

Abstract
Purpose:
To determine the full-length sequence of a gene with similarity to RP1 and to screen for mutations in this newly characterized gene, named retinitis pigmentosa 1-like 1(RP1L1). Since mutations in the RP1 gene cause autosomal dominant retinitis pigmentosa, it is possible that mutations in RP1's most sequence similar relative, RP1L1, may also be a cause of inherited retinal degeneration.
Methods:
A combination of cDNA clone sequencing, RACE, and database analysis were used to determine the RP1L1 mRNA sequence and its genomic organization. PCR analysis, semi-quantitative RT PCR, and in situ hybridization were used to determine the expression pattern of RP1L1. Single-strand conformational analysis and automated sequencing were used to screen probands from 60 adRP families for potential disease-causing mutations in RP1L1.
Results:
The human RP1L1 gene is encoded in 4 exons, which span 50 kb on chromosome 8p. The length of the RP1L1 mRNA is large, over 7 kb, but its exact length is variable between individuals due to the presence of several length polymorphisms, including a 48 bp repeat. RP1L1 encodes a protein with a minimal length of 2,400 amino acids and a predicted weight of 252 kDa. Expression of RP1L1 is limited to the retina and appears to be specific to photoreceptors. Mutational analysis of 60 autosomal dominant retinitis pigmentosa probands revealed the presence of 38 sequence substitutions in RP1L1. Over half of these substitutions result in alteration of the RP1L1 protein, but none of these substitutions appear to be pathogenic.
Conclusions:
The RP1L1 gene encodes a large, highly polymorphic, retinal-specific protein. No RP1L1 disease-causing mutations were identified in any of the samples tested, making it unlikely that mutations in RP1L1 are a frequent cause of autosomal dominant retinitis pigmentosa. Additional experiments will be needed to determine if mutations in RP1L1 cause other forms of inherited retinal degeneration.
Retinitis pigmentosa (RP) is a genetically heterogeneous inherited retinal degeneration which affects approximately 1 of 3,500 people worldwide. Individuals affected with retinitis pigmentosa exhibit night blindness, followed by a progressive reduction of visual field, which usually culminates in legal or complete blindness. Reduced or absent electroretinogram (ERG) and bone spicule-like pigmentary deposits accompany these symptoms [1]. Retinitis pigmentosa can be inherited in an autosomal dominant (adRP), autosomal recessive (arRP), X-linked (xlRP), or digenic form. To date, 12 adRP, 15 arRP, and 5 xlRP loci have been mapped and the disease-associated genes for 24 of these loci have been identified (RetNet).
Despite the large number of recent disease gene discoveries, much work still remains to completely understand the genetics of retinitis pigmentosa. Mutation analysis of the known disease-associated genes fails to identify mutations in at least 50% of cases and prevelences determined by linkage mapping are often inflated. For instance, despite the relatively large number of families originally mapped to the RP10 locus, mutations in the RP10 gene, IMPDH1, appear to account for less than 5% of adRP cases (unpublished data). The RP11 locus, estimated to be responsible for approximately 20% of adRP cases, also shows less than predicted mutation frequencies [2,3]. These data, along with the existence of large families in which the disease locus does not map to any of the known loci, suggest that a number of unidentified adRP loci still exist.
One strategy that can be used to find new adRP genes is to identify candidates that have sequence similarity to known adRP genes or that share functional pathways. For instance, three of the recently identified adRP disease-associated genes, HPRP3, PRPF8, and PRPF31, encode pre-mRNA splicing factors that participate in a common pathway. Using this strategy, we decided to characterize the nearest relative of RP1, and to determine if mutations in this newly characterized gene cause adRP.
Mutations in RP1 were first identified as a cause of adRP in 1999 and subsequent studies have determined that RP1 mutations account for 8-10% of adRP [4-7]. Our initial analysis of RP1 identified a retinal EST cluster, named eye2931, with significant sequence similarity. This EST cluster was incomplete, but showed 50% sequence identity to RP1 over the available 86 amino acids [4].
The purpose of this study was to characterize the full-length gene corresponding to eye2931 and to determine if mutations in this gene, subsequently named RP1L1, cause adRP. The RP1L1 mRNA is encoded in four exons and corresponds to a protein over 2,400 amino acids in length. Expression of RP1L1 appears to be limited to the retina, specifically, to photoreceptors. RP1L1 is an expressed gene, with conserved functional domains and a long open reading frame. That is, it is not a pseudogene.
