

Summary
Our inability to develop new therapeutic strategies to prevent meningitis due to Escherichia coli K1 is attributed to our incomplete understanding of the pathophysiology of the disease. Previously, we demonstrated that outer membrane protein A of E. coli interacts with a gp96 homologue, Ec-gp96, on human brain microvascular endothelial cells (HBMEC) for invasion. However, signalling events mediated by Ec-gp96 that allow internalization of E. coli are incompletely understood. Here, we demonstrate that signal transducer and activator of transcription 3 (Stat3) activation and its interaction with Ec-gp96 were critical for E. coli invasion. The activated Stat3 was colocalized with Ec-gp96 at the actin condensation sites, and overexpressing a dominant negative (DN) form of Stat3 in HBMEC significantly abrogated the invasion. Furthermore, overexpression of Ec-gp96Δ200, the C-terminal 214-amino-acid truncated Ec-gp96, prevented the invasion of E. coli in HBMEC. In contrast, lack of ATP binding by gp96 did not affect the invasion. Overexpression of DN forms of either phosphatidyl inositol-3 kinase (PI3-kinase) subunit p85 or protein kinase C-α (PKC-α) had no effect on the activation of Stat3 and its association with Ec-gp96, whereas overexpression of DN-Stat3 abolished the activation of both PI3-kinase and PKC-α. Together, our findings identified a novel interaction of Stat3 with Ec-gp96, upstream of PI3-kinase and PKC-α activation that is required for the invasion of E. coli into HBMEC.
Introduction
Neonatal meningitis due to Escherichia coli K1 (E. coli) is the most common central nervous system infection with unchanged mortality and morbidity rates over the last few decades despite the use of effective antibiotics. The disease is fatal in 5–40% of infected neonates and causes neurological sequelae in up to 30% of survivors, which include hearing impairment, mental retardation and seizure disorders. Importantly, the mortality rates are likely to increase significantly as the incidence of E. coli infections has been on the rise in recent years (Stoll et al., 2002). The poor outcome of this disease is due to incomplete understanding of the pathogenesis of meningitis to develop new modes of prevention. For example, the mechanisms utilized by various virulence factors that have been shown critical for the invasion of E. coli into human brain microvascular endothelial cells (HBMEC) remain elusive.
Our studies have demonstrated that outer membrane protein A (OmpA) expression is important for E. coli invasion of HBMEC in vitro as well as in the newborn rat model of meningitis (Prasadarao et al., 1996a,b). Subsequently, OmpA has been shown to interact with a 96 kDa receptor on HBMEC (named as Ec-gp96), a homologue of gp96, for invasion of HBMEC by E. coli (Prasadarao, 2002). Overexpression of either gp96 or Ec-gp96 in Chinese hamster ovary cells showed a significant increase in the invasion of E. coli, suggesting that minor changes in some amino acids between gp96 and Ec-gp96 do not affect the invasion (Prasadarao et al., 2003). Gp96, also referred to as glucose-regulated protein 94 (GRP94), is expressed ubiquitously, although we have shown that its expression is significantly lower in human umbilical vein endothelial cells when compared with HBMEC (Prasadarao et al., 2003). It is part of the Hsp90 protein family but it is an unusual member of this family, as it contains an N-terminal signal sequence and a C-terminal KDEL sequence for endoplasmic reticulum (ER) retention (Altmeyer et al., 1996; Castelli et al., 2004). Gp96 has previously been immunoprecipitated from cell surfaces and purified from plasma membrane (Jacques et al., 1999; Banerjee et al., 2002). Similarly, the ViP (virulence protein) of Listeria monocytogenes has been shown to interact with surface gp96 in Caco-2 and L2071 cells to invade (Cabanes et al., 2005). However, the mechanisms induced by OmpA–Ec-gp96 interaction for the invasion of E. coli are not completely understood.
The E. coli induces actin condensation underneath the bacterial entry site during the invasion of HBMEC (Prasadarao et al., 1999). Activation of both phosphatidyl inositol-3 kinase (PI3-kinase) and protein kinase C-α (PKC-α) is required for actin reorganization in HBMEC and thereby for E. coli entry (Sukumaran et al., 2002; 2003). Furthermore, the formation of a supra-molecular complex containing focal adhesion kinase and associated proteins in caveolae along with Ec-gp96 underneath the bacteria is necessary for internalization of E. coli. Nonetheless, the signalling molecules downstream of Ec-gp96 that transmit the signals to PI3-kinase or PKC-α for actin remodelling are not known. Caveolae, and more generally lipid rafts, can act as platforms for conducting a variety of cellular functions including signal transduction. Hsp90, caveolin-1 and signal transducer and activator of transcription 3 (Stat3) interact within lipid rafts during IL-6 signalling (Shah et al., 2002). Therefore, we speculate that Ec-gp96 might interact with Stat3 in caveolae during the E. coli invasion of HBMEC. The Stat proteins are a group of cytoplasmic transcription factors. The seven mammalian members of this family, Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b and Stat6, all share a conserved domain-like structure (Gamero et al., 2004). The phosphotyrosine-binding SH2 domain is required for receptor binding and dimerization. Within this domain is a conserved tyrosine residue (Tyr-705), which upon phosphorylation activates the Stat molecule, allowing it to interact with the SH2 domain of another Stat. In this study we demonstrate, for the first time, that the phosphorylated Stat3 interacts with the C-terminal domain of Ec-gp96 for the invasion of E. coli into HBMEC. The association of Stat3 with Ec-gp96 is important for the activation of both PKC-α and PI3-kinase, which are subsequently necessary for actin condensation in HBMEC.
Results
OmpA+ E. coli induces the interaction of Stat3 with Ec-gp96 in HBMEC
Our studies revealed that both Ec-gp96 and caveolin-1 colocalize in caveolae during the E. coli invasion of HBMEC (··· ···, unpubl. results). As Stat3 was also shown to be distributed to caveolae along with Hsp90, another homologue of gp96, in Hep3B cells (Yamashita et al., 1998), we hypothesized that Stat3 might be playing an important role in the signalling events required for E. coli invasion. Therefore, we initially examined the activation of Stat3 in HBMEC infected with either OmpA+ or OmpA−E. coli for varying periods. As shown in Fig. 1A, OmpA+ E. coli infection of HBMEC induced phosphorylation of Stat3, which peaked at 30 min. In contrast, OmpA−E. coli-infected HBMEC revealed no such activation. The differences in the phospho-Stat3 levels were not due to unequal loading of the proteins as β-actin levels were similar in all lanes. In addition, to examine whether phospho-Stat3 interacted with Ec-gp96, immunoprecipitation of the lysates of HBMEC infected with OmpA+ E. coli was performed using anti-Ec-gp96 antibody followed by immunoblotting with antiphospho-Stat3 antibody. The blot showed increased association of phospho-Stat3 (antibody specific to Tyr-705) with Ec-gp96 between 15 and 30 min post infection (Fig. 1B). When the blots were reprobed with an antibody to non-phosphorylated form of Stat3 total Stat3 interaction with Ec-gp96 also increased between 15 and 30 min. However, no serine-phosphorylated form of Stat3 was observed when a phosphoserine Stat3 antibody was used (data not shown). Furthermore, the blots were stripped and reprobed with phospho-specific antibodies to Stat1 and Stat5 to examine their association with Ec-gp96. Although phospho-Stat1 was associated with Ec-gp96 in control-uninfected cells, no changes in the interaction were observed over the time upon infection. In contrast, phospho-Stat5 did not show detectable levels of association with Ec-gp96 and this was not due to differences in loading the immune complexes, as the IgG levels on the blot appeared to be similar. These results indicate that OmpA+ E. coli interaction with HBMEC induces the phosphorylation of Stat3 at Tyr−705 and its association with Ec-gp96.
Fig. 1. Association of phospho-Stat3 with Ec-gp96 during the invasion of OmpA+ E. coli in HBMEC.
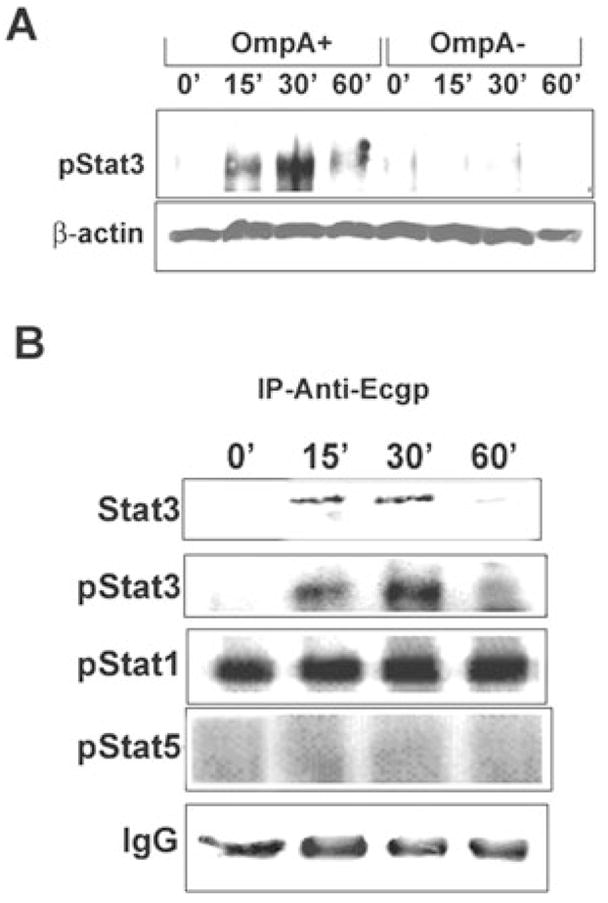
A. Confluent HBMEC monolayers were infected with either OmpA+ E. coli or OmpA− E. coli for varying periods, washed, and total lysates were prepared. Approximately 20 μg of total lysates was analysed by Western blotting using a phospho-Stat3 antibody. Protein loading in the samples was examined by assessing the amount of β-actin in the lysates.
