

Abstract
Macrophages express a spectrum of proinflammatory and regulatory mediators during African trypanosomiasis. Microarray analyses revealed similar profiles of induced genes in macrophages stimulated with the trypanosome soluble variant surface glycoprotein in vitro and in macrophages taken from infected mice. Genes associated with the acute phase response and with type I IFN responses were prominent components of the macrophage activation profiles expressed within 72 h in vitro and in vivo. Thus, induction of proinflammatory gene expression is a characteristic of early trypanosome infection that is driven primarily by soluble variant surface glycoprotein exposure, and it may be that IFN-α/β plays a central role in regulation of early resistance to trypanosomes. To test this hypothesis, we assessed parameters of infection in mouse strains with genetic alterations in the IFN-α/β response pathway. We found that Ifnar1−/− mice, which lack the receptor for type I IFNs, exhibited delayed control of parasite burden during the first week of infection and died earlier than did wild-type controls. However, infection of Ubp43−/− mice, which are hyperresponsive to type I IFNs, did not exhibit enhanced resistance to trypanosomes. Instead, these animals also failed to control parasite burden and were more susceptible than wild-type animals. Additionally, the Ubp43−/− mice exhibited a significant defect in IFN-γ production, which is definitively linked to host resistance in trypanosomiasis. These results show that type I IFNs play a role in early control of parasites in infected mice but may contribute to down-regulation of IFN-γ production and subsequent loss of host resistance later in infection.
African trypanosomiasis is a fatal infection of man and animals. B cell responses to the variant surface glycoprotein (VSG)3 molecules of the trypanosome surface coat result in clearance of organisms from the blood, but this response alone is not functionally or genetically linked to overall resistance to trypanosomiasis (1–6). In contrast, IFN-γ production by VSG-specific Th cells (and perhaps other cellular sources) has been linked definitively to relative resistance to disease (7–9); it is presumed that macrophage activation by IFN-γ results in the production of factors such as TNF-α, reactive nitrogen intermediates, and reactive oxygen intermediates into the extravascular tissues that are known to be cytolytic for trypanosomes (10–13). However, the sequential expression of new VSG genes and surface coat determinants, coupled with immunomodulatory effects of trypanosome infection, ultimately lead to escape from adaptive immunity and a fatal outcome for the host (8, 14–19).
Establishment of an IFN-γ response and relative host resistance are intimately linked to parasite factors that impact the innate immune system early in infection. Exposure of infected animals to the glycosylinositolphosphate (GIP) residues of soluble VSG (sVSG) molecules and also to parasite CpG DNA results in early macrophage activation in a MyD88-dependent manner; these events are linked through IL-12 production to establishment of polarized Th1 cell responses to parasite Ag (8, 20–24). Macrophage activation in experimental African trypanosomiasis has been well documented in previous studies, and lymphoid organs become markedly enlarged during infection due to a disproportionate increase in macrophages within these tissues (25–27). Studies from our laboratory and those of others have documented that macrophages from trypanosome-infected hosts exhibit increased expression of proinflammatory and immunoregulatory molecules such as IL-12, inducible NO synthase, TNF-α, IL-1, and IL-10 (22, 28, 29). Despite evidence for activation, however, there are also data implicating macrophages in the immune dysfunction and down-regulation of adaptive immunity characteristic of African trypanosomiasis (17, 18, 30–33). Thus, it is clear that the role of macrophage activation and other elements of the innate immune system in regulating host resistance is complex and remains to be fully elucidated.