We tested probands from 60 adRP families for mutations in the RP1L1 gene using a combination of SSCA and sequencing. Our analysis determined that RP1L1 contains a polymorphic 16 amino acid repeat, a common 21 bp deletion, and numerous polymorphic missense and silent substitutions. Despite the large amount of variation seen in RP1L1, no disease-causing mutations were identified in any of the individuals tested.
METHODS
cDNA clones
cDNA clones corresponding to the ESTs from the eye2931 cluster were purchased from Research Genetics (Carlsbad, CA). Bacteria containing cDNA clones were grown overnight and the plasmids purified using the QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA). Plasmid DNAs were sequenced according to the manufacturer's protocol using the ABI BigDye cycle sequencing dye terminator kit. Sequence reactions were purified using Centri Sep 96 columns (Princeton Separations Inc., Freehold, NJ) and run on an ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence from the cDNA clones was assembled using AutoAssembler (Applied Biosystems).
Rapid Amplification of cDNA ends (RACE)
RACE analysis was conducted using Human Retina Marathon-Ready cDNA (Clontech, Palo Alto, CA). rTthDNA polymerase, cDNA adapter primers, and several different gene-specific primers were used to amplify additional 5′ and 3′ regions of the RP1L1 cDNA. Resulting PCR products were separated on agarose gels, and the bands excised and purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). Purified PCR product was sequenced using the methods described above.
Database analysis
The Celera (Rockville, MD) database was searched at several different times using the incomplete human RP1L1 cDNA sequence. Genomic sequence 3′ of the incomplete cDNA sequence was analyzed for continuation of an open reading frame using MacVector (Accelrys, San Diego, CA). The complete human RP1L1 protein sequence was compared by BLAST analysis against the Celera mouse genome database to determine the mouse RP1L1 coding sequence.
Expression analysis
DNAs from human and mouse tissues (human and mouse MTC panels; Clontech) were amplified by PCR using RP1L1 specific primers, Amplitaq Gold (Applied Biosystems), and standard cycling conditions. Aliquots of PCR product were removed and visualized on agarose gels after 25, 30, and 35 cycles.
RNAs at postnatal day P0-21 were isolated from mouse retinas using RNA Trizol (Invitrogen, Carlsbad, CA). RNAs at embryonic day E7-14.5 were isolated from whole embryos, and RNA at E15.5 was isolated from whole brain using the same protocol. RT-PCR was conducted (Omni-Script RT kit, Qiagen, Hilden, Germany) to reverse-transcribe 2 μg of RNA using random hexamer primers. PCR conditions were as follows: hot start at 95 °C for 5 min followed by 33 cycles at 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min. PCR products were analyzed on 1% agarose gels. For RP1L1, two pairs of primers were used: RP1L1-Ex2F (5′-gca tga agc tcc aga aac atc cta ttc-3′) and RP1L1-Ex3R (5′-cca tct cca gac atc tga agg cct cgt-3′); and RP1L1-Ex3F (5′-gtg gac tct ctg cag aca ctc ctt ga-3′) and RP1L1-Ex4R (5′-gaa gtt gga agc gga ctt tca tct cca-3′). Both pairs gave identical results. As a control, primers for Myb P42POP (NM-145579) were used: Myb-F1 (5′-tgg tag ctg ctg ttg tgg atg-3′) and Myb-R1 (5′-att tca cgg aga ctt cca gcg-3′).
Semi-quantitative RT-PCR was performed using first strand cDNA which was synthesized from 2 mg of total retinal RNA from 4 month old wild type and rho−/− mice. Semi-quantitative PCR was performed on a Roche LightCycler using the Quantitech PCR kit (Qiagen, Valencia, CA). Serial dilutions of cDNA were used to generate standard curves of crossing-cycle number v. the logarithm of concentration for RP1L1. Relative transcript levels in both RNA samples were determined using a linear regression line calculated from standard curves. Values were normalized to the relative amounts of GAPDH present in the same cDNA preparations.
In situ hybridization was carried out using sense and antisense DIG-labelled riboprobes generated from PCR templates that incorporated T7 and T3 promoters. Frozen cryosections were fixed in para-formaldehyde post cutting, treated with active DEPC, and hybridized with both sense and antisense probes overnight at 58 °C. Stringent washes with SSC were performed and then the cryosections were incubated with an AP coupled anti-DIG antibody. Binding of the probes was detected using NBT/BCIP solution. Sections were mounted and analyzed using a Zeiss Axioplan 2 microscope.
Subjects
Subjects tested in this study were diagnosed at the Jules Stein Eye Institute, UCLA School of Medicine, Los Angeles, CA or at the Anderson Vision Research Center, Retina Foundation of the Southwest, Dallas, TX. Informed consent was obtained from all subjects tested.