B. Approximately 300 μg of the lysates was subjected to immunoprecipitation with anti-Ec-gp96 antibody and the resulting immune complexes were analysed for association of phosphorylated Stat1, Stat3 or Stat5. Equality of protein loading in the gel was assessed by the amount of IgG present in each lane.
Colocalization of Stat3 and phospho-Stat3 with Ec-gp96 at the actin condensation sites upon infection with OmpA+ E. coli
We hypothesized that Ec-gp96 and Stat3 associate within the caveolae, the initiation points for the signalling for actin condensation during E. coli invasion of HBMEC. Therefore, to visualize the association of Stat3 with Ec-gp96 upon infection with E. coli, HBMEC were plated in eight-well chamber slides, incubated for 15–60 min with either OmpA+ or OmpA− E. coli, fixed and then stained with antibodies to either Stat3, phospho-Stat3 or Ec-gp96 followed by incubation with specific fluorescent conjugates. The cells were also stained by rhodamine phalloidin to examine the condensation of actin filaments. Uninfected HBMEC stained with anti-Ec-gp96 antibody showed that the molecule is present throughout the cell with polarization in some areas (Fig. 2B). Optical sectioning of the cells revealed that Ec-gp96 was also present around the cell membrane (data not shown). However, colocalization of actin and Ec-gp96 was not observed in these cells. Upon infection with OmpA+ E. coli, Ec-gp96 was clustered near the bacterial binding sites and showed colocalization with actin (yellow colour in overlay image, Fig. 2H). Also, staining with antibodies to Stat3 and phospho-Stat3 revealed that these molecules were concentrated at the points of bacterial entry between 15 and 30 min post infection along with actin condensation. In contrast, HBMEC infected with OmpA− E. coli revealed no accumulation of either of these molecules (data not shown). These results suggest that Stat3 accumulation, Ec-gp96 localization and actin condensation occurred at sites of E. coli entry into HBMEC in OmpA-dependent fashion.
Fig. 2.
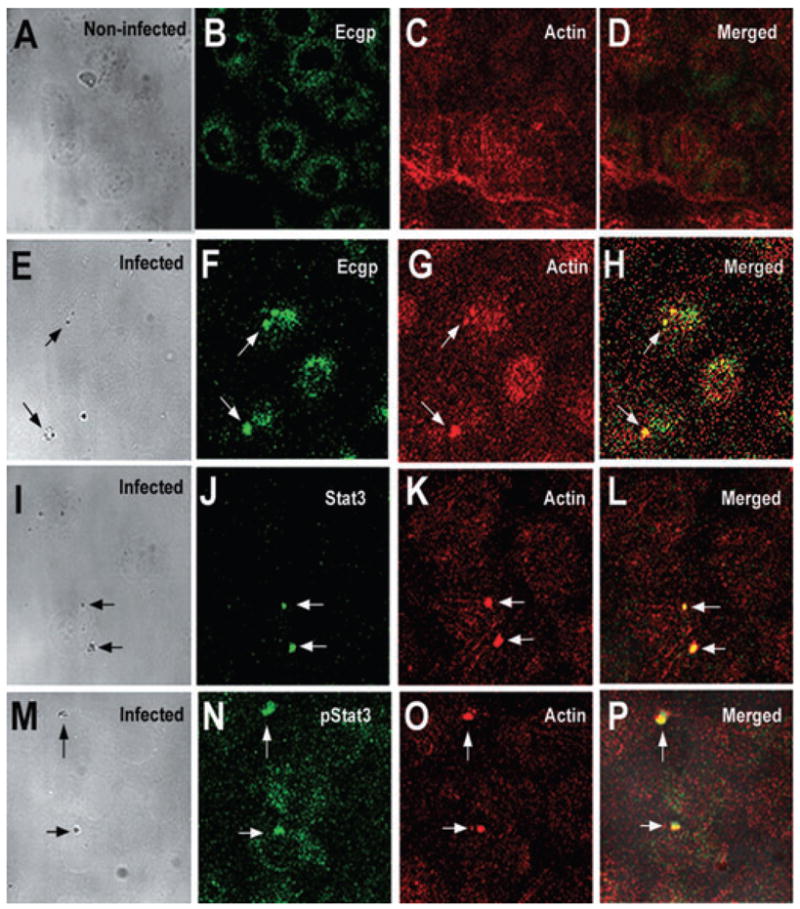
Colocalization of Ec-gp96, Stat3 or phospho-Stat3 and actin at the sites of OmpA+ E. coli binding. HBMEC seeded in eight-well chamber slides were incubated with OmpA+ or OmpA− E. coli for 15–60 min, washed, fixed and then stained for Ec-gp96, Stat3 or phospho-Stat3 using specific antibodies followed by a secondary antibody conjugated to Alexa 488 (green). Actin condensation was analysed by counterstaining the cells with rhodamine phalloidin (red). Cell morphology along with bacteria was viewed under transmitted light. Arrows indicate the position of bacteria and the corresponding association of Ec-gp96, Stat3, phospho-Stat3 or actin condensation (magnification: 60×).
E. coli invasion of HBMEC was significantly reduced when the cells overexpress dominant negative-Stat3, but not dominant negative-Stat5
To confirm the role of Stat3 activation in E. coli invasion of HBMEC, the cells were transiently transfected with a dominant negative (DN) form of Stat3, which is a splice variant of Stat3β that lacks the C-terminal region, and used for invasion assays. In addition, HBMEC transfected with a DN form of Stat5, in whichAla713 was mutated toAsp713, and plasmids alone were used as controls. The results showed that OmpA+ E. coli entry was decreased by ~60% in DN-Stat3/HBMEC when compared with either normal HBMEC or pcDNA3/HBMEC (5.3 × 103 ± 0.5 × 103 cfu per well for control versus 1.6 × 103 ± 0.3 × 103 cfu per well for DN-Stat3/HBMEC, P < 0.02 by two-tailed t-test) (Fig. 3A). However, no significant decrease in the binding of bacteria to these cells was observed. In contrast, the invasion of E. coli was reduced only by 20% in DN-Stat5/HBMEC (5.7 ± 0.6 × 103 cfu per well for plasmid control versus 4.1 ± 0.3 × 103 cfu per well in DN-Stat5/HBMEC), suggesting that the suppression of Stat5 activation did not affect invasion, whereas Stat3 activation was required for invasion of E. coli into HBMEC. To further evaluate whether the interaction of phospho-Stat3 with Ec-gp96 is inhibited in DN-Stat3/HBMEC, immunoprecipitation of HBMEC lysates after infecting the cells with OmpA+ E. coli using anti-Ec-gp96 antibody was performed. As predicted, the association of anti-phospho-Stat3 with Ec-gp96 was not observed in DN-Stat3/HBMEC, but was preserved in DN-Stat5/HBMEC lysates (Fig. 3B), at levels similar to that of non-transfected HBMEC (data not shown). Reprobing the blots with anti-Stat3 antibodies also showed no association of Stat3 with Ec-gp96 in DN-Stat3/HBMEC. These results suggest that specific association of Stat3 with Ec-gp96 and its activation was important for E. coli invasion of HBMEC. Next, we examined the effect of overexpression of DN-Stat3 on the actin condensation induced by entering E. coli by immunocytochemistry. As shown in Fig. 3C, control pcDNA3/HBMEC infected with OmpA+ E. coli revealed the attachment of a group of bacteria to the cell surface under which accumulation of both phospho-Stat3 and actin was observed (Fig. 3Ca–d). In contrast, DN-Stat3/HBMEC showed no such association of phospho-Stat3 and actin even after 60 min post infection with E. coli (Fig. 3Ce–h), whereas DN-Stat5/HBMEC revealed the colocalization of these two molecules (Fig. 3Ci–l). The intensity of immunofluorescence in these cells depends on the number of bacteria entering into the cells, as more bacteria appear entering in Fig. 3Ca, the density of pStat3 or actin is greater. Only one bacterium appears entering into HBMEC in Fig. 3Ci in which the density of these molecules is moderate. These results suggest that the interaction of Ec-gp96 with Stat3 is critical for regulating actin remodelling for subsequent entry of OmpA+ E. coli into HBMEC.
Fig. 3. Effect of overexpression of DN-Stat3 in HBMEC on the E. coli invasion, Stat3 activation and actin condensation.