A key requirement for dissecting the complex role of macrophages during infection is to understand how parasite molecules activate or regulate cells of the innate immune system and the extent to which these factors control the disease process. In this regard, the activating properties of trypanosome sVSG have been well documented, in which the GPI-anchored membrane form of the VSG molecule (mfVSG) is cleaved from the parasite membrane by an endogenous phospholipase C (22, 23, 28), and sVSG with its GIP residues is released into blood and other tissues of infected mice (34). Several lines of evidence indicate that sVSG modulates macrophage function during infection. For example, Magez et al. (22) observed that mfVSG and sVSG activate macrophages to produce TNF-α and other cytokines, with the GIP portion of sVSG responsible for its biological activity. Studies conducted in our laboratory yielded additional insights into the immunomodulatory properties of sVSG, demonstrating that sVSG has both stimulatory and suppressive effects on macrophages, and that these effects are dependent on the timing of exposure and the amount of sVSG as well as IFN-γ accessible to macrophages (34). These observations suggest that it is the balance of host and parasite molecules that determines the predominant macrophage activation profile at different time points and in different tissues during infection.
Therefore, we employed microarray analyses of infected macrophages ex vivo as well as of macrophages treated in vitro with sVSG to define the spectrum of host innate immune response genes that are induced during early trypanosome infection. The results of these studies demonstrated that induction of proinflammatory and acute phase response gene expression is characteristic of early infection and that genes associated with type I IFN signaling were induced both in vitro by sVSG and in vivo as a result of infection. Subsequently, in vitro and in vivo studies employing genetically deficient IFNAR1 and UBP43 knockout mouse strains were used to further determine the effects of host type I IFN responses on host resistance to infection. These studies clearly demonstrate that type I IFNs are important during early infection for efficient control of parasite burden but that excessive reactivity to type I IFNs can lead to detrimental effects for the host by modulating IFN-γ production.
Materials and Methods
Animals
Age-matched Swiss Webster and C57BL/6J mice were obtained from The Jackson Laboratory. Male and female matched sets of C57BL/6 wild-type (wt) and C57BL/6-Ifnar1 (IFNAR1 KO) mice, and C57BL/6 wt and C57BL/6-Ubp43 (UBP43 KO) mice, were kindly provided by Dr. Dong-Er Zhang at the Scripps Research Institute. These mice served as the founder strains for breeding and selection of animals to be used for experimentation purposes in the present study. All mice were housed in pathogen-free facilities at University of Wisconsin–Madison and were handled strictly according to National Institutes of Health and American Association for the Accreditation of Laboratory Animal Care guidelines. The C57BL/6 wt and genetically modified mice were used for all experimental infections; mice were 8–12 wk of age at the time of infection. Swiss Webster mice were used for growth of primary trypanosome stabilates from which experimental infections of mice were established. Swiss Webster mice were also used for the growth of large numbers of trypanosomes necessary for preparation and purification of sVSG.
Trypanosomes
Stabilates of Trypanosoma brucei rhodesiense (clone LouTat 1) frozen at −85°C were thawed and injected i.p. into mice immunosuppressed with cyclophosphamide (300 mg/kg body weight) to allow outgrowth of trypanosomes in the absence of immune selection. Trypanosomes were then isolated from the blood by cardiac puncture and the blood was diluted 1/10 in PBS containing 1 mM EDTA and 1% glucose (PBSG) with 10 U/ml heparin added; parasites were counted in a hemacytometer and infected blood subsequently was diluted as necessary in PBSG without heparin for infection of experimental mouse strains.
Experimental infections
Mice were infected by i.p. injection of 1 × 105 trypanosomes. Throughout the infection levels of parasitemia, IFN-γ, and VSG-specific Abs were monitored in the blood by sampling 12 μl of tail blood diluted in 96 μl PBSG containing heparin. Parasites were counted in a hemacytometer using 1 μl of this sample diluted in PBSG. The remaining portion of the sample was gently centrifuged at 4°C to pellet the trypanosomes and RBC. The cell-free supernatant was recovered and frozen at −70°C until analysis. Levels of IFN-γ in the blood of infected mice were quantified by sandwich ELISA as per the manufacturer’s instructions (BD Pharmingen). Ab levels in the blood of infected mice were also assessed by sandwich ELISA as we have described previously (9) and according to the manufacturer’s instructions (Zymed Laboratories).