SSCA and sequencing analysis
Genomic DNA was extracted from peripheral blood using previously reported methods [8]. Exons 2, 3 and amplimers 4A-4K of exon 4 were tested for the presence of disease-causing mutations by SSCA. The remainder of exon 4 (amplimers 4M-4P), and all SSCA variants were tested using automated PCR product sequencing. Amplimer 4L contains a polymorphic 48 bp repeat which made analysis of this region of the gene very difficult. This region was analyzed by sequencing in selected homozygotes only.
For SSCA, genomic DNA was amplified using the primers listed in Table 1 and AmpliTaq Gold polymerase (Applied Biosystems, Foster City, CA). Reactions were cycled as follows: 95 °C for 5 min, 35 cycles of 95 °C for 1 min, annealing temperature for 1 min, and 72 °C for 1 min, with a final extension of 75 °C for 5 min. PCR products were radiolabelled by incorporating 1 μCi of [32P]-dCTP (Amersham BioSciences, Piscataway, NJ). The majority of PCR products were then digested with restrictions enzymes. PCR products were denatured and separated overnight on 0.6x MDE gels (BioWhittaker, Rockland, ME) at room temperature and 4 °C. The gels were dried and exposed to film after electrophoresis.
Table 1.
PCR primers and restrictions enzymes used to analyze RP1L1
|
Amplimer |
Primer sequences 5′-3′ temperature |
Annealing temperature |
Product size (bp) |
Restriction enzyme |
|---|---|---|---|---|
| 2A | GCCAATCCCCCAAGCTG | 60 °C | 321 | BamHI |
| GGGTGTGGTGACAGAGCG | ||||
| 2B | CGTGCCTCTCTCCTTTGG | 60 °C | 444 | HinfI |
| AGGTCTAAAGAACCTTTTCAAGG | ||||
| 3 | TGGTGAGACTGGATCCTTCC | 60 °C | 292 | HpaII |
| CAGCCCTACTGAACCACCAT | ||||
| 4A | CTGTTTTATTCCTTTATCCTGACGC | 54 °C | 417 | AluI |
| CTACCTCCCCCAGAACGG | ||||
| 4B | GCTTCCACCTGGTCGGCG | 54 °C | 386 | EaeI |
| GGCTGGGCTGGCACTGTC | ||||
| 4C | GGAAGAGGTGGGGACTGG | 62 °C | 411 | DdeI |
| TTGCCTTGCCTGGACAGC | ||||
| 4D | AGCGAATGGGGTGGGCGG | 54 °C | 404 | MboII |
| GAGTCCAGTGGGCTGTGG | ||||
| 4E | TCCCAGGCATTCTCACTACC | 54 °C | 362 | BsaI |
| AGCAGGAGTCGGATGTGTG | ||||
| 4F | CGGCCCCATACCTCCCCAC | 60 °C | 229 | None |
| TGAGCAGCAGTGGCTTCG | ||||
| 4G | GCCTCAGCCCCTCCTCACC | 60 °C | 392 | RsaI |
| TCCTCAAGGTCTTCTCCTCG | ||||
| 4H | CAGTGCCAGCCAGGGTGC | 54 °C | 207 | None |
| GTGGTCTCGTCCGCCAAC | ||||
| 4I | ATGGCTGGACAACATTCCA | 54 °C | 393 | StuI |
| ATCAGCGCCCTCATGATCT | ||||
| 4J | CCGGAGCAGACAGAGAGG | 54 °C | 410 | BsaI |
| CGTGAAGTTCTCCGTCATGG | ||||
| 4K | GATGTGACGTTGGGGAAGAC | 54 °C | 401 | MslI |
| AGCTAACTGCTCCAGGTTCG | ||||
| 4L | AGAGACAGTGAGGAGCAGAGG | 60 °C | Variable | None |
| TCTCCTTGCAGTCCTCCTTC | ||||
| 4M | GAGGGGGTGCAGTTAGAGG | 62 °C | 874 | None |
| CCTCGCAGGGACAGAACTC | ||||
| 4N | CTCTCCTTCACCCTGGAGG | 65 °C | 829 | None |
| CTTCTGACTCTGGCTGGACC | ||||
| 4O | GTAAAACGACGGCCAGTCCCCAGAGGCAGAAGGAG | 69 °C | 861 | None |
| CCTTTGTCGATACTGGTACTGCCTCTACACCTTCTGACTCAGG | ||||
| 4P | GAAGGGGAGGCCCAGAAG | 62 °C | 919 | None |
| CCAAGCTCGTGATTGTTTTC |
Amplimer 4N PCR product was sequenced using the following nested sequencing primers: 5′-CCT TCA CCC TGG AGG ACG-3′; 5′-GGC TGG ACC TCC CAT TC-3′. Amplimer 4O primers incorporated M13 sequences into the PCR products. M13 sequencing primers were then used to sequence this amplimer.