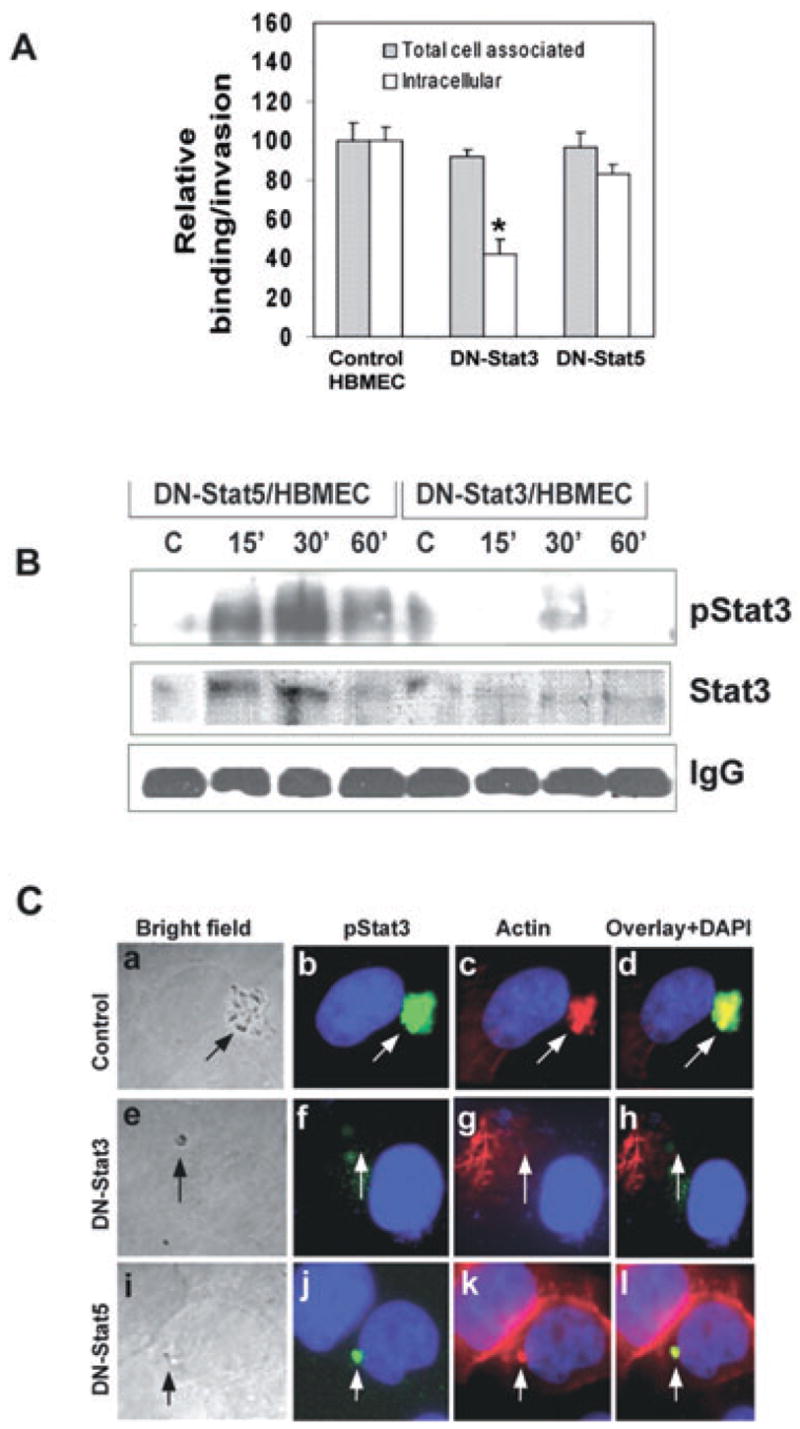
A. HBMEC expressing DN-Stat3 or DN-Stat5 were incubated with OmpA+ E. coli and the invasion and binding assays were performed as described in Experimental procedures. The data represent mean ± SD values from three different experiments carried out in triplicate and expressed as relative binding/invasion being taken OmpA+ E. coli invasion in HBMEC as 100%. The invasion in DN-Stat3/HBMEC is significantly lower when compared with control or DN-Stat5/HBMEC (P < 0.02 by two-tailed t-test).
B. To examine the association of Ec-gp96 with phospho-Stat3, lysates from HBMEC transfected with DN-Stat3 or DN-Stat5 and infected with OmpA+ E. coli for varying time points were immunoprecipitated with anti-Ec-gp96 antibody and analysed by Western blotting using anti-phospho-Stat3 antibody. Equality of protein loading was assessed by the amount of IgG used in each sample.
C. HBMEC (control), DN-Stat3/HBMEC or DN-Stat5/HBMEC were cultured in eight-well chambers and infected with OmpA+ E. coli for 30–60 min. The cells were washed and incubated with anti-phospho-Stat3 antibody followed by Alexa 488-conjugated secondary antibody and rhodamine phalloidin. Cell morphology along with bacteria was viewed under transmitted light (magnification: 100×). Arrows indicate the position of bacteria and the corresponding association of Ec-gp96, pStat3 or actin condensation.
The C-terminal domain of Ec-gp96, but not its ATP binding site, is necessary for E. coli invasion of HBMEC
To examine whether the C-terminal domain of Ec-gp96 is necessary for the binding of Stat3, 214 amino acids from the C-terminal region (589–803 amino acids) of Ec-gp96 were deleted (Ec-gp96Δ200). In addition, a gp96 construct that lacks ATP binding domain (termed Ec-gp96ΔATP) was included in these experiments to evaluate the role of ATP binding in Stat3 association. HBMEC were transfected with Ec-gp96Δ200, Ec-gp96ΔATP, full-length Ec-gp96 (FL-Ec-gp96) and pcDNA3.1 independently. Overexpression of FL-Ec-gp96 and Ec-gp96Δ200 containing FLAG epitope was verified by Western blotting of the total lysates with anti-FLAG antibody. The blot revealed a 65 kDa protein in Ec-gp96Δ200, whereas 96 and 72 kDa proteins in FL-Ec-gp96 (Fig. 4A). In contrast, no proteins were observed in both non-transfected HBMEC and in Ec-gp96ΔATP-transfected cells, as expected, because the Ec-gp96ΔATP is untagged. To further examine whether the overexpressed Ec-gp96 proteins recruited to the cell surface, lipid rafts of HBMEC transfectants were prepared. Western blotting of these microdomains with anti-Ec-gp96 antibody revealed that significant amounts of the overexpressed Ec-gp96 were present in the lipid rafts. The blot when reprobed with anti-caveolin-1 antibody revealed equal quantities of caveolin-1 protein, a marker for lipid rafts in all lanes, suggesting that both Ec-gp96 and caveolin-1 resides in lipid microdomains. Densitometric analysis of the bands normalized to caveolin-1 levels revealed that approximately 30% higher levels of Ec-gp96 were expressed in FL-Ec-gp96 and Ec-gp96ΔATP rafts when compared with control cells. A greater amount of 65 kDa protein was present in Ec-gp96Δ200 rafts although a similar molecular mass protein was present in Ec-gp96ΔATP rafts in very small quantities.
Fig. 4. Effect of overexpression of FL-Ec-gp96, Ec-gp96Δ200 and Ec-gp96ΔATP on the binding to and invasion of OmpA+ E. coli in HBMEC.
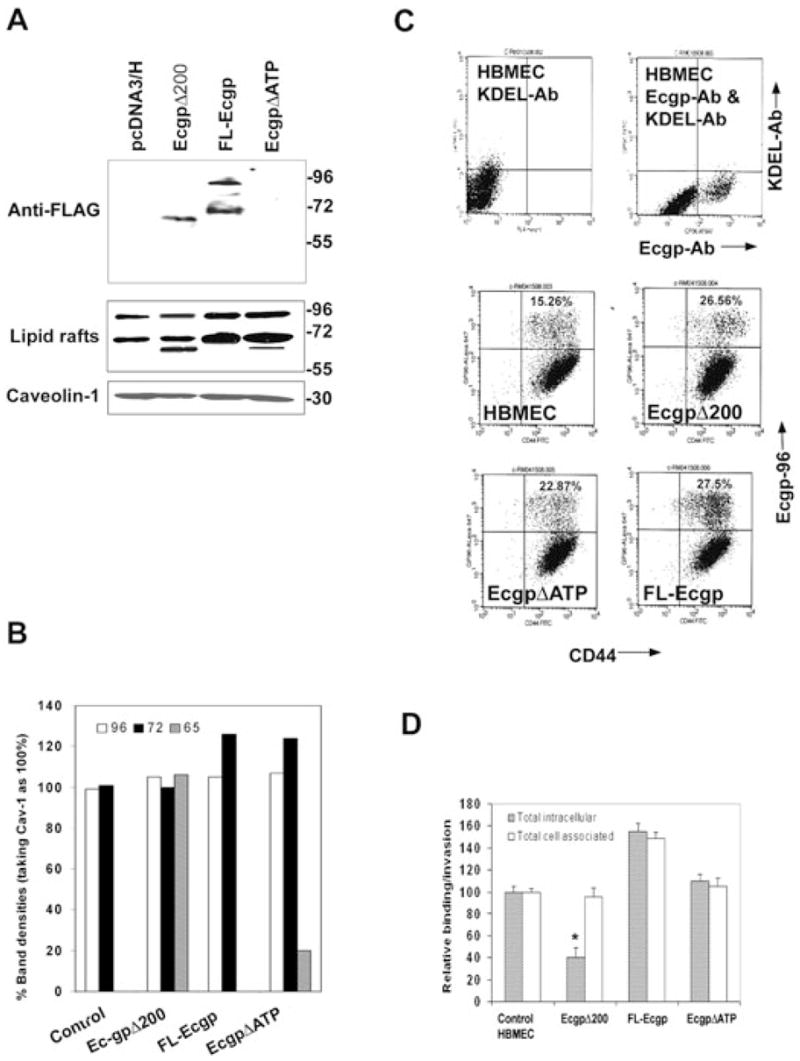
A. Total cell lysates of HBMEC expressing Ec-gp96Δ200, FL-Ec-gp96 and Ec-gp96ΔATP were prepared and examined by Western blotting using anti-FLAG antibody. Similarly, proteins from lipid raft fractions were subjected to Western blotting with anti-Ec-gp96 antibody. The amounts of protein loaded from lipid rafts were assessed by the presence of caveolin-1.