Preparation and purification of sVSG
For in vitro studies of macrophage activation, sVSG was prepared according to established procedures (35). Briefly, trypanosomes were isolated aseptically from the blood of mice at the peak of parasitemia, washed in PBSG, and concentrated by centrifugation. Trypanosomes were lysed hypotonically (109 cells/ml) in ice-cold 0.3 mM zinc acetate containing protease inhibitors. The cell lysate was centrifuged at 3000 × g for 10 min at 4°C and the supernatant fluid was placed on ice. The pellet was resuspended in an equal volume of 10 mM phosphate buffer containing protease inhibitors and incubated at 37°C for 20 min to allow GPI-phospholipase C cleavage of mfVSG, followed by centrifugation at 10,000 × g for 15 min at 4°C. Supernatant fluids from these two steps were combined and centrifuged at 300,000 × g for 1 h at 4°C, dialyzed against PBS, and concentrated using Amicon Ultra-15 centrifugal tubes (Millipore). Subsequently, the combined fractions were passed over a DEAE Sephadex column equilibrated with 10 mM phosphate buffer. Eluate containing sVSG was dialyzed against RPMI 1640 medium and concentrated using Amicon Ultra-15 centrifugal tubes. Coomassie blue staining and Western blot analysis were employed to confirm the purity of sVSG after resolving purified protein by SDS-PAGE under reducing conditions using 10% polyacrylamide gels; sVSG appeared as a single band of 60 kDa in both Coomassie staining and in Western blot analysis using Abs to a cross-reactive determinant expressed on GIP (data not shown) (36). Potential environmental contamination of sVSG preparations with endotoxin was controlled for by using polymyxin B columns to remove any trace amounts of endotoxin that may have been introduced during animal blood collection; the levels of endotoxin in all preparations were confirmed to be below detection (<0.05 EU) by the Limulus amebocyte lysate assay (Associates of Cape Cod).
Reagents
Murine rIFN-γ (provided by the American Cancer Society, Schering, Bloomfield, NJ; sp. act. 1.7 × 106 U/mg) was used at a concentration of 20 U/ml for in vitro studies of macrophage activation. Synthetic CpG-containing oligodeoxynucleotides (ODNs) (Cell Sciences) and LPS (Escherichia coli serotype 026:B6, Sigma-Aldrich) were used as positive controls. Polymyxin B (Sigma-Aldrich) 10 μg/ml was routinely used in some macrophage cell cultures (except when LPS was the stimulant) to eliminate potential effects of any contaminating endotoxin. In none of the present or past experiments, however, was an effect of endotoxin (or of polymyxin B alone) noted for macrophages.
Cell cultures
The RAW 264.7 murine macrophage cell line obtained from the American Type Culture Collection (ATCC) was cultured at 37°C, 7% CO2 in RPMI 1640 supplemented with 10% FBS (Invitrogen), 16 mM HEPES buffer, 0.3 mg/ml L-glutamine and 50 μg/ml gentamicin, as we have previously described (34).
Peritoneal (PEC) and spleen (SPC) cells were isolated from mice at various time points during infection according to established procedures (8, 17). Briefly, cells were washed several times with RPMI 1640 medium and enumerated in a hemacytometer using trypan blue dye exclusion to determine cell viability. Adherent PEC and SPC were harvested and cultured at a cell density of 1 × 106 cells/ml at 37°C with 7% CO2 in 12-well tissue culture plates. Cells were cultured 24–48 h, at which time cell-free culture supernatant fluids were harvested and stored at −70°C until further analysis.