For sequencing, PCR was performed using the primers listed in Table 1, Amplitaq Gold polymerase (Applied Biosystems, Foster City, CA) and with the exception of amplimer 4O, the standard cycling parameters described above. Amplimer 4O was cycled as follows: 95 °C for 5 min, 35 cycles of 95 °C for 1 min, annealing temperature for 1 min, and 72 °C for 2 min, with a final extension of 75 °C for 5 min. The PCR products were purified using either ExoSAP-IT (USB, Cleveland, OH) and the manufacturer's protocol or the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). Purified PCR product was sequenced according to the manufacturer's protocol using the ABI BigDye cycle sequencing dye terminator kit or the ABI dGTP dye terminator kit. Sequence reactions were purified as described above and run on an ABI Prism 310 or 3100 Genetic Analyzer.
Each of the protein altering variants observed in the adRP probands was tested in control individuals. DNA was tested using either PCR product sequencing or restriction digest to determine if the variants were present.
RESULTS
RP1L1 sequence
The full-length mRNA sequence corresponding to the human EST cluster eye2931 was determined using a combination of laboratory and database analyses. Initially, cDNA clones corresponding to the ESTs in eye2931 were sequenced to obtain additional expressed sequence. Repetitive rounds of RACE and Celera database analyses were then used to determine the remaining mRNA sequence.
Comparison of RP1L1 mRNA sequence with the human genome sequence from the Celera database determined that RP1L1 is organized into 4 exons with the initiation codon in exon 2. Like RP1, the majority of RP1L1 coding sequence is located in a very large fourth exon. In fact, all of the intron/exon boundary locations of RP1L1 are notably similar to those of RP1 (Figure 1).
Figure 1.
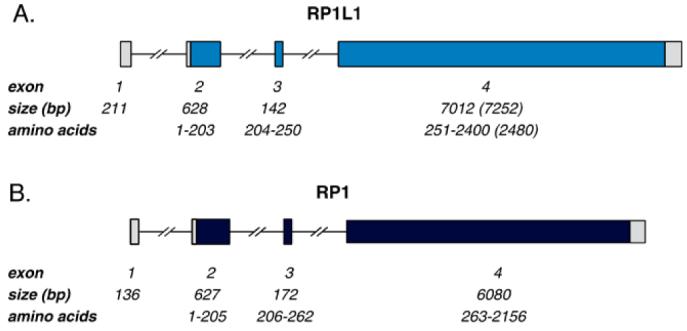
Comparison of the genomic structure of RP1L1 and RP1. A: RP1L1 is composed of four exons, three of which (2-4) contain coding sequence. Exon 4 is of variable length, with minimum size shown and maximum size in parentheses. B: RP1 is also composed of four exons, three of which (2-4) contain coding sequence. RP1L1 and RP1 have similar genomic organization, especially the location of the intron/exon junctions.
The exact size of the RP1L1 mRNA is variable due to the presence of a 48 bp polymorphic coding repeat (Figure 2). The shortest common mRNA sequence of RP1L1 (GenBank entry AY168341) is 7,993 bp in length and encodes a 2,400 amino acid protein. This version contains one of the 48 bp repeats, but as many as six 48 bp repeats have been observed in normal controls (GenBank entries AY168341, AY168342, AY168343, AY168344, AY168345, and AY168346). In addition to the polymorphic repeat, another large repetitive region is present near the 3′ end of RP1L1. Both of these repetitive regions contain an unusually high percentage of glutamine and glutamic acid residues. RACE analysis also suggests that RP1L1 uses 3 different polyadenlyation signals (data not shown).
Figure 2.

RP1L1 protein sequence. A: RP1L1 contains a region of homology to doublecortin (DC) and to RP1 (RP1). RP1L1 also contains a 16 amino acid polymorphism and large low complexity repeat. B: Comparison of the four shortest RP1L1 repeat polymorphisms. Repeat sequences are in red with an arrow depicted for each repeat. Sequence surrounding the repeats is very similar to that contained in one copy of the repeat. C: Partial alignment of human RP1L1 and RP1 protein sequences. The sequences corresponding to the doublecortin domains and RP1 domain are identified with bars. The asterisk (“*”) represents identical amino acids and the period (“.”) represents similar amino acid residues.