B. The densities of the protein bands in blots in Fig. 4A were determined, normalized to caveolin-1 and expressed as per cent densities being taken caveolin-1 levels as 100%.
C. HBMEC were subjected to flow cytometry after staining the cells with anti-Ec-gp96 antibody and with a monoclonal anti-gp96 antibody, which identifies a C-terminal portion of Ec-gp96 or monoclonal anti-KDEL antibody, which interacts with the KDEL region of Ec-gp96. In separate experiments, HBMEC overexpressing FL-Ec-gp96, Ec-gp96Δ200 and Ec-gp96ΔATP were subjected to flow cytometry after labelled with anti-Ec-gp96 and anti-CD44 antibodies followed by secondary antibodies coupled to Alexa 488 and 647. D. HBMEC expressing plasmid alone (control), FL-Ec-gp96, Ec-gp96Δ200 or Ec-gp96ΔATP were subjected to FACS, the Ec-gp96-positive populations were plated in 24-well culture dishes until they reach confluence and used for binding and invasion assays with OmpA+ E. coli as described in Methods. The data represent mean ± SD from three different experiments carried out in triplicate and expressed as relative binding/invasion being taken OmpA+ E. coli parameters in control HBMEC as 100%.
To confirm the expression of Ec-gp96 on the cell surface, we next analysed non-transfected HBMEC by flow cytometry using anti-Ec-gp96, anti-gp96 and anti-KDEL antibodies. The anti-Ec-gp96 antibody recognizes the N-terminal portion of Ec-gp96, whereas both anti-gp96 and anti-KDEL antibodies recognize the C-terminal portions of Ec-gp96 and the KDEL region respectively (Ménoret et al., 2001). Both anti-gp96 and anti-KDEL antibodies did not show any reactivity to cell-surface molecules by flow cytometry, suggesting that neither the KDEL nor the C-terminal regions of the gp96 was exposed at the surface of the HBMEC (Fig. 4C). However, staining with anti-Ec-gp96 antibody revealed that a significant amount of the protein was on the HBMEC surface, suggesting that the N-terminus was accessible on the cells exterior. In addition, the cell-surface expression of FL-Ec-gp96, Ec-gp96Δ200 and Ec-gp96ΔATP was also examined by flow cytometry in HBMEC transfectants. An antibody to HBMEC surface marker CD44 was also used in these studies. Approximately 15% of cells showed positive staining with anti-Ec-gp96 antibody whereas 85% of the cells were positive to the expression of CD44 in control HBMEC (Fig. 4C). The Ec-gp96-positive cells were increased by ~7–12% in transfected HBMEC, suggesting that 50–80% more cells expressed Ec-gp96 on their surface, when compared with non-transfected cells. Next, to analyse the effect of overexpressed Ec-gp96 or its constructs on the binding and invasion of OmpA+ E. coli, the control and Ecgp-96-transfected HBMEC were subjected to FACS to selectively increase the populations of cells positive for Ecgp-96 and cultured for a period of 3 days before conducting binding and invasion experiments. Both binding and the invasion of OmpA+ E. coli was increased by 10-fold in control non-transfected HBMEC when compared with unsorted cells (Fig. 4D). FL-Ec-gp96/HBMEC showed an increase of E. coli binding and invasion by ~50–60%, whereas the invasion in Ec-gp96Δ200/HBMEC was reduced by ~60% when compared with non-transfected and sorted cells despite binding equally well. Of note, the invasion of E. coli in Ec-gp96ΔATP/HBMEC was decreased although by only 20%. These data suggest that the C-terminal portion of Ec-gp96 may be critical and that the ATP binding domain may not play a significant role in the invasion.
The C-terminal truncated Ec-gp96 does not associate with phospho-Stat3 and prevents actin condensation upon infection with OmpA+ E. coli
As the invasion of E. coli in Ec-gp96Δ200/HBMEC was significantly reduced, we reasoned that the truncated Ec-gp96 might not be interacting with Stat3. Therefore, HBMEC transfectants were infected with OmpA+ E. coli for 15–60 min. Total lysates were immunoprecipitated with anti-Ec-gp96 antibody, and were then analysed by Western blotting with antibodies to Stat3 and phospho-Stat3. Due to the presence of multiple molecular sizes of Ec-gp96 in cell lysates, the equality of loading in these blots was verified by the presence of IgG levels in each lane. Consistent with the data shown in Fig. 3, pcDNA3/HBMEC revealed association of Stat3 and phospho-Stat3 with Ec-gp96 between 15 and 30 min (Fig. 5A). Similar increase in the association of Stat3 was also observed in FL-Ec-gp96/HBMEC and Ec-gp96ΔATP/HBMEC. In contrast, Ec-gp96Δ200/HBMEC showed neither Stat3 nor phospho-Stat3 association, suggesting that the C-terminal 214-amino acid region might be responsible for the interaction with Stat3. Furthermore, colocalization of Ec-gp96 with Stat3 and actin condensation sites was also examined by immunocytochemistry. Confocal microscopy images of pcDNA3/HBMEC infected with OmpA+ E. coli revealed that both Ec-gp96 and phospho-Stat3 were colocalized with actin condensation sites (Fig. 5Ba–h). In contrast, Ec-gp96Δ200/HBMEC showed very little or no condensation of actin despite the presence of significant amounts of Ec-gp96 on the surface. The distribution of phospho-Stat3 was diffused throughout the cells and no significant actin remodelling was observed in these cells. These data provided further evidence that binding of phospho-Stat3 to the C-terminal domain of Ec-gp96 is required for actin remodelling and for subsequent entry of OmpA+ E. coli into HBMEC.
Fig. 5. The C-terminal truncated Ec-gp96 does not bind Stat3/phospho-Stat3 upon infection with OmpA+ E. coli.
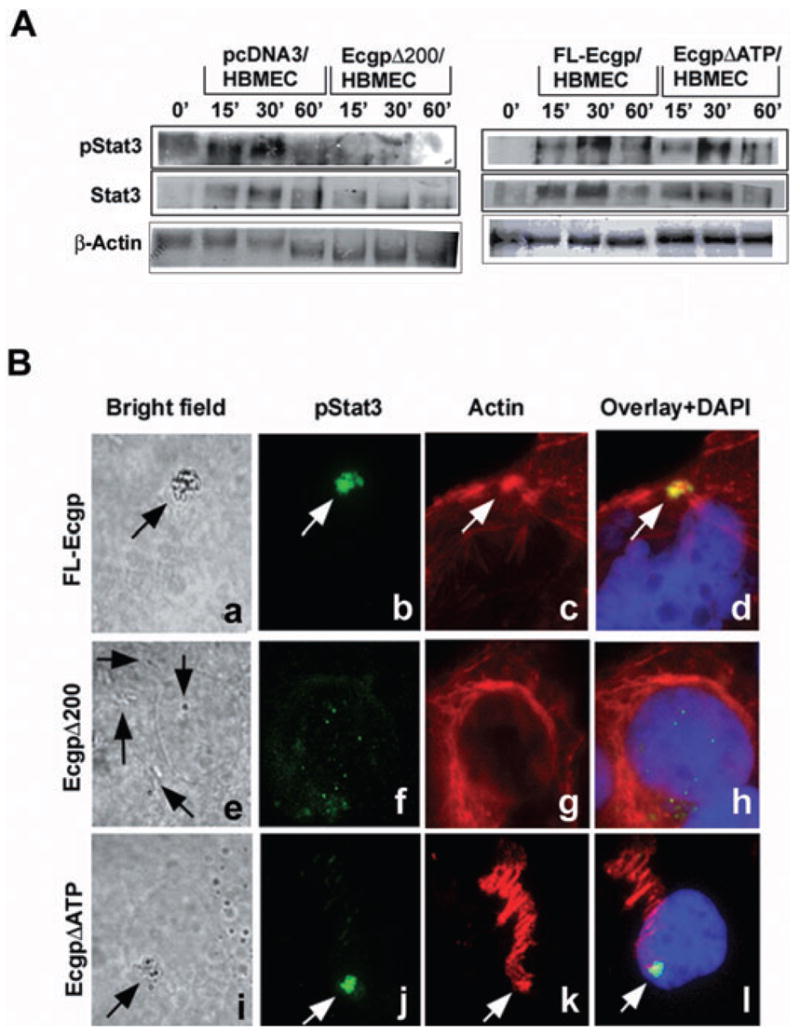
A. HBMEC expressing control plasmid, FL-Ec-gp96, Ec-gp96Δ200 or Ec-gp96ΔATP were infected with OmpA+ E. coli for various time points. Total lysates were then immunoprecipitated with anti-Ec-gp96 antibody followed by immunoblotting with antibodies to Stat3 or phospho-Stat3. Equal amounts of proteins (20 μg) from the lysates were also subjected to Western blotting with β-actin.