For the purposes of determining T cell IFN-γ responses following Ag-specific stimulation with sVSG, CD4 T cells were harvested from wt and trypanosome-infected mice and stimulated in vitro as we have previously described (7–9). Specifically, T cells were enriched from SPC of wt and UBP43KO mice by adherence to nylon wool; cell depletion was subsequently performed on the T cell-enriched population using Low-Tox rabbit complement (Cedarlane Laboratories) and mAbs 31M (anti-CD8α) and M1/70 (TIB128, ATCC). Live cells were collected on a Percoll (Sigma-Aldrich) gradient, washed twice in complete DMEM, and examined by flow cytometry for CD4 expression before use. APCs (irradiated naive splenocytes; 106 cells) were combined with CD4 T cells (105 cells) plus 50 μg/ml VSG at 37°C, 5% CO2 in 96-well ELISPOT plates in complete RPMI 1640 medium. The numbers of IFN-γ-secreting VSG-specific T cells present in wt and UBP43KO cell cultures were determined by ELISPOT assay after 24 h of culture using ELISPOT kits purchased from R&D Systems. The ELISPOT assay was developed according to the manufacturer’s instructions: plates were washed four times with PBS, and 100 μl of diluted IFN-γ detection Ab was added to each well; after an overnight incubation at 4°C, the plates were washed four times in PBS; diluted streptavidin-alkaline phosphatase (100 μl/well) was added and plates were incubated for 2 h at room temperature; after washing, 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/NBT chromogen (100 μl/well) was added and the plates incubated at room temperature for 1 h in the dark; and the chromogen solution was washed from plates using double-distilled water and plates were air dried. Spot-forming cells were enumerated using a dissection microscope.
Viral plaque assays
IFN-α/β was measured in serum or culture supernatants using L929 cells in a standard vesicular stomatitis virus plaque reduction assay (38). Cells were cultured overnight in 24- or 48-well plates with dilutions of a recombinant IFN-α standard (Chemicon International) ranging from 2000 IU/ml to 15 IU/ml, or with dilutions of the experimental samples. Subsequently, the medium was aspirated, cell monolayers were washed with PBS, and vesicular stomatitis virus was added at 80–100 PFU/well. After a 1-h incubation at 37°C, the viral inoculum was removed, cell monolayers were washed with PBS, and a carboxymethylcellulose overlay containing medium was added. Cells were incubated at 37°C, 5% CO2 for an additional 1–2 days, at which time the cells were fixed with 1% crystal violet and viral plaques were enumerated microscopically. IFN titers in experimental samples were determined by extrapolation based on the standard curve generated using the recombinant IFN-α standard.
Western blot analysis
RAW 264.7 macrophages were cultured overnight in 12-well tissue culture dishes at 7.5 × 105 cells per well in RPMI 1640 medium. Cells were then primed with 20 U/ml IFN-γ or left untreated for 24 h before stimulation with various concentrations of sVSG for 30 min. Cells were lysed in 200 μl ice-cold Nonidet P-40 lysis buffer (150 mM NaCl, 2 mM EDTA, 50 mM Tris (pH 7.4), 1% Nonidet P-40, 0.02% NaN3, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 1 mM PMSF, and 1 mM Na3VO4). Total protein was quantified with the BCA protein assay (Pierce) using BSA as the standard.
Proteins were resolved by SDS-PAGE (25 μg protein per lane) under reducing conditions in 10% polyacrylamide gels and transferred to an Immobilon polyvinylidene difluoride membrane. Target proteins were detected using the following primary Abs: IκB-α (Santa Cruz Biotechnologies), anti-active MAPK (Promega), phosphop38 (Promega), phospho-JNK1/2 (Promega), ERK1/2 (Promega), panJNK (R&D Systems), p38 (Santa Cruz Biotechnologies), and actin (Sigma-Aldrich). Proteins were visualized by chemiluminescence using goat anti-rabbit Ab conjugated to HRP (Molecular Probes) and with chemiluminescent SuperSignal reagents (Pierce). All results were quantitated by densitometric analysis of detectable bands on the blots.
