treated with TEV protease to release the ZZ module from tagged TERT prior to immunoblot analysis (Fig. 6B, lanes 5 to 8). This increased the electrophoretic mobility and reduced the detection signal of formerly ZZ-tagged proteins, leaving only a small mobility difference between FLAG3-tagged and untagged TERTs. 293T cells also express endogenous TERT, but because the recombinant proteins expressed by transient transfection accumulate at much higher than endogenous levels, they overshadow the immunoblot signal of the endogenous protein.
FIG. 6.

Tandem affinity purification of active RNP using tagged TERTs. 293T cells were transfected to express combinations of tagged TERTs. Input extracts harbored similar levels of telomerase activity by TRAP (A) and similar levels of TERT proteins by immunoblotting (B). IC, internal control. The immunoblot is quantitative only after removal of the ZZ module from ZZFL-TERT and ZZ-TERT by TEV protease addition to extract (lanes 5 to 8), because the ZZ tag binds to the rabbit IgG used to detect TERT. Different numbers of asterisks are used to designate different TERT polypeptides: ZZFL-TERT (****), ZZ-TERT (***), FL-TERT (**), and TERT (*). The same extracts were used for tandem steps of affinity purification binding first to IgG (C) or FLAG antibody (D). All TRAP assays used serial 5-fold dilutions of sample to provide 25-fold range of detection level for telomerase activity. A weak background of FL-TERT binding to IgG resin was detected in panel D whether or not ZZ-TERT was coexpressed, due to the combination of relatively purified first-step elution samples and relatively background-absorbing IgG agarose as a second step.
TERTs with a ZZ tag were bound to and eluted from IgG agarose (Fig. 6C, left), and complexes in the first-step elutions harboring a FLAG tag were then bound to and eluted from FLAG antibody resin (Fig. 6C, right). No activity was recovered in the first-step purification from extract with only FL-TERT, and activity in the first-step elution using extract with only ZZ-TERT was not recovered by the second-step purification as expected. These controls verify the specificity of affinity enrichment. On the other hand, robust telomerase activity was recovered by tandem affinity purification from extract with ZZFL-TERT, providing a benchmark for the efficiency of tandem affinity purification. If active telomerase RNPs are dimeric for TERT, tandem affinity purification from extract with coexpressed ZZ-TERT and FL-TERT could recover up to 50% of the activity level obtained from extract with ZZFL-TERT. For higher-order multimers, maximal yield would increase. Instead, no telomerase activity was recovered by tandem affinity purification from extract with coexpressed ZZ-TERT and FL-TERT. Importantly, activity was enriched by the first step of affinity purification from this extract to a level roughly comparable to that obtained by first-step purification from cell extract with ZZ-TERT or ZZFL-TERT.
It was important to test the same extracts for tandem affinity purification using the reverse order of enrichment steps. This controls for an unlikely but possible difference in the active RNP assembly efficiency of ZZ-TERT and FL-TERT. If one tagged TERT assembled much more efficiently into active RNP, first-step purification for that tagged TERT would recover activity but the tandem step might fail to recover activity even in the case of a TERT multimer. However, in this case, the population of active RNP recovered in first-step purification for the disadvantaged subunit would be enriched in the tandem second step with particularly high yield. Thus, performing tandem affinity purification using both orders of purification steps controls for any differences in expression level or RNP assembly efficiency of the differentially tagged TERTs.
For reciprocal tandem affinity purification using aliquots of the same transfected cell extracts, TERTs with a FLAG tag were bound to and eluted from FLAG antibody resin (Fig. 6D, left) and complexes in the first-step elutions harboring the ZZ tag were then bound to and eluted from IgG agarose (Fig. 6D, right). No activity was recovered in the first-step purification from extract with only ZZ-TERT, and activity enriched in the first-step elution using extract with only FL-TERT was not recovered by the second step of purification. Again, tandem affinity purification from extract with ZZFL-TERT enriched robust telomerase activity. Telomerase activity was also recovered from extract with coexpressed ZZ-TERT and FL-TERT after the first step of affinity purification, at a level roughly comparable to that of purification from extracts with FL-TERT or ZZFL-TERT, but this activity was not enriched by the second step of affinity purification. A weak background of FL-TERT binding to IgG resin was detected with relatively purified first-step elution samples, whether or not ZZ-TERT was coexpressed. All of these tandem affinity purification results were highly reproducible. Overall, these findings imply that the physiologically assembled human telomerase holoenzymes recovered in cell extract harbor monomers rather than multimers of TERT and TER.