B. HBMEC expressing FL-Ec-gp96, Ec-gp96Δ200 and Ec-gp96ΔATP upon infection with OmpA+ E. coli for 30 min in eight-well chamber slides were stained with anti-phospho-Stat3 antibodies followed by Alexa 488 coupled secondary antibody. Actin was stained with rhodamine phalloidin. The cells were examined under a fluorescent microscope (magnification: 100×). Arrows indicate the bacteria attached to cells, the condensation of phospho-Stat3 or actin.
The association of Stat3 with Ec-gp96 is an upstream event of PKC-α and PI3-kinase activation
Our earlier studies demonstrated that PKC-α and PI3-kinase were involved in actin condensation during E. coli entry. However, the link between Ec-gp96 and these signalling molecules is not known. Therefore, we next examined the effect of overexpression of DN-Stat3 on the activation of PKC-α and PI3-kinase. HBMEC transfected with DN-Stat3 were infected with OmpA+ E. coli for 15–60 min and the total cell lysates were subjected to Western blotting with antibodies to phospho-PKC-α and phospho-Akt, a downstream effector protein of PI3-kinase. As illustrated in Fig. 6A, phosphorylation of Akt and PKC-α increased transiently between 15 and 30 min post infection in control HBMEC; however, the activation of these two signalling molecules were not detected in DN-Stat3/HBMEC. These observations suggest that both PKC-α and PI3-kinase act downstream of Stat3 activation.
Fig. 6. Activation of Stat3, PI3-kinase or PKC-α in HBMEC that express various DN molecules infected with OmpA+ E. coli.
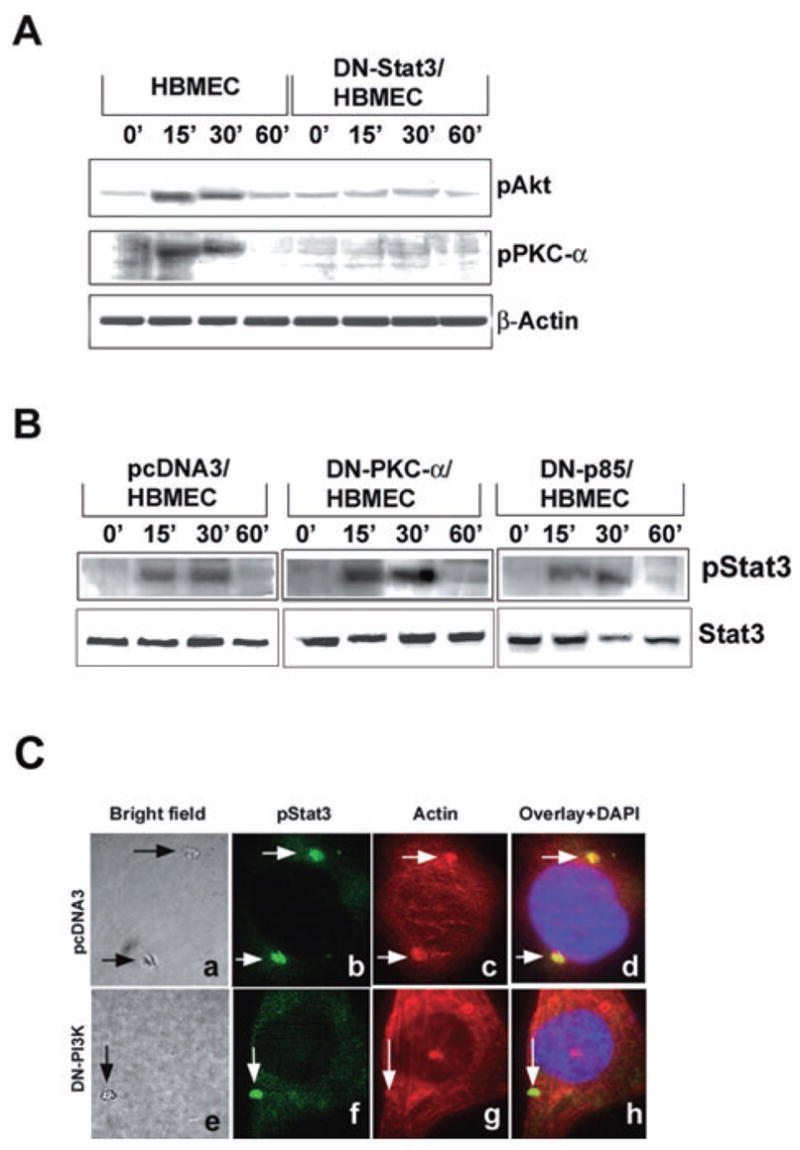
A. HBMEC either non-transfected or transfected with DN-Stat3 were infected with OmpA+ E. coli for varying time points, total cell lysates prepared and equal amounts of proteins were analysed by immunoblotting with anti-phospho-Akt (downstream product of PI3-kinase) or anti-phospho PKC-α antibodies. The total lysates (20 μg of proteins) were also subjected to Western blotting with anti-β-actin.
B. HBMEC expressing DN forms of either PKC-α or p85, a subunit of PI3-kinase, were infected with OmpA+ E. coli for varying time points, total cell lysates prepared and then immunoprecipitated with anti-Ec-gp96 antibody. The immune complexes were then analysed by Western blotting using anti-phospho-Stat3 or anti-Stat3 antibody. C. HBMEC transfectants were cultured in eight-well chamber slides and infected with OmpA+ E. coli for 30 min, washed and stained with anti-phospho-Stat3 antibody and rhodamine phalloidin. Arrows indicate the position of the bacteria, phospho-Stat3 or actin condensation (magnification: 100×).
To confirm this observation, HBMEC were transfected with a DN form of PI3-kinase subunit, p85 or PKC-CAT/KR, a DN form of PKC (Reddy et al., 2000; Sukumaran et al., 2002). The DN/p85 mutant contains a defective iSH2 region, fails to bind to PI3-kinase catalytic subunit p110 and dominantly inhibits the activation of PI3-kinase by titrating out the signalling molecules that interact with PI3-kinase. The PKC-CAT/KR construct encodes a truncated protein in which the catalytic domain (CAT) containing amino acids 326–672 of PKC is preserved, with a point mutation that abolishes ATP binding ability, while the regulatory N-terminal domain is deleted (Soh et al., 1999). The PKC-CAT/KR construct has been shown to dominantly inhibit PKC-α activation. HBMEC trans-fected with pcDNA3 vector alone were used as a control. Both DN-p85/HBMEC and PKC-CAT/KR/HBMEC were infected with OmpA+ E. coli for varying times and the total cell lysates were subjected to immunoblotting with anti-phospho-Stat3 antibodies. It was observed that phospho-Stat3 levels in DN-p85/HBMEC and PKC-CAT/KR/HBMEC increased between 15 and 30 min post infection similar to the response in pcDNA3/HBMEC (Fig. 6B). The above data suggest that Stat3 acts as an upstream linking molecule that relays signal from Ec-gp96 to activate PKC-α and PI3-kinase. To examine whether the activation of both PKC-α and PI3-kinase links Stat3 activation to actin rearrangements, immunocytochemistry was also performed using DN-p85/HBMEC and PKC-CAT/KR/HBMEC. The phospho-Stat3 colocalization was observed beneath the bacterial attachment site with actin condensation in pcDNA3/HBMEC (Fig. 6Cb), whereas only phospho-Stat3 was accumulated at the E. coli binding sites without actin condensation in DN-p85/HBMEC (Fig. 6Cf and g). Similar results were also observed with PKC-CAT/KR/HBMEC (data not shown). Taken together these results suggest that Stat3 activation and its association with Ec-gp96 is an upstream event of both PKC-α and PI3-kinase and actin arrangements.
Discussion
The stress protein gp96, also known as GRP94, has long been known for its ability to induce T cell immunity in addition to chaperoning a host of proteins in the cell (Udono et al., 1994; Yamazaki et al., 1999). Gp96 binds to a number of molecules on the host cell as well as bacterial proteins simultaneously and helps in antigen presentation. We have identified a gp96 homologue on HBMEC (Ec-gp96) as a receptor for OmpA during the invasion of E. coli (Prasadarao, 2002). Subsequently, other reports have shown that gp96 acts as a receptor for other pathogenic microbes (Banerjee et al., 2002). Of note, the expression of Ec-gp96 is significantly greater in HBMEC when compared with endothelial cells of different origins. The invasion of OmpA+ E. coli into HBMEC is regulated by several intracellular signalling molecules, such as PKC-α and PI3-kinase (Reddy et al., 2000; Sukumaran and Prasadarao, 2002). Furthermore, preventing the interaction of OmpA with Ec-gp96 by using antibodies to either ligand or receptor significantly reduced the activation of these signalling molecules, suggesting that Ec-gp96 relays signals to activate PKC-α and PI3-kinase upon interacting with OmpA. However, the connection between Ec-gp96 and the downstream signalling events in the host cell that accompany bacterial entry is not known.
