

The crystal structure of the MH2 domain of Mad, which closely matches those of other Smad MH2 domains, is reported at 3.2 Å resolution.
Keywords: decapentaplegic, mother against Dpp, Drosophila, MH2, recognition site, TGF-β
Abstract
In Drosophila, decapentaplegic (Dpp), a member of the TGF-β superfamily, plays a pivotal role in control of proliferation, global patterning and induction of specific cell fates. Together with Medea, mother against Dpp (Mad), the founding member of the Smad family, specifically transduces the Dpp signal from the plasma membrane to the nucleus. Here, the crystal structure of the MH2 domain of Mad, which closely matches those of other Smad MH2 domains, is reported at 3.2 Å resolution. The conservation of Smad protein structures is consistent with their evolutionary conserved and significant function. Furthermore, sequence alignment revealed that most of the variant amino acids in Smad proteins specific to the BMP pathway (Smad1, Smad5 and Mad) were clustered at the surface. In particular, Ser296 and Asp297 of Mad introduced a negative patch into the positive surface observed in the surface electrostatic potential of Smad1 MH2.
1. Introduction
In Drosophila, decapentaplegic (Dpp) signalling has been implicated in many developmental processes, including the determination of the dorsal–ventral axis, the dorsal closure of the embryo, tracheal cell migration and the patterning of the imaginal discs (Patterson & Padgett, 2000 ▶; Ten Dijke et al., 2002 ▶; Siegel & Massague, 2003 ▶). Signalling from the membrane to the nucleus is mediated by the Smad family of proteins, which are divided into three functional classes: the co-mediator Smads (co-Smads), the receptor-regulated Smads (R-Smads) and the inhibitory Smad (I-Smad) (Shi & Massague, 2003 ▶). In the Dpp pathway, the Dpp ligand binds to a complex of the type I and II receptors Thickveins (Tkv) and Punt, which are both transmembrane serine/threonine kinase receptors and undergo phosphorylation/activation. Activation of the type I receptor leads to C-terminal phosphorylation of R-Smad, which corresponds to Mad in the Dpp/BMP pathway and Smox (DSmad2) in the TGF-β/activin pathway. Activated R-Smad forms a heteromeric Smad complex with Co-Smad (Medea in Drosophila), which then translocates into the nucleus. In the nucleus, the complex can bind to a cis-acting element in target genes and activate or repress transcription (Affolter et al., 2001 ▶; Raftery & Sutherland, 1999 ▶). The Smad proteins contain two conserved structural domains: an N-terminal MH1 (Mad-homology 1) and a C-terminal MH2 domain. The MH1 domain exhibits sequence-specific DNA-binding activity and negatively regulates the functions of the MH2 domain, while the MH2 domain can act as a transcriptional activator/repressor and mediates the formation of oligomeric complexes. Importantly, both the MH1 and MH2 domains interact with a large number of proteins in the nucleus, effecting transcription. The R-Smads have a characteristic ‘SXS’ motif at their extreme C-termini, with both Ser residues phosphorylatable by the counterpart receptor (Massague & Wotton, 2000 ▶; Shi & Massague, 2003 ▶; Shi, 2001 ▶).
Although several structures of vertebrate Smad proteins have been solved (Shi et al., 1997 ▶, 1998 ▶; Wu et al., 2000 ▶, 2001 ▶, 2002 ▶; Qin et al., 2001 ▶; Chacko et al., 2004 ▶), little is known about Drosophila Smads. In order to address the molecular basis of Mad, we crystallized the MH2 domain (residues 237–455) of Mad and determined its structure at 3.2 Å resolution. We found that Mad MH2 shares both sequence and structural homology with vertebrate Smads, suggesting evolutionary conservation among Smad proteins. Furthermore, most of the variant amino acids in the R-Smad proteins of the BMP pathway (Smad1, Smad5 and Mad) were on the surface. In particular, when viewed as surface electrostatic potentials, Ser296 and Asp297 of Mad cause the only difference between Mad MH2 and Smad1 MH2, suggesting a specific recognition site.