The mouse RP1L1 coding sequence was predicted by comparing the human coding sequence to the Celera mouse genome sequence. Interestingly, the predicted mouse protein is only 1,859 amino acids in length due to the absence of both the 48 bp polymorphic repeat and the repetitive region found in human RP1L1 (Figure 3).
Figure 3.
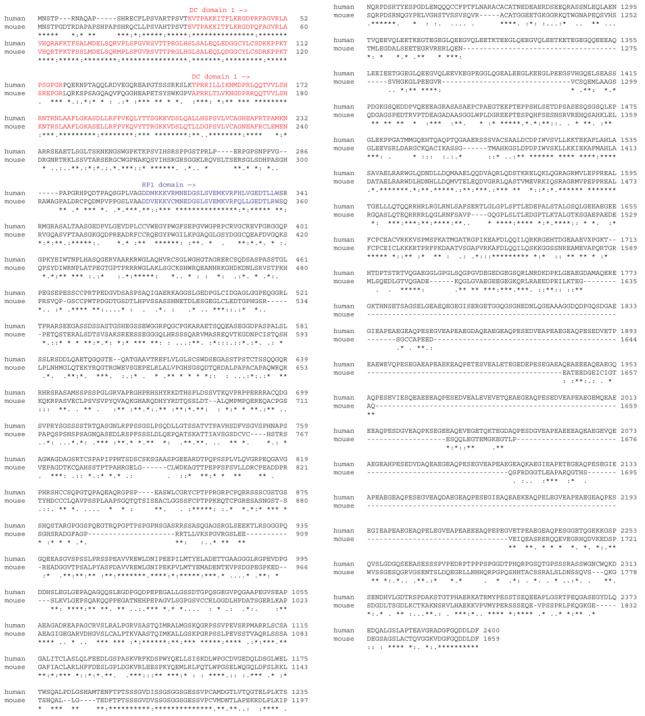
Alignment of human and mouse RP1L1 sequences. The mouse sequence lacks the polymorphic repeat and low complexity repeat found in human RP1L1. The doublecortin and RP1 regions of the proteins are designated with solid bars.
Human RP1L1 is located on chromosome 8p23.1 while mouse RP1L1 is located on chromosome 14E1. Neither the human nor mouse RP1L1 gene maps to a chromosomal region previously associated with inherited retinal degeneration.
RP1L1 expression
ESTs that matched the RP1L1 cDNA originated from retina or eye tissue sources, suggesting a retinal-restricted expression pattern. To test this hypothesis, we screened human and mouse multiple-tissue cDNA panels for the presence of RP1L1 expression. No expression was detected in any of the tissues tested except retina (Figure 4A). The developmental pattern of RP1L1 retinal expression was subsequently analyzed using RT-PCR of retinas from mice of different ages. RP1L1 is present in mouse retinas at birth, and was not found at detectable levels at any of the prenatal time points analyzed (Figure 4B). This suggests that RP1L1 expression is turned on sometime between prenatal day 15.5 and birth, which is earlier than any RP1 expression has been detected via Northern blot analysis (P5), and also happens to coincide with the development of photoreceptors [5].
Figure 4.

Expression analyses of RP1L1. A: PCR analyses of RP1L1 expression in mouse and human tissues. Human tissues are to the left of the molecular weight ladder in lanes 1-11 and mouse tissues are to the right in lanes 13-25. B: Pattern of RP1L1 expression in different aged mouse retinas. RP1L1 is found in the retinas of mice at birth, but does not appear before embryonic day 15.5. The control probe is mybP42POP. The band underneath RP1L1 is primer-dimer and the last lane is marker. P: postnatal day; E: embryonic day; NO is no DNA negative control. C: In situ hybridization of mouse retina with RP1L1 antisense (right) and sense (left) probes. Arrow shows hybridization of antisense probe in inner segments of photoreceptors.
Semi-quantitative PCR was used to compare the expression of RP1L1 in retinas from normal and rho−/− mice. Since rho−/− mice lack photoreceptors, any reduction in gene expression is likely to result from loss of photoreceptors only. RP1L1 showed a 16-fold reduction in the retinas from rho−/− mice. Interestingly, this reduction is greater than that seen in other genes associated with degenerative retinal disorders such as CRX (2.8 fold) and IMPDH1 (7 fold), but not as great a reduction as its most sequence-similar relative, RP1 (26 fold). Due to the different efficiencies of the primers used, the relative fold reductions must be taken as suggestive in regards to expression levels, not as absolute values. Despite this complication, these data do strongly imply that expression of RP1L1 is largely or exclusively limited to photoreceptors within the retina.