RT-PCR
Confluent cultures of RAW 264.7 and primary macrophages were split into 12-well tissue culture dishes at 5.5 × 105 cells per well and cultured overnight. Cells were treated with 20 U/ml IFN-γ or were untreated for 24 h before stimulation with sVSG or positive control reagents (CpG-ODN, LPS) for 6, 10, or 24 h. Total RNA was isolated from cells using RNA STAT-60 (Tel-Test B) according to the manufacturer’s instructions. cDNA was synthesized from each sample by reverse transcription using oligo(dT) primers (IDT). The resulting cDNA was used as a template for gene-specific PCR. PCR amplification was conducted in a 96-well thermocycler (MJR Research). Amplification of the housekeeping gene G3PDH served as a control to ensure equal loading of cDNA template in each reaction. All primers except G3PDH (Clontech Laboratories) were designed using OLIGO 6.2 software (National Biosciences) and synthesized by IDT. PCR-amplified samples were resolved by electrophoresis in 1% agarose gels and visualized using ethidium bromide staining. Selected results were quantitated by densitometric analysis of detectable bands on the gels.
Microarray analysis
In vitro array analyses were performed as follows: confluent cultures of RAW 264.7 macrophages were split into 12-well tissue culture dishes at 7.5 × 105 cells per well and cultured overnight. Cells were then treated in triplicate with sVSG (8 μM; this concentration was chosen to approximate the levels of sVSG that may be encountered by cells in infected mice, below, at the 72 h time point of sampling for ex vivo microarray analyses) or medium alone for 24 h. Total RNA was isolated from cells using Qiagen’s RNeasy Mini kit according to the manufacturer’s instructions. RNA samples were submitted to NimbleGen, where samples were processed to cDNA, which was then hybridized to NimbleGen’s mouse (24-mer) array consisting of 9400 genes. Hybridizations were conducted in triplicate for each treatment regimen, and the resulting data were analyzed using Gene-Spring microarray analysis software (Silicon Genetics/Agilent Technologies). Log-transformed data were screened for 2-fold variance or greater over the control using a two-sample t test (p-value cutoff of 0.05).
In vivo microarray analyses were performed as follows: age-matched C57BL/6J female mice were infected with 1 × 105 trypanosomes; control mice were injected with PBS alone. RNA was isolated from adherent liver, spleen, and resident peritoneal macrophages 72 h postinfection using Qiagen’s RNeasy Mini kit. RNA samples from these three different cell populations were pooled, cDNA was prepared, and this material was hybridized to NimbleGen’s mouse (60-mer) array consisting of 35,497 genes. Normalized replicates were screened for 2-fold variance over the control using GeneSpring microarray analysis software as described above.
Statistical analysis
The statistical significance of the differences observed was assessed by Student’s t test. Results are expressed as means ± SEM. Differences were considered significant when p-values of <0.05 were obtained. All experiments were performed at least three times unless noted otherwise; a representative experiment is shown in all figures except where mean values are presented.
Results
sVSG activates signaling intermediates in RAW 264.7 macrophage cells
Previous results from our laboratory and others have demonstrated that specific activation events occur in response to sVSG stimulation (22, 23, 28, 34). It was important to verify for each sVSG preparation that similar biological activities were associated with the sVSG molecules to be used in the planned experiments. Thus, an assessment of changes in signaling intermediates was used to provide both confirmation as well as new insights into the activation capacity of sVSG for macrophages. Confluent cultures of RAW 264.7 cells or primary macrophages were treated with medium alone or different concentrations of sVSG for 30 min; phosphorylation of signaling intermediates in the MAPK signaling pathway and degradation of the NF-κB inhibitor, IκBα, were assessed via Western blot and revealed that treatment with sVSG (16 μM) in vitro leads to activation of ERK1/2, JNK1/2, p38, and degradation of IκBα (Fig. 1). The degradation of IκBα is a hallmark of sVSG activation of macrophages and is regulated by both TNFR-associated factor (TRAF)6-dependent and -independent mechanisms (24).
FIGURE 1.