In this study, we have shown that Stat3 interacts with Ec-gp96 upon infection with OmpA+ E. coli and that the bound Stat3 was activated by phosphorylation of its Tyr-705. In contrast, Stat5 did not show significant association with Ec-gp96. Although Stat1 was also associated with Ec-gp96, even in non-infected cells, its phosphorylation levels did not change upon infection with OmpA+ E. coli, suggesting that Stat1 does not play a role in the invasion process. The requirement of Stat3 in E. coli invasion was further confirmed by the inhibition of invasion by overexpression of DN-Stat3. We were unsuccessful in generating stable Stat3-transfected HBMEC, presumably because Stats are very important for cell migration (Takeda et al., 1997; Takeda and Akira, 2000; Xie et al., 2001). Thus, inhibition of E. coli invasion beyond 60% could not be achieved. Of note, overexpression of DN-Stat5 also showed 20% inhibition of E. coli invasion and could be due to the effect of DN-Stat5 on other signalling pathways. Association of Stat3 with Hsp90, similar to the one reported here, was observed in IL-6-treated Hep3B hepatocytes (Yamashita et al., 1998; Xie et al., 2001). However, in that case, the ATP binding site of Hsp90 was critical for the association, unlike the association of Stat3 with Ec-gp96. Recent studies have shown that lipopolysaccharide treatment of macrophages induces the activation of Stat3, which in turn induces the expression of IL-10 (Carl et al., 2004). Nonetheless, the mode of activation of Stat3 in HBMEC must be different because OmpA− E. coli, having as much lipopolysaccha-ride as OmpA+ E. coli, did not activate Stat3 when bound to the cell surface.
Due to the presence of ER retention KDEL amino acid signal at the C-terminal end, gp96 is predicted to reside in the lumen of the ER. Previous studies have shown that gp96 is sometimes expressed on cell surfaces (Altmeyer et al., 1996). Two scenarios can be envisioned for cell-surface expression of this molecule. First, the entire gp96 is transported to the extracellular space where it is attached non-covalently to other proteins, glycoproteins or glycolipids. Second, it itself is expressed as a plasma membrane spanning protein. Besides a signal peptide sequence, prediction for transmembrane sequences showed a weak hydrophobic 10-amino-acid domain in gp96 or Ec-gp96 (Prasadarao et al., 2003). As our flow cytometry analysis on HBMEC that overexpress FL-Ec-gp96 or Ec-gp96ΔATP demonstrated that the C-terminal and KDEL portion of the molecule appeared to be intra-cellular, it is likely that Ec-gp96 is expressed on the plasma membrane of HBMEC as a transmembrane protein. However, due to the weak transmembrane domain, it is also possible that the insertion of Ec-gp96 into the lipid bilayer is further strengthened by other molecules such as toll-like receptors (TLRs). Previous studies have shown that gp96 acts as a chaperone for TLR2 and TLR4 and that the N-terminal portion of gp96 can interact with TLRs (Vabulas et al., 2002; Warger et al., 2006). The possibility of TLR association with Ec-gp96 in HBMEC remains to be determined. Lack of Stat3 association with Ec-gp96Δ200 indicates that OmpA binding to the N-terminal portion of Ec-gp96 induces a spatial reorganization within the receptor, thereby triggering the interaction of the C-terminal domain with proximal Stat3, which is present in the caveolae (Fig. 7).
Fig. 7.
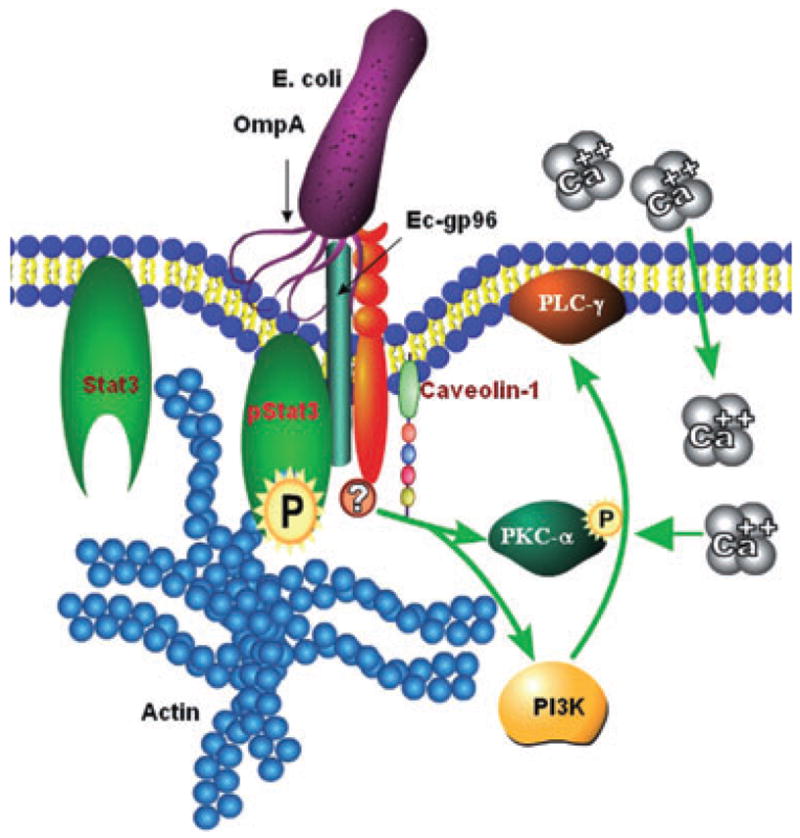
A schematic representation of the mechanisms involved in E. coli invasion of HBMEC. The interaction of OmpA with Ec-gp96 present in caveolae induces conformational changes in Ec-gp96, which allows the interaction of phosphorylated Stat3 with Ec-gp96. Subsequently, the activated Stat3 relays the signals either directly or indirectly to PKC-α or PI3-kinase to induce actin condensation. The activation of PKC-α and PI3-kinase was also dependent on other molecules such as caveolin-1 and PLC-γ to stimulate the influx of Ca2+ into the cytosol of HBMEC.
Actin rearrangements are important for E. coli invasion of HBMEC (Prasadarao et al., 2003). Our previous studies utilizing DN PI3-kinase or PKC-α mutants showed that actin accumulation was absent from the bacterial entry site, thus blocking the invasion (Prasadarao et al., 1999). Similarly, overexpression of DN-Stat3 in HBMEC significantly inhibited actin condensation underneath the E. coli binding sites, suggesting cross-talk between the Stat3 and actin polymerization pathways in E. coli invasion of HBMEC. In addition, HBMEC that express DN-Stat3 did not show activation of either PI3-kinase or PKC-α upon infection with OmpA+ E. coli, placing Stat3 activation as an intermediate step between PI3-kinase and PKC-α-mediated actin remodelling. Using Stat3-null cells, Stat3 was shown to regulate multiple cellular functions including actin cytoskeleton reorganization via RhoA, Rac1 and Cdc42 (Debidda et al., 2005). In these studies, RhoA, Rac1 and Cdc42 can all stimulate Stat3 Tyr-705 and Ser-727 phosphorylation while our studies revealed only Tyr-705 phosphorylation but not Ser-727 phosphorylation. Of note, the overexpression of constitutively active Rac1 in HBMEC showed significant decrease in E. coli invasion of HBMEC, suggesting that Stat3 activation and Rac1 activation are independent events that occur during E. coli invasion (Rudrabhatla et al., 2006). In several other bacteria, the effector molecules are injected into host cells by a specialized protein secretion system and translocation apparatus type III. SopE, a Salmonella typhimurium-secreted protein was characterized as a guanidine exchange factor for Rho-GTPases by its ability to stimulate conformational change in Cdc42, thereby promotes guanine nucleotide release (Patel and Galán, 2006). ExoT of Pseudomonas aeruginosa, on the other hand, acts as a GTPase-activating protein for RhoA, Rac1 and Cdc42 (Kazmierczak and Engel, 2002). However, no Type III secretion system has been identified in E. coli till date. The cytotoxic necrotizing factor 1 (CNF1) from E. coli activates members of the Rho family by deamidation of glutamine 61/63 and was shown to be necessary for E. coli invasion of HBMEC (Khan et al., 2002; Brest et al., 2004). It was demonstrated that the level of CNF1-activated Rac is rapidly diminished in CNF1-treated cells by proteolytic degradation (Pop et al., 2004). Therefore, our present view is that two different signalling pathways operate in HBMEC upon contact with E. coli, one actin rearrangement-related (Stat3, FAK, PI3K and PKC-α) due to OmpA interaction with Ec-gp96 and another myosin-related (Rac1, PAK1 and MLC) modulated by CNF1, whose convergence is required at the E. coli entry site for efficient internalization of bacteria. Nonetheless, the release of CNF1 into HBMEC did not occur until E. coli interacts with HBMEC through OmpA (··· ···, unpubl. results).
In summary, our results identified two novel phenomena: Stat3 involvement in bacterial pathogenesis and Ec-gp96 interaction with Stat3. Importantly, we also demonstrated that the interaction of Stat3 with Ec-gp96 via the C-terminal 214 amino acids is critical for OmpA+ E. coli invasion of HBMEC. By placing Stat3 activation as an intermediate step before PI3-kinase and PKC-α, we have progressed closer to the definition of the missing link between the E. coli receptor and the downstream actin condensation that is required for bacterial entry.
Experimental procedures
Bacterial strains and plasmids
The E. coli strains E44 (OmpA+) and E91 (OmpA−) used in this study are derivatives of the E. coli K1 strain RS218 (serotype 018:K1:H7) (Prasadarao et al., 1996). Strain E44 is a spontaneous rifampicin-resistant mutant and invades HBMEC. E91 is a non-invasive mutant lacking the entire ompA gene and was used as a negative control. The strains were grown in brain heart infusion medium supplemented with antibiotics rifampicin (100 μg ml−1) for E44 and tetracycline (12.5 μg ml−1) for E91.