DISCUSSION
Loss of function for disease-associated human TER variants in vivo.
Human disease has been linked to diverse molecular changes in TER. The few available cell cultures from X-linked or AD DC patients have genetic alterations that reduce mature TER accumulation. In these cells, expression of additional TER precursor compensates for the TER accumulation deficiency and can stimulate telomere elongation (45, 49). Here we sought to evaluate the mechanism of dysfunction of a different class of TER variants that are predicted to accumulate normally but be defective in catalytic activation of TERT. In the absence of cell cultures from representative patients, we exploited a recently developed biological assay for recombinant TER function in X-linked DC patient cells (49). X-linked DC patient cells accumulate a reduced level of mature TER, due to less efficient processing of the TER precursor or reduced assembly or stability of telomerase RNP. Recombinant TER expression compensates for this defect, increasing mature TER accumulation and allowing telomere elongation in the presence of TERT. We note that there is no impact of X-linked DC dyskerin sequence substitutions on telomerase holoenzyme catalytic activity or biological function at telomeres in vivo (49).
Most TER variants investigated here increased the steady-state level of mature TER when stably expressed in primary cells. To our surprise, stable expression of the A48G TER variant did not lead to the expected increase in steady-state TER accumulation. Among many possible models to account for this finding, the A48G substitution could interfere with a tertiary structure important for telomerase RNP stability (38) or it could promote a nonnative folding that destabilizes telomerase RNP. Important confirmation of disease significance of the observed defect in recombinant A48G TER accumulation awaits the analysis of primary cells from a representative AD DC patient.
Cell cultures expressing TER variants that increased the pool of mature TER could be evaluated for catalytic activity and biological function of the altered TER. In cell cultures expressing the GC107AG and Δ52-55 TER variants, telomere lengths remained indistinguishable from telomere lengths in the negative control cell cultures. In cell cultures expressing the C72G variant, a limited but reproducible extent of telomere elongation was detected. It is tempting to speculate about the physiological consequence of partial rather than complete loss of function for the C72G TER, but we note that the weak biological activity of C72G TER shown here was detected in the highly sensitized context of the extremely short telomeres of X-linked DC patient cells constitutively expressing TERT. In the telomerase-positive somatic tissues of a patient with AA, the limited function of C72G TER may or may not influence telomere structure.
Although the disease-associated TER variants showed little or no biological function, they did not detectably compromise the function of coexpressed wild-type TER. Despite the accumulation of TER variants GC107AG, C72G, and Δ52-55 in substantial excess of the wild-type TER, we observed no dominant-negative impact of these TER variants on wild-type TER in vitro activity or in vivo function. We note that although there is no “positive control” for the exaggerated telomere shortening and/or senescence that would be expected for a dominant-negative impact, the lack of impact on telomerase catalytic activity supports the lack of a telomere phenotype. In addition, our findings are in accord with the general similarity of disease phenotypes imposed by heterozygous expression of alleles that reduce TER accumulation versus alleles that reduce telomerase catalytic activation. Furthermore, if the altered template of some TER variants was actually copied into telomeric repeats as part of the dominant-negative mechanism, exacerbated disease phenotypes might be expected in telomerase-positive cells of patients and cell cultures without critically short telomeres, due to the toxicity of telomere mutagenesis.
Disease phenotypes from TER haploinsufficiency suggest that TER is frequently limiting for telomerase function in the human organism. Most human somatic cells are telomerase-negative for telomere elongation due to transcriptional or posttranscriptional regulation of TERT (10, 12). However, disease phenotypes arise from the impact of telomerase deficiency on the subset of cells that do activate telomerase (11). In this minority of endogenously telomerase-positive somatic cells, TER rather than TERT may be most limiting for telomerase function at telomeres. TERT gene mutations have been linked to bone marrow failure and other diseases (18, 40). We suggest that most of these TERT variants, particularly the variants associated with DC and AA, could impose telomere shortening indirectly through sequestration of TER. If TERT variants are compromised in telomere elongation function without compromised affinity for TER, inactive TERT will compete with active TERT for the limiting pool of telomerase RNP. One recently identified TERT mutation forces a frameshift before translation of the TERT high-affinity RNA binding domain (25) and thus would be unlikely to bind TER; interestingly, this mutation is associated with pulmonary fibrosis rather than DC (18).