To further examine the expression pattern of RP1L1 within the retina, we performed in situ hybridization. Mouse retina cryosections were hybridized with DIG labeled sense and antisense RP1L1 probes. Signal from the RP1L1 antisense probe was detected only in the inner segments of the photoreceptors, further demonstrating that RP1L1 is photoreceptor specific (Figure 4C).
AdRP testing
Based on the expression pattern of RP1L1, and its homology to the adRP-associated gene RP1, we decided to screen 60 unrelated probands for disease-causing mutations. All 60 probands were members of American families with autosomal dominant RP who tested negative for mutations in rhodopsin, peripherin/RDS, RP1 and IMPDH1 [7,8; unpublished data]. Individuals were tested using a combination of SSCA and sequencing.
SSCA and sequencing of the adRP probands identified 38 sequence variants (Table 2, Table 3, Table 4). Numbering of these variants is based on GenBank entry AY168341. Ten of the RP1L1 sequence variants are either silent or intronic and do not affect the RP1L1 protein. These benign variants are listed in Table 2. Table 3 and Table 4 describe the remaining 28 variants that do cause alteration in the RP1L1 protein. Table 3 describes 22 of these variants that were found in at least one unaffected control DNA sample and therefore, are nonpathogenic. The remaining six protein altering variants observed in the adRP probands are described in Table 4. These variants were not seen in the 60 control individuals tested. Based on the large number of benign sequence variants seen in RP1L1 during patient screening, and the presence of additional sequence variants in the control DNA samples analyzed in this project (data not shown), it is likely that the variants listed in Table 4 are rare family variants that are not associated with adRP. Based on our testing, the frequency of pathogenic mutations in RP1L1 must be 3% or less in our cohort of adRP families (95% confidence interval).
Table 2.
Silent and non-coding sequence variants found in RP1L1
|
Nucleotide change |
Codon change |
Protein change |
Frequency |
|---|---|---|---|
| 32G->A | CCG->CCA | Pro11Pro | 0.01 |
| 501A->G | ACA->ACG | Thr167Thr | 0.02 |
| 609-13G->A | None | (intronic) | 0.15 |
| 1791T->C | GGT->GGC | Gly597Gly | 0.19 |
| 1842C->A | GGC->GGA | Gly614Gly | 0.01 |
| 2238G->A | TCG->TCA | Ser746Ser | 0.01 |
| 2268C->T | AAC->AAT | Asn756Asn | 0.10 |
| 2316G->A | TCG->TCA | Ser772Ser | 0.17 |
| 3405T->G | CCT->CCG | Pro1135Pro | 0.03 |
| 4440G->A | CCG->CCA | Pro1480Pro | 0.01 |
Ten nucleotide substitutions which do not alter the RP1L1 protein sequence were identified in the 60 adRP probands. Because they do not appear to affect the protein, these changes are not likely to be pathogenic. Numbering is based on GenBank entry AY168341. Frequencies are based on the testing of 60 individuals with adRP.
Table 3.
Nonpathogenic RP1L1 variants
|
Nucleotide change |
Codon change |
Protein change |
Frequency |
|---|---|---|---|
| 130C->G | CCA->GCA | Pro44Ala | 0.02 |
| 335C->G | ACC->AGC | Thr112Ser | 0.03 |
| 407G->A | CGT->CAT | Arg136His | 0.01 |
| 1460C->T | GCC->GTC | Ala487Val | 0.01 |
| 2375C->T | CCG->CTG | Pro792Leu | 0.44 |
| 2578C->T | CGG->TGG | Arg860Trp | 0.10 |
| 3436T->C | TGG->CGG | Trp1146Arg | 0.31 |
| 4448C->T | GCC->GTC | Ala1483Val | 0.10 |
| 4484C->G | CCC->CGC | Pro1495Arg | 0.24 |
| 5126C->T | GCC->GTC | Ala1709Val | 0.45 |
| 5447G->A | GGT->GAT | Gly1816Asp | 0.02 |
| 5584_5604del | Glu1862_Gln1868del | 0.10 | |
| 5666A->T | GAT->GTT | Asp1889Val | 0.19 |
| 5860G->A | GCC->ACC | Ala1954Thr | 0.37 |
| 6209A->T | GAG->GTG | Gly2070Val | 0.17 |
| 6264G->T | CAG->CAT | Gln2088His | 0.19 |
| 6418G->A | GAG->AAG | Glu2140Lys | 0.17 |
| 6511A->G | AAG->GAG | Lys2171Glu | 0.48 |
| 6596C->T | CCA->CTA | Pro2199Leu | 0.11 |
| 6725G->A | GGA->GAA | Gly2242Glu | 0.48 |
| 6853G->A | GGA->AGA | Gly2285Arg | 0.29 |
| 7004A->G | CAT->CGT | His2335Arg | 0.08 |
Twenty-two variants within the RP1L1 coding region which alter the protein sequence were identified in the 60 adRP probands. Each of these variants was found in at least one unaffected control individual and therefore, each is believed to be nonpathogenic. Numbering is based on GenBank entry AY168341. Frequencies are based on the testing of 60 individuals with adRP. Each variant was found in at least one unaffected control individual and therefore, each is believed to be nonpathogenic.