2. Materials and methods
2.1. Protein expression and purification
The cDNA of Mad was provided by Dr Richard W. Padgett at Rutgers University. The MH2 domain of Mad (residues 237–455) was cloned into expression vector pET21b and the C-terminal His6-tagged fusion protein was expressed in Escherichia coli strain BL21 (DE3). The protein was purified using Ni–NTA agarose (Qiagen) and ion-exchange chromatography (Source-15Q; GE Healthcare). After concentration, the protein was further purified by gel-filtration chromatography (Superdex-200; GE Healthcare) eluted with a buffer containing 10 mM Tris pH 8.0, 150 mM NaCl, 2 mM DTT. The final concentration of this protein was about 20 mg ml−1 and its purity was greater than 95% as judged by SDS–PAGE.
2.2. Crystallization and data collection
Crystallization was performed by the hanging-drop vapour-diffusion method at room temperature. 2 µl protein solution was mixed with 2 µl reservoir solution containing 0.1 M HEPES pH 7.5, 0.35 M sodium formate, 2% glycerol. Crystals appeared after 1 d and grew to maximum size (0.1 × 0.1 × 0.3 mm) after 5 d. The crystals belonged to space group P32, with unit-cell parameters a = b = 95.864, c = 66.726 Å, α = β = 90, γ = 120°, and contained three molecules per asymmetric unit. The diffraction data set was collected to 3.2 Å resolution at 100 K using a Rigaku R-AXIS IV++ image-plate system mounted on a Rigaku MM007X generator and was processed with the HKL package (Otwinowski & Minor, 1997 ▶).
2.3. Structure determination
The structure was determined by molecular replacement using Phaser (McCoy et al., 2007 ▶), with the structure of human Smad1 MH2 (PDB code 1khu) as a search model. The molecular-replacement procedure was performed in the 35.25–3.20 Å resolution range and the final log-likelihood gain was 858.778. The structure was further refined using CNS (Brünger et al., 1998 ▶) and Coot (Emsley & Cowtan, 2004 ▶). The final structure had an R work of 24.4% (R free = 31.0%). The Ramachandran plot of Mad MH2 generated by PROCHECK (Laskowski et al., 1993 ▶) contained only one violating residue, Val407, which is positioned similarily to its counterpart in other Smad structures, indicating that the final structure has good stereochemistry. All structural representations were prepared using PyMOL (http://www.pymol.org).
3. Results and discussion
3.1. Overall structural of Mad MH2
In order to address the molecular basis for its function, we crystallized the MH2 domain (residues 237–455) of Mad. The structure was determined by molecular replacement and refined to 3.2 Å resolution (Table 1 ▶). In general, the electron density is clear for most residues (Fig. 1 ▶ a). As shown in Fig. 1 ▶(b), the monomeric structure of Mad MH2, similar to those of other Smad MH2 domains (Qin et al., 2001 ▶; Wu et al., 2001 ▶; Shi et al., 1997 ▶), contains a central β-sandwich with a three-helix bundle (H3, H4 and H5) at one end and a loop–helix region (L1, L2, L3 and H1) at the other. Residues 237–258 are part of the flexible linker connecting the MH1 and MH2 domains, which has no stable secondary structure. Although there are three molecules in each asymmetric unit, they have no direct interactions and are arranged differently from the functional trimer (Fig. 1 ▶ c), which is consistent with our gel-filtration results (data not shown). The trimerization of Mad MH2, similar to that observed in the trimer of phosphorylated Smad2 MH2, may require interaction between the phosphorylated C-terminal ‘SXS’ motif and the L3/B8 pocket. However, the Mad MH2 protein we used in this research was unphosphorylated and the extreme C-terminus (residues 447–455) is not visible owing to structural disorder (Fig. 1 ▶ b). Therefore, the Mad trimer observed in the asymmetric unit is most likely to be an effect of crystal-packing forces.