Physical basis for functional independence of coexpressed TERs.
Affinity purification assays using tagged TER and/or tagged TERT(s) coexpressed in vivo suggest that catalytically active human telomerase holoenzyme harbors a monomer of TER and a monomer of TERT. Likewise, subunit physical interaction studies using endogenously assembled Tetrahymena and budding yeast telomerase holoenzymes, or the reconstituted Tetrahymena telomerase minimal catalytic core of TERT and TER, indicate that active telomerase complexes in these single-celled organisms are monomeric for TER and TERT (6, 14, 29, 47). Coexpression of mutant-template and wild-type TERs in vivo can give rise to alternating mutant and wild-type telomeric repeats (54), which led to the hypothesis that there could be frequent substrate switching between cooperating telomerase active sites. The interspersed telomeric repeats generated in vivo could indeed arise from processive repeat synthesis by a telomerase particle that is multimeric for TER, but they could also reflect biological restraint of the inherent repeat addition processivity of a telomerase particle that is monomeric for TER.
Numerous telomerase-interacting proteins and transiently associated chaperones provide assembly instructions for human TER and TERT in vivo. The absence or altered specificity of these events in vitro may be responsible for allowing human TERT and TER to assemble as multimers. At least two physically separable interactions are established between human TERT and TER (32), as also shown for ciliate TERT and TER (24). Our results suggest that when an active telomerase RNP is assembled in vivo, these multiple interactions occur between one molecule of TERT and one molecule of TER. In contrast, in vitro, two different molecules of human TERT can complement to reconstitute catalytic activity and two physically separate fragments of TER can be combined to reconstitute activity as well (3, 32, 35). In vitro reconstitution of human TERT and TER may allow or even encourage the formation of nonnative multisubunit complexes, depending on subunit stoichiometry and other conditions of reconstitution. In addition, affinity purification strategies that target a TERT-TER contact surface could preferentially enrich complexes of altered stoichiometry, because RNPs assembled with subunit monomers would most effectively bury the interaction interfaces and make them inaccessible for oligonucleotide hybridization or antibody binding. We suggest that heterogeneous architectures of RNP assembly in vitro can account for some of the contradictory conclusions about dominant-negative interference by mutant-template TERs.
Acknowledgments
We thank Catherine O'Connor for initial studies of telomerase RNP affinity purification using PP7-tagged TER and Emily Egan for testing retroviral expression constructs by transient transfection.
This work was funded by Public Health Service grant HL079585 from the National Heart, Lung, and Blood Institute and a BIG award from the American Federation for Aging Research.
Footnotes
Published ahead of print on 18 August 2008.
REFERENCES
- 1.Alves, D., H. Li, R. Codrington, A. Orte, X. Ren, D. Klenerman, and S. Balasubramanian. 2008. Single-molecule analysis of human telomerase monomer. Nat. Chem. Biol. 4287-289. [DOI] [PubMed] [Google Scholar]
- 2.Autexier, C., and N. F. Lue. 2006. The structure and function of telomerase reverse transcriptase. Annu. Rev. Biochem. 75493-517. [DOI] [PubMed] [Google Scholar]
- 3.Beattie, T. L., W. Zhou, M. O. Robinson, and L. Harrington. 2001. Functional multimerization of the human telomerase reverse transcriptase. Mol. Cell. Biol. 216151-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackburn, E. H., C. W. Greider, and J. W. Szostak. 2006. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 121133-1138. [DOI] [PubMed] [Google Scholar]
- 5.Bodnar, A. G., N. W. Kim, R. B. Effros, and C.-P. Chiu. 1996. Mechanism of telomerase induction during T cell activation. Exp. Cell Res. 22858-64. [DOI] [PubMed] [Google Scholar]
- 6.Bryan, T. M., K. J. Goodrich, and T. R. Cech. 2003. Tetrahymena telomerase is active as a monomer. Mol. Biol. Cell 144794-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cech, T. R. 2004. Beginning to understand the end of the chromosome. Cell 116273-279. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, S. B., M. E. Graham, G. O. Lovrecz, N. Bache, P. J. Robinson, and R. R. Reddel. 2007. Protein composition of catalytically active human telomerase from immortal cells. Science 3151850-1853. [DOI] [PubMed] [Google Scholar]