Table 4.
Rare RP1L1 variants
|
Nucleotide change |
Codon change |
Protein change |
Frequency |
|---|---|---|---|
| 166C->T | CGC->TGC | Arg56Cys | 0.01 |
| 1870G->A | GCC->ACC | Ala624Thr | 0.01 |
| 2383G->A | GAG->AAG | Glu795Lys | 0.01 |
| 4514C->T | TCG->TTG | Ser1505Leu | 0.02 |
| 4731_4733dupAAG | 1578Lysdup | 0.01 | |
| 5837A->C | GAG->GCG | Glu1946Ala | 0.02 |
Six additional variants within the RP1L1 coding region which alter the protein sequence were identified in the 60 adRP probands. These variants were not found in 120 normal chromosomes. Numbering is based on GenBank entry AY168341. Frequencies are based on the testing of 60 individuals with adRP.
The large amount of RP1L1 variation is further compounded by the polymorphic 48 bp epeat found in exon 4. Testing of the adRP probands identified all six of the repeat alleles found in normal controls. The frequency of each allele found in the patients did not differ significantly from the frequency in normal controls, although preferential amplification of the shorter alleles did complicate individual genotyping. Average allele frequencies are given in Table 5. Sequencing of the repeats in select individuals demonstrated the existence of polymorphic base substitutions in alleles of the same repeat number. These polymorphic sites are represented as ambiguous bases in the corresponding GenBank entry.
Table 5.
RP1L1 repeat allele frequencies
|
Allele |
Number of repeats |
GenBank entry |
Frequency |
|---|---|---|---|
| 1 | 1 | AY168341 | 0.18 |
| 2 | 2 | AY168342 | 0.61 |
| 3 | 3 | AY168343 | 0.02 |
| 4 | 4 | AY168344 | 0.07 |
| 5 | 5 | AY168345 | 0.08 |
| 6 | 6 | AY168346 | 0.03 |
The 16 amino acid repeat polymorphism was typed in all 60 adRP samples as well as in 60 control DNAs. The number of observed copies of the repeated sequence varies from 1 to 6 in both adRP and control populations. Frequencies were calculated based on the combined 120 individuals.
DISCUSSION
We have determined the full-length mRNA sequence and genomic structure of human RP1L1. This gene corresponds to the RP1 sequence-similar EST cluster eye2931, which we described previously [4]. We have determined that RP1L1 encodes a large retinal-specific transcript which, to date, has the highest sequence similarity to RP1. Despite its similarity to RP1, our analysis did not identify any adRP disease-associated mutations in the 60 probands tested.
Sequence analysis
The sequence similarity between RP1L1 and RP1 extends from amino acid 1 through 350, with no significant similarity over the rest of the protein. However, the majority of this similarity can be attributed to the presence of two tandem doublecortin (DC) domains, which extend from amino acids 29-115 and 150-231 within the RP1L1 protein (Figure 2C). RP1L1 and RP1 are 39% identical through their DC regions, with 63% overall similarity. A number of proteins that contain doublecortin domains have been identified and there is evidence that the function of this domain involves interaction with microtubules. This has been shown for RP1 [9] as well as for doublecortin itself [10,11].
The sequence similarity between RP1L1 and RP1 is not limited to the DC domains, as it reappears 86 amino acids beyond them, defining what might be called the “RP1 domain”(Figure 2C). This domain is 34 amino acids in length, and through this region RP1L1 and RP1 are 47% identical and 79% similar. This 34 amino acid domain is not present in any other DC domain-containing protein nor can it be identified elsewhere in the human genome. Although not very large, this domain clearly shows that RP1L1 and RP1 are more closely related to each other than either is to any other members of the doublecortin family. Additionally, the entire region of sequence similarity extends through all three coding exons, suggesting that it is not the result of exon shuffling between genes, but is instead the result of an ancient gene duplication.
Outside of the doublecortin and RP1 domains there are no other identifiable protein domains in RP1L1 that might suggest its function in the retina. RP1 has been localized to the connecting cilium of rod and cone photoreceptors and has been shown to bind to microtubules via its DC domain. It is tempting to speculate that RP1L1 may have a similar localization.