Table 1. Summary of crystal analysis for Mad MH2.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P32 |
| Unit-cell parameters (Å, °) | a = b = 95.864, c = 66.726, α = β = 90, γ = 120 |
| Resolution (Å) | 50.0–3.20 (3.31–3.20) |
| Observed reflections | 72297 (7995) |
| Unique reflections | 10321 (1126) |
| Completeness (%) | 91.3 (100) |
| Redundancy | 7.0 (7.1) |
| Rmerge† (%) | 11.6 (57.5) |
| Average I/σ(I) | 19.2 (4.0) |
| Refinement | |
| Resolution (Å) | 50.0–3.20 (3.31–3.20) |
| No. of reflections used | 9604 |
| No. of reflections in working set | 9095 |
| No. of reflections in test set | 509 |
| Rwork‡ (%) | 24.4 |
| Rfree‡ (%) | 31.0 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.551 |
| No. of protein atoms | 4458 |
| No. of water molecules | 89 |
| Average B value (Å2) | 63.52 |
| Ramachandran angles (%) | |
| Most favoured | 79.4 |
| Additionally allowed | 18.0 |
| Generously allowed | 2 |
| Disallowed | 0.6 |
R
merge = 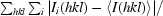
 , where 〈I(hkl)〉 is the mean of the observations Ii(hkl) of reflection I(hkl).
, where 〈I(hkl)〉 is the mean of the observations Ii(hkl) of reflection I(hkl).
R
work = 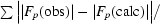
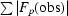 ; R
free is the R factor for a selected subset (4.5%) of the reflections that were not included in prior refinement calculations.
; R
free is the R factor for a selected subset (4.5%) of the reflections that were not included in prior refinement calculations.
Figure 1.

(a) Quality of the electron-density map. The map is contoured at the 1.5σ level. (b) Three Mad MH2 molecules in the asymmetric unit. (c) Schematic representation of a Mad MH2 monomer. The two views of the structure are related by a 90° rotation around a vertical axis. The secondary-structure elements are annotated.
3.2. Sequence alignment
All of the determined MH2 structures, that of Drosophila Mad and those of vertebrate Smad proteins including Smad4 MH2 with an insertion of ∼40 amino acids in the middle (Shi et al., 1997 ▶), share the same fold, demonstrating the evolutionary conservation and functional significance of Smad proteins. Sequence alignment of the Smad proteins revealed that the MH2 domain is highly conserved, especially the residues involved in the formation of the hydrophobic core and those at the interface of the homo- and hetero-trimerization interfaces (Fig. 2 ▶).
Figure 2.

Sequence alignment of Smad MH2 domains from Drosophila and human. The secondary-structural elements are indicated above the alignment. The residues different in Smads that transduce the BMP pathway are highlighted in yellow and the subtype-specific residues in H2 and L3 are shown in magenta and blue, respectively.
R-Smad proteins have signalling specificity: Smad1, Smad5, Smad8 and Mad mediate the BMP cascade, while Smad2, Smad3 and dSmad2 transduce the TGF-β/activin signal. These two subclasses of Smad proteins regulate the transcription of different target genes. Sequence alignment shows that the subtype-specific residues are mostly located on the L3 loop and H2 helix (Fig. 2 ▶), which provide specificity for receptor recognition and cofactor interaction, respectively (Lo et al., 1998 ▶; Chen et al., 1998 ▶).
3.3. Comparison between Mad MH2 and Smad1 MH2
Mad and Smad1 are homologues that specifically transduce BMP pathways in Drosophila and mammals, respectively. The similarity between the full-length proteins is 73.8%, while the two MH2 domains share an even higher similarity of 87.8%. As shown in the sequence alignment (Fig. 2 ▶), the Mad MH2 residues that differ from those of Smad1 MH2 are mostly located in the N-terminal region (Fig. 3 ▶ a). Interestingly, there is a negative patch on the surface of Mad MH2 that disrupts the continuous positive surface observed in the Smad1 MH2 structure (Fig. 3 ▶ b). This negatively charged surface patch results from a cluster of variant amino acids located at the N-terminus of the MH2 domain, especially Ser296 and Asp297 in the L1 loop connecting β3 and β4 (Figs. 2 ▶ and 3 ▶ a).
Figure 3.

Structural features of Mad MH2. (a) The variant residues of Mad MH2 indicated in Fig. 2 ▶ are coloured magenta. The two views of the structure are related by a 90° rotation around a horizontal axis. (b) Surface representations of Mad MH2 (left) and Smad1 MH2 (right), coloured according to electrostatic potential, with positive and negative charges in blue and red, respectively.