- 9.Collins, K. 2006. The biogenesis and regulation of telomerase holoenzymes. Nat. Rev. Mol. Cell Biol. 7484-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins, K. 2008. Physiological assembly and activity of human telomerase complexes. Mech. Ageing Dev. 12991-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins, K., and J. R. Mitchell. 2002. Telomerase in the human organism. Oncogene 21564-579. [DOI] [PubMed] [Google Scholar]
- 12.Cong, Y.-S., W. E. Wright, and J. W. Shay. 2002. Human telomerase and its regulation. Microbiol. Mol. Biol. Rev. 66407-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cristofari, G., and J. Lingner. 2006. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 25565-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham, D. D., and K. Collins. 2005. Biological and biochemical functions of RNA in the Tetrahymena telomerase holoenzyme. Mol. Cell. Biol. 254442-4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng, J., W. D. Funk, S. Wang, S. L. Weinrich, A. A. Avilion, C. Chiu, R. R. Adams, E. Chang, R. C. Allsopp, J. Yu, S. Le, M. West, C. B. Harley, W. H. Andrews, C. W. Greider, and B. Villeponteau. 1995. The RNA component of human telomerase. Science 2691236-1241. [DOI] [PubMed] [Google Scholar]
- 16.Fu, D., and K. Collins. 2003. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol. Cell 111361-1372. [DOI] [PubMed] [Google Scholar]
- 17.Fu, D., and K. Collins. 2007. Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell 28773-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia, C. K., W. E. Wright, and J. W. Shay. 2007. Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Res. 357406-7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilson, E., and V. Geli. 2007. How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 8825-838. [DOI] [PubMed] [Google Scholar]
- 20.Greider, C. W. 2006. Telomerase RNA levels limit the telomere length equilibrium. Cold Spring Harbor Symp. Quant. Biol. 71225-229. [DOI] [PubMed] [Google Scholar]
- 21.Hogg, J. R., and K. Collins. 2007. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13868-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hug, N., and J. Lingner. 2006. Telomere length homeostasis. Chromosoma 115413-425. [DOI] [PubMed] [Google Scholar]
- 23.Kapust, R. B., J. Tozser, J. D. Fox, D. E. Anderson, S. Cherry, T. D. Copeland, and D. S. Waugh. 2001. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14993-1000. [DOI] [PubMed] [Google Scholar]
- 24.Lai, C. K., M. C. Miller, and K. Collins. 2003. Roles for RNA in telomerase nucleotide and repeat addition processivity. Mol. Cell 111673-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai, C. K., J. R. Mitchell, and K. Collins. 2001. RNA binding domain of telomerase reverse transcriptase. Mol. Cell. Biol. 21990-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeCuyer, K. A., L. S. Behlen, and O. C. Uhlenbeck. 1995. Mutants of the bacteriophage MS2 coat protein that alter its cooperative binding to RNA. Biochemistry 3410600-10606. [DOI] [PubMed] [Google Scholar]
- 27.Li, S., and E. H. Blackburn. 2006. Expression and suppression of human telomerase RNA. Cold Spring Harbor Symp. Quant. Biol. 71211-215. [DOI] [PubMed] [Google Scholar]
- 28.Lim, F., and D. S. Peabody. 2002. RNA recognition site of PP7 coat protein. Nucleic Acids Res. 304138-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livengood, A. J., A. J. Zaug, and T. R. Cech. 2002. Essential regions of Saccharomyces cerevisiae telomerase RNA: separate elements for Est1p and Est2p interaction. Mol. Cell. Biol. 222366-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marrone, A., D. Stevens, T. Vulliamy, I. Dokal, and P. J. Mason. 2004. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood 1043936-3942. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell, J. R., J. Cheng, and K. Collins. 1999. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol. 19567-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell, J. R., and K. Collins. 2000. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase in vivo and in vitro. Mol. Cell 6361-371. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell, J. R., E. Wood, and K. Collins. 1999. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402551-555. [DOI] [PubMed] [Google Scholar]