Sequence analysis of the mouse homologue shows that RP1L1 is even less conserved than RP1, with only 48% identity and 61% similarity between the human and mouse sequences. The mouse protein is considerably shorter than its human counterpart, with a length of only 1,859 amino acids. In order to align the human and mouse sequences (Figure 3), several large gaps must be introduced, and these gaps appear to center around the two regions of the human protein that are both highly repetitive and unusually polymorphic. The mouse protein does not appear to have either the polymorphic 16 amino acid repeat or the repetitive Glu-rich region found in humans.
Polymorphic variation
Our analysis demonstrated a large amount of variation in RP1L1. The most striking variability in RP1L1 was the presence of a polymorphic, 48 bp coding repeat in exon 4. In humans, one to six copies of this repeat are present, while mouse RP1L1 only contains one imperfect copy of the repeat (which is also present in humans).
The presence of a coding repeat of this size is unusual, though not unique. For example, several of the human dopamine receptors contain polymorphic coding repeats. One in particular, the D4 receptor, contains a 48 bp coding repeat which, despite being the same length as the RP1L1 repeat, differs substantially in sequence. The D4 receptor repeats are also imperfect in nature and result in alleles the same length coding for different proteins [12].
Several studies have shown associations between different D4 allele sizes and behavioral phenotypes such as novelty seeking and attention-deficit/hyperactivity disorder (ADHD) [12,13]. We did not observe any significant differences in RP1L1 allele frequencies between the normal controls and adRP probands, but it is possible that certain RP1L1 alleles may be associated with other multifactorial forms of retinal degeneration such as age related macular degeneration (ARMD) or may modify clinical expression of inherited adRP caused by mutations in RP1.
ACKNOWLEDGEMENTS
This work was supported by the Foundation Fighting Blindness, the Hermann Eye Fund, Alfred W. Lasher, III, Research to Prevent Blindness, and by grants EYO5235, EY07142, and EY12950 from the National Eye Institute-National Institutes of Health. The work at Trinity College Dublin was supported by the Higher Education Authority, Ireland.
REFERENCES
- 1.Heckenlively JR, Daiger SP. Hereditary retinal and choroidal degenerations. In: Rimoin DL, Connor M, Pyeritz RE, Korf BR, Emery AE, editors. Emery & Rimoin's principals and practice of medical genetics. 4th ed. Vol. 3. Churchill Livingstone; New York: 2002. pp. 3555–3593. [Google Scholar]
- 2.Inglehearn CF, Tarttelin EE, Plant C, Peacock RE, al-Maghtheh M, Vithana E, Bird AC, Bhattacharya SS. A linkage survey of 20 dominant retinitis pigmentosa families: frequencies of the nine known loci and evidence for further heterogeneity. J Med Genet. 1998;35:1–5. doi: 10.1136/jmg.35.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan LS, Heckenlively JR, Bowne SJ, Zuo J, Hide WA, Gal A, Denton M, Inglehearn CF, Blanton SH, Daiger SP. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–9. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–54. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 6.Guillonneau X, Piriev NI, Danciger M, Kozak CA, Cideciyan AV, Jacobson SG, Farber DB. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum Mol Genet. 1999;8:1541–6. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- 7.Bowne SJ, Daiger SP, Hims MM, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DG, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–8. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierce EA. Use of oligonucleotides and single-stranded DNA to introduce mutations into mouse embyronic stem cells; ARVO Annual Meeting; Fort Lauderdale, FL. 2002 May 5-10. [Google Scholar]
- 10.Yoshiura K, Noda Y, Kinoshita A, Niikawa N. Colocalization of doublecortin with the microtubules: an ex vivo colocalization study of mutant doublecortin. J Neurobiol. 2000;43:132–9. [PubMed] [Google Scholar]
- 11.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275:34442–50. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]
- 12.Chang FM, Kidd JR, Livak KJ, Pakstis AJ, Kidd KK. The world-wide distribution of allele frequencies at the human dopamine D4 receptor locus. Hum Genet. 1996;98:91–101. doi: 10.1007/s004390050166. [DOI] [PubMed] [Google Scholar]
- 13.Ding YC, Chi HC, Grady DL, Morishima A, Kidd JR, Kidd KK, Flodman P, Spence MA, Schuck S, Swanson JM, Zhang YP, Moyzis RK. Evidence of positive selection acting at the human dopamine receptor D4 gene locus. Proc Natl Acad Sci U S A. 2002;99:309–14. doi: 10.1073/pnas.012464099. [DOI] [PMC free article] [PubMed] [Google Scholar]