Cell culture, DNA transfection and invasion assays
Human brain capillaries were isolated from small fragments of cerebral cortex, which were obtained from surgical resections of 4- to 7-year-old children with seizure disorders at Children’s Hospital Los Angeles. Microvascular endothelial cells (HBMEC) were isolated from these capillaries and cultured as described previously (Stins et al., 1994). HBMEC were maintained at 37°C in a humidified atmosphere of 5% CO2 in medium containing M199-Ham F-12 (1:1) supplemented with 10% fetal bovine serum, sodium pyruvate and 2 mM glutamine. HBMEC were used between 12 and 16 passages for all the experiments. For stable transfection, HBMEC were transfected with either Ec-gp96 mutants or empty vector using Lipofectamine reagent, allowed to recover for 24 h, and the transfected cells were selected using 300 μg ml−1 of G418. For transient transfection, HBMEC at 30% confluence were transfected with the Stat plasmids or the empty vectors using Lipofectamine for 6 h, washed, allowed to recover overnight and continued in culture for additional 3 days till they were 90% confluent. For invasion assays, HBMEC grown in 24-well cell culture plates to 90–95% confluence were infected with 107 cfu of E. coli strains in experimental medium (1:1 mixture of Ham F-12 and M-199 containing 5% heat-inactivated fetal bovine serum) and incubated for 90 min at 37°C in an atmosphere containing 5% CO2. The monolayers were washed three times with RPMI 1640 after which experimental medium containing gentamicin (100 μg ml−1) was added to the wells, and further incubated for 1 h at 37°C. The cells were then washed three times with RPMI 1640 and lysed with 0.3% of Triton X-100. The released bacteria were diluted with saline and enumerated by plating on blood agar. In duplicate experiments, the total cell-associated bacteria were determined as described for invasion, except that the gentamicin step was omitted.
Ec-gp96, DN-Stat3 and DN-Stat5 constructs
To amplify the full-length Ec-gp96 gene (FL-Ec-gp) coding for 803 amino acids, RT-PCR was performed on total mRNAfrom HBMEC using the following primer set – FP, 5′-TAAGAATTCCGCCAT GAGGGCCCTGTG-3′ and RP, 5′-CCTCTAGACTACTTGTCGT CATCGTCTTTGTAGTCTTCAGCTGTAGATT-3′. EcgpΔ200, a mutant of FL-Ecgp that is devoid of 214 amino acids from the C-terminal was amplified using the FP, 5′-TAAGAATTCCG CCATGAGGGCCCTTG-3′ and the RP, 5′-CCTCTAGACTACT TGTCGTCATCGTCTTTGTAGTCCTGGAACCTCTTC-3′. Both constructs contained a FLAG epitope at the C-terminal end. The amplified products were cloned into the restriction sites EcoRI and XbaI of pcDNA3.1. The resulting plasmids were transformed into DH5-α and the purified plasmids subjected to DNA sequencing to verify sequence integrity. EcgpΔATP was constructed by first cloning cDNA for murine GRP94 in the expression plasmid pcDNA3.1.Amino acid substitutions were introduced by the Quick-Change method of site-directed mutagenesis (Stratagene, CA), according to the manufacturer’s instructions. The EcgpΔATP mutant is a double substitution of two neighbouring amino acids predicted to be involved in nucleotide binding, D128N and G132A, using a single oligonucleotide. The mutations were verified by sequencing. The double mutant cannot bind ATP or the inhibitors geldanamycin and radicicol, but is functional in other activities of the protein (Vogen et al., 2002; Gidalevitz et al., 2004). DN-Stat3 was a kind gift from Dr. Hua Yu (City of Hope, CA). Human Stat3β (pIRES-Stat3b) or the empty vector encoding only EGFP (pIRES-EGFP) was used in these studies. Stat3β is a naturally occurring splice variant of Stat3 that lacks the COOH-terminal transcriptional activation domain and hence functions as a DN form of Stat3 in many cellular contexts (Catlett-Falcone et al., 1999; Niu et al., 1999). DN-Stat5 was provided from Dr. H. Yamashita (Ibaraki Prefectural Central Hospital, Ibaraki, Japan). Expression vector for Stat5aΔ713 were generated by truncation after amino-acid residues Ala713 of pXM-Stat5a (Yamashita et al., 1998).
Antibodies and reagents
Antibodies to Stat3, pStat3 and caveolin-1 were obtained from Cell Signalling (Danvers, MA). Rhodamine phalloidin, goat anti-rabbit and anti-mouse antibodies coupled to Alexa 488 and Alexa 648 respectively, were from Invitrogen (Carlsbad, CA). Protein A/G agarose beads were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit antibodies to Ec-gp96 were raised in our lab as described previously and recognizes the N-terminal region of Ec-gp96 (Prasadarao et al., 1996). Monoclonal anti-KDEL and anti-gp96 antibodies were obtained from StressGen (Ann Arbor, MI).
Flow cytometry
The HBMEC, non-transfected or transfected with the Ec-gp96 plasmids, cultured in 100 mm dishes were released from the plates using 1% BSA/10 mM EDTA, washed with PBS and then incubated with anti-Ec-gp96 antibody at a concentration of 1:1000 for 45 min followed by goat anti-rabbit fluorescent antibody (1:1000 dilution) for 30 min. In separate experiments, the cells were also incubated with either anti-KDEL or anti-gp96 antibody, washed and further incubated with goat anti-mouse antibody coupled to Alexa 648. Cells were washed three times in PBS and then analysed by flow cytometry using a FACS Calibur (BD BioSciences). Forward scatter was analysed using CellQuest software and approximately 10 000 flow events were recorded. Cells treated only with secondary antibody served as controls for gating. To enrich the population of HBMEC expressing Ecgp, cells transfected with Ecgp-96 constructs were sorted on Becton Dickinson’s Diva FACSVantage flow sorter to obtain approximately 5 × 105 cells per sample. The cells were plated in 24-well plates, cultured for 3 days and analysed for bacterial binding and invasion as mentioned earlier.
Western blotting and immunoprecipitations
Confluent cell cultures in 100 mm dishes were incubated with either OmpA+ or OmpA− E. coli for varying time points at 37°C. The cells were then rinsed with ice-cold PBS and scraped off into 500 μl of RIPA buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg ml−1 PMSF, Aprotinin and 1 mM sodium orthovanadate). The lysates were clarified by centrifugation for 10 min at 10 000 g to remove cell debris. The supernatants were collected and approximately 20 μg of total proteins was fractionated by electrophoresis on a 4.0% stacking-12% separating SDS polyacrylamide gel. Proteins were then transferred to a nitrocellulose membrane using a Bio-Rad semidry transfer apparatus. The blot was incubated with blocking buffer (100 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Tween 20; 2% bovine serum albumin and 2% milk) for 1 h at room temperature and then probed with the appropriate antibody. The same blot was stripped and re-blocked for subsequent probing with additional antibodies.
For immunoprecipitation, HBMEC were grown in 100 mm dishes and incubated with E. coli (1:100 cells to bacteria ratio) at 37°C for varying time periods. Cells were washed in cold PBS before lysis in RIPA Buffer. Lysates were clarified by centrifugation at 12 000 g for 10 min and approximately 300 μg of proteins was incubated with anti-Ec-gp96 antibody at 4°C overnight. The lysates were then incubated with Protein A/G-agarose beads for 1 h at 4°C. The beads were washed three times in ice-cold lysis buffer and the bound proteins were released by boiling the beads in SDS sample buffer, resolved by SDS-PAGE, and analysed by Western blotting with various antibodies. The proteins were detected using a Super Signal chemiluminescence detection kit (Pierce, Chicago, IL) followed by exposure to an X-ray film.
Preparation of membrane rafts
The HBMEC, either non-transfected or transfected with Ec-gp96 constructs, cultured in 100 mm dishes were scraped off into PBS, pH 7.2, containing a cocktail of protease inhibitors and 0.5% Triton X-100, and incubated on ice for 30 min. Lipid rafts were prepared from the total lysates using the caveolae/raft isolation kit (Sigma, St Louis, MO). The lysates were then applied to an Optiprep density gradient of 5–35% and centrifuged at 100 000 g in a Beckman ultracentrifuge. The fractions containing lipid rafts were then carefully removed from the top layers, estimated the protein content and analysed for the presence of Ec-gp96 by immunoblotting with anti-Ec-gp96 antibody. The blots were stripped and reprobed with either KDEL or caveolin-1 antibodies.
Immunocytochemistry
The HBMEC were grown in eight-well chamber slides, infected with OmpA+ or OmpA− E. coli for varying periods and then washed three times with RPMI. Cells were fixed in 2% paraform-aldehyde solution for 15–20 min at room temperature. Non-specific binding sites on the cells were blocked with 2% BSA in PBS for 30 min. The cells were then incubated with anti-Ec-gp96, anti-Stat3 or anti-phospho-Stat3 rabbit antibodies followed by goat anti-rabbit antibody conjugated to Alexa 488 at a concentration of 1:1, 500 along with rhodamine phalloidin (to stain actin) for 30 min. The slides were washed and then mounted with Vectashield (Vector Laboratories, Burlingame, CA) containing DAPI to stain the nucleus. Cells were viewed with a Leica (Wetzlar, Germany) fluorescent microscope using Plan-Apochromat oil immersion objective lens with a magnification of 100×. Images were acquired by EasyFish software and arranged into a composite panel using Adobe Photoshop 7.0.