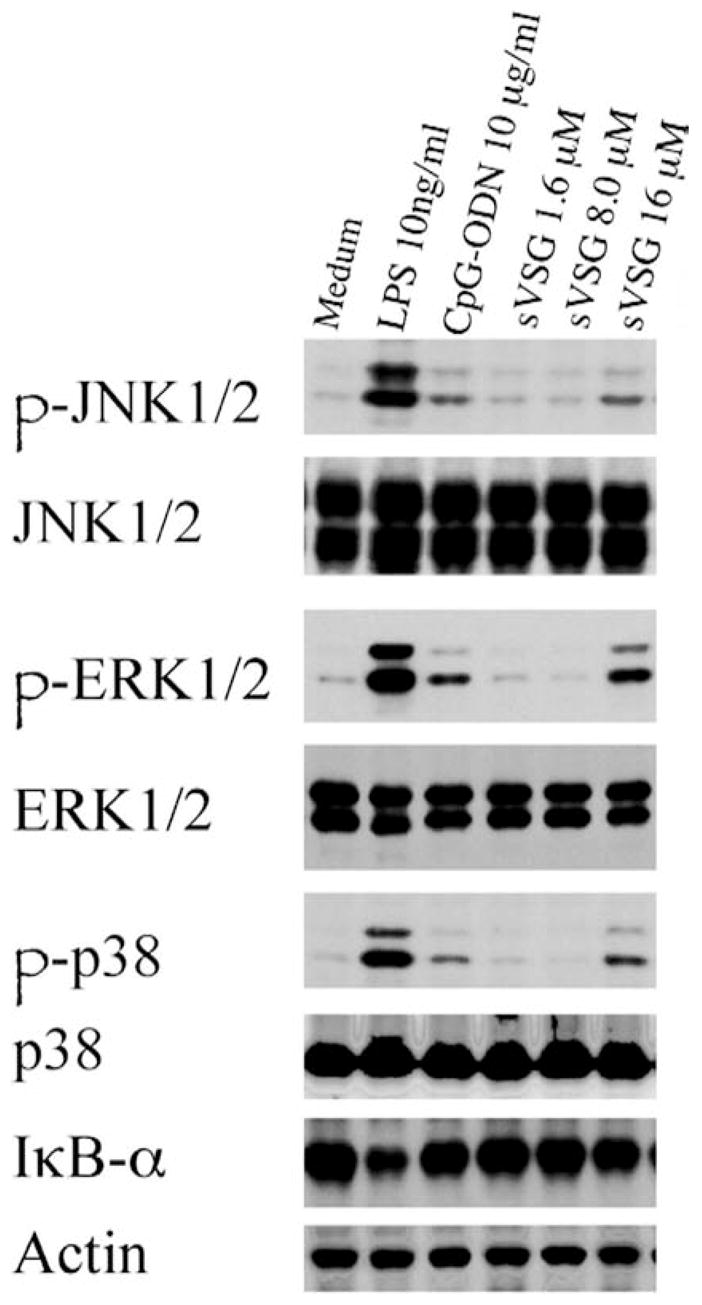
sVSG activation of the NF-κB and MAPK signaling pathways in macrophages is dose-dependent. RAW 264.7 macrophages were treated with medium alone for 24 h, followed by treatment with LPS (10 ng/ml), CpG-ODN (10 μg/ml), or different concentrations of sVSG for 30 min, as shown. Whole-cell lysates were harvested and 25 μg of total protein from each sample was resolved by SDS-PAGE and assessed via Western blot for phosphorylation or degradation of the signaling intermediates indicated. To verify equal loading, blots were reprobed using Abs that recognize both phosphorylated and unphosphorylated forms of JNK1/2, ERK1/2, and p38. In the case of IκBα blots, an actin reprobe was used as the loading control. Results from one of three independent and representative experiments are shown, and results were confirmed by densitometric analyses.