Smad proteins are conserved across species, particularly the C-terminal MH2 domain to which most of the tumour-derived mutations have been mapped. In the last decade, several structures of vertebrate Smad proteins have been solved, most of which are of MH2 domains. However, little was known about the structures of Drosophila Smad proteins. In this paper, we have solved the crystal structure of the Mad MH2 domain, which shares strong similarity to other Smad MH2 domains. This suggests that Smad proteins possess high sequence and structure conservation during protein evolution. Sequence alignment of the Smad proteins reveals that the amino acids involved in the formation of the hydrophobic structure core are conserved and the variant residues involved in interaction with other proteins are mostly located on the surface. Furthermore, most of the different amino acids in Smad proteins of the BMP pathway (Smad1, Smad5 and Mad) were in the N-terminal portion, suggesting that these residues may contribute to the sub-pathway characteristics of BMP Smads. Interestingly, the variant residues Ser296 and Asp297 of Mad (Lys306 and Gln307 of Smad1) cause an obvious difference in the surface electrostatic potential, suggesting a possible recognition site for specific regulators.
Supplementary Material
PDB reference: Drosophila Mad MH2 domain, 3dit, r3ditsf
Acknowledgments
We thank Dr Zhi-Yong Lou at Tsinghua University for help with data collection and processing. This work was supported in part by grants 2006CB503900 and 2007CB914400 from the Ministry of Science and Technology of China and grants 30425005 and 30770476 from the National Natural Science Foundation of China.
References
- Affolter, M., Marty, T., Vigano, M. A. & Jazwinska, A. (2001). EMBO J.20, 3298–3305. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chacko, B. M., Qin, B. Y., Tiwari, A., Shi, G., Lam, S., Hayward, L. J., De Caestecker, M. & Lin, K. (2004). Mol. Cell, 15, 813–823. [DOI] [PubMed]
- Chen, Y. G., Hata, A., Lo, R. S., Wotton, D., Shi, Y., Pavletich, N. & Massague, J. (1998). Genes Dev.12, 2144–2152. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lo, R. S., Chen, Y. G., Shi, Y., Pavletich, N. P. & Massague, J. (1998). EMBO J.17, 996–1005. [DOI] [PMC free article] [PubMed]
- Massague, J. & Wotton, D. (2000). EMBO J.19, 1745–1754. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Patterson, G. I. & Padgett, R. W. (2000). Trends Genet.16, 27–33. [DOI] [PubMed]
- Qin, B. Y., Chacko, B. M., Lam, S. S., de Caestecker, M. P., Correia, J. J. & Lin, K. (2001). Mol. Cell, 8, 1303–1312. [DOI] [PubMed]
- Raftery, L. A. & Sutherland, D. J. (1999). Dev. Biol.210, 251–268. [DOI] [PubMed]
- Shi, Y. (2001). Bioessays, 23, 223–232. [DOI] [PubMed]
- Shi, Y., Hata, A., Lo, R. S., Massague, J. & Pavletich, N. P. (1997). Nature (London), 388, 87–93. [DOI] [PubMed]
- Shi, Y. & Massague, J. (2003). Cell, 113, 685–700. [DOI] [PubMed]
- Shi, Y., Wang, Y. F., Jayaraman, L., Yang, H., Massagué, J. & Pavletich, N. P. (1998). Cell, 94, 585–594. [DOI] [PubMed]
- Siegel, P. M. & Massague, J. (2003). Nature Rev. Cancer, 11, 807–821. [DOI] [PubMed]
- Ten Dijke, P., Goumans, M. J., Itoh, F. & Itoh, S. (2002). J. Cell. Physiol.191, 1–16. [DOI] [PubMed]
- Wu, G., Chen, Y. G., Ozdamar, B., Gyuricza, C. A., Chong, P. A., Wrana, J. L., Massague, J. & Shi, Y. (2000). Science, 287, 92–97. [DOI] [PubMed]
- Wu, J. W., Hu, M., Chai, J., Seoane, J., Huse, M., Li, C., Rigotti, D. J., Kyin, S., Muir, T. W., Fairman, R., Massague, J. & Shi, Y. (2001). Mol. Cell, 8, 1277–1289. [DOI] [PubMed]
- Wu, J. W., Krawitz, A. R., Chai, J., Li, W., Zhang, F., Luo, K. & Shi, Y. (2002). Cell, 111, 357–367. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Drosophila Mad MH2 domain, 3dit, r3ditsf