- 34.Prowse, K. R., and C. W. Greider. 1995. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl. Acad. Sci. USA 924818-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tesmer, V. M., L. P. Ford, S. E. Holt, B. C. Frank, X. Yi, D. L. Aisner, M. Ouellette, J. W. Shay, and W. E. Wright. 1999. Two inactive fragments of the integral RNA cooperate to assemble active telomerase with the human protein catalytic subunit (hTERT) in vitro. Mol. Cell. Biol. 196207-6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theimer, C. A., C. A. Blois, and J. Feigon. 2005. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell 17671-682. [DOI] [PubMed] [Google Scholar]
- 37.Theimer, C. A., and J. Feigon. 2006. Structure and function of telomerase RNA. Curr. Opin. Struct. Biol. 16307-318. [DOI] [PubMed] [Google Scholar]
- 38.Ueda, C. T., and R. W. Roberts. 2004. Analysis of a long-range interaction between conserved domains of human telomerase RNA. RNA 10139-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verdun, R. E., and J. Karlseder. 2007. Replication and protection of telomeres. Nature 447924-931. [DOI] [PubMed] [Google Scholar]
- 40.Vulliamy, T. J., and I. Dokal. 2008. Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie 90122-130. [DOI] [PubMed] [Google Scholar]
- 41.Vulliamy, T. J., A. Marrone, I. Dokal, and P. J. Mason. 2002. Association between aplastic anemia and mutations in telomerase RNA. Lancet 3592168-2170. [DOI] [PubMed] [Google Scholar]
- 42.Vulliamy, T. J., A. Marrone, S. W. Knight, A. Walne, P. J. Mason, and I. Dokal. 2006. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 1072680-2685. [DOI] [PubMed] [Google Scholar]
- 43.Weinrich, S. L., R. Pruzan, L. Ma, M. Ouellette, V. M. Tesmer, S. E. Holt, A. G. Bodnar, S. Lichsteiner, N. W. Kim, J. B. Trager, R. D. Taylor, R. Carlos, W. H. Andrews, W. E. Wright, J. W. Shay, C. B. Harley, and G. B. Morin. 1997. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 17498-502. [DOI] [PubMed] [Google Scholar]
- 44.Wenz, C., B. Enenkel, M. Amacker, C. Kelleher, K. Damm, and J. Lingner. 2001. Human telomerase contains two cooperating telomerase RNA molecules. EMBO J. 203526-3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westin, E. R., E. Chavez, K. M. Lee, F. A. Gourronc, S. Riley, P. M. Lansdorp, F. D. Goldman, and A. J. Klingelhutz. 2007. Telomere restoration and extension of proliferative lifespan in dyskeratosis congenita fibroblasts. Aging Cell 6383-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson, D. B., J. Ivanovich, A. Whelan, P. J. Goodfellow, and M. Bessler. 2003. Human telomerase RNA mutations and bone marrow failure. Lancet 3611993-1994. [DOI] [PubMed] [Google Scholar]
- 47.Witkin, K. L., and K. Collins. 2004. Holoenzyme proteins required for the physiological assembly and activity of telomerase. Genes Dev. 181107-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong, J. M., M. J. Kyasa, L. Hutchins, and K. Collins. 2004. Telomerase RNA deficiency in peripheral blood mononuclear cells in X-linked dyskeratosis congenita. Hum. Genet. 115448-455. [DOI] [PubMed] [Google Scholar]
- 49.Wong, J. M. Y., and K. Collins. 2006. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 202848-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong, J. M. Y., and K. Collins. 2003. Telomere maintenance and disease. Lancet 362983-988. [DOI] [PubMed] [Google Scholar]
- 51.Xin, Z. T., A. D. Beauchamp, R. T. Calado, J. W. Bradford, J. A. Regal, A. Shenoy, Y. Liang, P. M. Lansdorp, N. S. Young, and H. Ly. 2007. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood 109524-532. [DOI] [PubMed] [Google Scholar]
- 52.Yamaguchi, H., G. M. Baerlocher, P. M. Lansdorp, S. J. Chanock, O. Nunez, E. Sloand, and N. S. Young. 2003. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 102916-918. [DOI] [PubMed] [Google Scholar]
- 53.Yi, X., V. M. Tesmer, I. Savre-Train, J. W. Shay, and W. E. Wright. 1999. Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol. Cell. Biol. 193989-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu, G., and E. H. Blackburn. 1991. Developmentally programmed healing of chromosomes by telomerase in Tetrahymena. Cell 67823-832. [DOI] [PubMed] [Google Scholar]