Acknowledgments
The authors thank Drs Hiroko Yamashita, Nagoya City University, Japan and Hua Yu, City of Hope, Duarte, CA for providing DN forms of Stat5 and Stat3 respectively. Our sincere thanks to Dr. Yannan Ouyang, Childrens Hospital Los Angeles Research Institute Image Core, for assistance with fluorescence imaging. This work was supported by NIH Grant AI40567 (N.V.P.).
References
- Altmeyer A, Maki RG, Feldweg AM, Heike M, Proto-popov VP, Masur SK, Srivastava PK. Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int J Cancer. 1996;69:340–349. doi: 10.1002/(SICI)1097-0215(19960822)69:4<340::AID-IJC18>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Banerjee PP, Dass SV, Mathew A, Raje M, Parekh V, Prasad DVR, et al. Evidence that glycoprotein 96 (B2), a stress protein, functions as a Th2-Specific co-stimulatory molecule. J Immunol. 2002;169:3507–3518. doi: 10.4049/jimmunol.169.7.3507. [DOI] [PubMed] [Google Scholar]
- Brest P, Turchi L, Le’Negrate G, Berto F, Moreilhon C, Mari B, et al. Escherichia coli cytotoxic necrotizing factor 1 inhibits intestinal epithelial wound healing in vitro after mechanical injury. Infect Immun. 2004;72:5733–5740. doi: 10.1128/IAI.72.10.5733-5740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanes D, Sousa S, Cebriá A, Lecuit M, Garcíadel Portillo F, Cossart P. Gp96 is a receptor for a novel Listeria monocytogenes virulence factor, Vip, a surface protein. EMBO J. 2005;24:2827–2838. doi: 10.1038/sj.emboj.7600750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl VS, Gautam JK, Comeau LD, Smith MF., Jr Role of endogenous IL-10 in LPS-induced STAT3 activation and IL-1 receptor antagonist gene expression. J Leukoc Biol. 2004;76:735–742. doi: 10.1189/jlb.1003526. [DOI] [PubMed] [Google Scholar]
- Castelli C, Rivoltini L, Rini F, Belli F, Testori A, Maio M, et al. Heat shock proteins: biological functions and clinical application as personalized vaccines for human cancer. Cancer Immunol Immunother. 2004;53:227–233. doi: 10.1007/s00262-003-0481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signalling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Debidda M, Wang L, Zang H, Poli V, Zheng Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J Biol Chem. 2005;29:17275–17285. doi: 10.1074/jbc.M413187200. [DOI] [PubMed] [Google Scholar]
- Gamero AM, Sakamoto S, Montenegro J, Larner AC. Identification of a novel conserved motif in the STAT family that is required for tyrosine phosphorylation. J Biol Chem. 2004;279:12379–12385. doi: 10.1074/jbc.M310787200. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Biswas C, Ding H, Schneidman-Duhovny D, Wolfson HJ, Stevens F, et al. Identification of the N-terminal peptide binding site of glucose-regulated protein 94. J Biol Chem. 2004;279:16543–16552. doi: 10.1074/jbc.M313060200. [DOI] [PubMed] [Google Scholar]
- Jacques R, Antoine M, Cohen N. Cell surface expression of the endoplasmic reticular heat shock protein gp96: is phylogenetically conserved. J Immunol. 1999;163:4133–4139. [PubMed] [Google Scholar]
- Kazmierczak I, Engel JN. Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun. 2002;70:2198–2205. doi: 10.1128/IAI.70.4.2198-2205.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Wang Y, Kim KJ, Chung JW, Wass CA, Kim KS. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2002;277:15607–15612. [Google Scholar]
- Ménoret A, Li Z, Niswonger ML, Altmeyer A, Srivastava PK. An endoplasmic reticulum protein implicated in chaperoning peptides to major histocompatibility of class I is an aminopeptidase. J Biol Chem. 2001;276:33313–33318. doi: 10.1074/jbc.M103383200. [DOI] [PubMed] [Google Scholar]
- Niu G, Heller R, Catlett-Falcone R, Coppola D, Jaroszeski M, Dalton W, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999;59:5059–5063. [PubMed] [Google Scholar]
- Patel JC, Galán JE. Differential activation and function of Rho GTPases during Salmonella–host cell interactions. J Cell Biol. 2006;175:453–463. doi: 10.1083/jcb.200605144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop M, Aktories K, Schmidt G. Isotype-specific degradation of Rac activated by the Cytotoxic Necrotizing Factor 1. J Biol Chem. 2004;279:35840–35848. doi: 10.1074/jbc.M404346200. [DOI] [PubMed] [Google Scholar]
- Prasadarao NV. Identification of Escherichia coli outer membrane protein a receptor on human brain microvascular endothelial cells. Infect Immun. 2002;70:4556–4563. doi: 10.1128/IAI.70.8.4556-4563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996a;64:146–153. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasadarao NV, Wass CA, Kim KS. Endothelial cell GlcNAc-1–4 GlcNAc epitopes for outer membrane protein A enhance traversal of Escherichia coli across the blood–brain barrier. Infect Immun. 1996b;64:154–160. doi: 10.1128/iai.64.1.154-160.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasadarao NV, Wass CA, Stins MF, Shimada H, Kim KS. Outer membrane protein A-promoted actin condensation of brain microvascular endothelial cells is required for Escherichia coli invasion. Infect Immun. 1999;67:5775–5783. doi: 10.1128/iai.67.11.5775-5783.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasadarao NV, Srivastava PK, Rudrabhatla RS, Kim KS, Huang SH, Sukumaran SK. Cloning and expression of the Escherichia coli K1 outer membrane protein a receptor, a gp96 homologue. Infect Immun. 2003;71:1680–1688. doi: 10.1128/IAI.71.4.1680-1688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MA, Prasadarao NV, Wass CA, Kim KS. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2000;275:36769–36774. doi: 10.1074/jbc.M007382200. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla RS, Selvaraj SK, Prasadarao NV. Role of Rac1 in Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microbes Infect. 2006;8:460–469. doi: 10.1016/j.micinf.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Patel K, Victor A, Fried VA, Sehgal PB. Interactions of STAT3 with Caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. J Biol Chem. 2002;277:45662–45669. doi: 10.1074/jbc.M205935200. [DOI] [PubMed] [Google Scholar]
- Soh JW, Lee EH, Prywes R, Weistein IB. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol. 1999;19:1313–1324. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stins MF, Prasadarao NV, Ibric L, Wass CA, Luckett P, Kim KS. Binding characteristics of S fimbriated Escherichia coli to isolated brain microvascular endothelial cells. Am J Pathol. 1994;145:1228–1236. [PMC free article] [PubMed] [Google Scholar]
- Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N Engl J Med. 2002;347:240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- Sukumaran SK, Prasadarao NV. Regulation of protein kinase C in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2002;277:12253–12262. doi: 10.1074/jbc.M110740200. [DOI] [PubMed] [Google Scholar]
- Sukumaran SK, Quon MJ, Prasadarao NV. Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase C-α interaction in human brain microvascular endothelial cells. J Biol Chem. 2002;277:50716–50724. doi: 10.1074/jbc.M208830200. [DOI] [PubMed] [Google Scholar]
- Sukumaran SK, McNamara G, Prasadarao NV. Escherichia coli K-1 interaction with human brain microvascular endothelial cells triggers phospholipase C-γ1 activation downstream of phosphatidylinositol 3-Kinase. J Biol Chem. 2003;278:45753–45762. doi: 10.1074/jbc.M307374200. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udono H, Levey DL, Srivastava PK. Cellular requirements for tumor-specific immunity elicited by heat shock proteins: tumor rejection antigen gp96 primes CD8+ T cells in vivo. Proc Natl Acad Sci USA. 1994;91:3077–3081. doi: 10.1073/pnas.91.8.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Braedel S, Hilf N, Jasuja HS, Herter S, Nejad PA, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–20853. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- Vogen SM, Gidalevitz T, Biswas C, Simen BS, Stein E, Gulmen F, Argon Y. Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J Biol Chem. 2002;277:40742–40750. doi: 10.1074/jbc.M205323200. [DOI] [PubMed] [Google Scholar]
- Warger T, Hilf N, Rechtsteiner G, HaselmayerPCarrick DM, Jonuleit H, et al. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem. 2006;281:22545–22553. doi: 10.1074/jbc.M502900200. [DOI] [PubMed] [Google Scholar]
- Xie B, Zhao J, Kitagawa M, Durbin J, Madri JA, Guan JL, Fu XY. Focal adhesion kinase activates Stat1 in integrin-mediated cell migration and adhesion. J Biol Chem. 2001;276:19512–19523. doi: 10.1074/jbc.M009063200. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Xu J, Erwin RA, Farrar WA, Kirken RA, Rui H. Differential control of the phosphorylation state of Proline-juxtaposed serine residues Ser725 of Stat5a and Ser730 of Stat5b in Prolactin-sensitive cells. J Biol Chem. 1998;273:30218–30224. doi: 10.1074/jbc.273.46.30218. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Nguyen T, Podack ER. Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol. 1999;163:5178–5151. [PubMed] [Google Scholar]