sVSG induces changes in global gene expression in macrophages
The effects of sVSG on gene expression in macrophages were explored using microarray analysis to provide a global view of the downstream events associated with activation by this molecule. For these experiments, RAW 264.7 cells were stimulated with sVSG or medium alone for 24 h. Total RNA was extracted and the cDNA produced was used to probe NimbleGen’s standard mouse gene microarray. The resulting data revealed 103 differentially expressed genes. Further analysis demonstrated that 44 of these genes were up-regulated to a much greater degree in sVSG-treated samples compared with unstimulated controls; these encoded molecules associated with the acute phase response, such as mannose-binding lectin A and serum amyloid proteins (Table I), providing additional evidence that sVSG readily stimulates an acute inflammatory response in macrophages. Of interest was that several genes associated with type I IFN signaling in sVSG-stimulated cells (Table I) were induced. These genes included IFN regulatory factor 7 (IRF-7) and IFN-α responsive gene 15, which were induced 630-fold and 400-fold, respectively. Using a less stringent p-value cutoff of 0.1 to screen differentially expressed genes in sVSG-treated samples revealed additional IFN-α/β gene targets that were up-regulated 2-fold or greater over background, including 2′-5′ oligoadenylate synthetase 1G, and double-stranded RNA-activated protein kinase (PKR) (data not shown). These results suggested type I IFNs may be key downstream gene targets of sVSG-mediated signaling in macrophages.
Table I.
Purified sVSG induces the up-regulation of genes associated with type I IFN signaling in macrophages: in vitro microarray analysisa
| Accession No. | Fold Change | Description |
|---|---|---|
| Inflammatory/stress and immune responses | ||
| Acute phase response | ||
| NM_010775 | 656.9 | Mannose binding lectin liver (A) (Mbl1) |
| NM_011315 | 13 | Serum amyloid A3 (Saa3) |
| NM_011316 | 343.8 | Serum amyloid A4 (Saa4) |
| NM_031167 | 177 | IL-1R antagonist (Il1rn) |
| IFN-inducible | ||
| NM_016850 | 634 | IFN regulatory factor 7 (Irf7) |
| NM_022329 | 393.9 | IFN-α-responsive gene, 15 kDa (Ifrg15) |
Confluent cultures of RAW 264.7 macrophages were treated with medium alone or with 8 μM sVSG for 24 h. Total RNA was harvested and assessed for changes in gene expression via microarray analysis. Shown is a subset of genes up-regulated 2-fold or more over background in macrophages treated with sVSG.
IFN-γ potentiates sVSG-induced transcription of IRF-7 and IFN-β in macrophages
To verify that sVSG induced type I IFNs at the transcript level in vitro, RAW 264.7 macrophages were primed with IFN-γ or medium alone for 24 h, followed by treatment with different concentrations of sVSG for 6 h. As shown in Fig. 2, treatment of RAW 264.7 cells with sVSG alone induced transcription of IRF-7, thus confirming the microarray data. Induction of IFN-β mRNA clearly required IFN-γ priming, and IFN-γ priming also appeared by densitometric analysis to enhance the transcription of IRF-7, but only in response to low concentrations of sVSG. Thus, IFN-γ may potentiate sVSG-induced transcription of both IRF-7 and IFN-β in macrophages, especially during early infection when parasite burden and sVSG concentrations are relatively low. These results demonstrate the importance of both host and parasite molecules in achieving full macrophage activation (20, 21, 34).
FIGURE 2.

IFN-γ synergizes with sVSG to induce transcription of IRF-7 and IFN-β in macrophages. RAW 264.7 macrophages were pretreated with IFN-γ (γ, 20 U/ml) or medium alone (M) for 24 h followed by treatment with CpG-ODN (O, 10 μg/ml), IFN-γ alone, or different concentrations of sVSG for 6 h as shown. Total RNA was harvested from cell monolayers by phenol-chloroform extraction and each sample was analyzed by RT-PCR for changes in IRF-7 and IFN-β expression. Changes in gene expression were assessed by resolving 10 μl of sample on 1% agarose gels containing ethidium bromide. The housekeeping gene G3PDH served as a loading control, and densitometric analysis was used to verify differences. Results shown are from one of four representative and independent experiments.
Gene expression patterns in vivo during infection mimic those of macrophages stimulated with sVSG in vitro
We next determined whether the pattern of infection-induced gene expression in vivo was similar to the pattern of gene expression observed in vitro with macrophages treated only with sVSG. Importantly, such comparisons may be challenging if the factors inducing gene expression in vitro and in vivo are significantly different. For the in vivo study, changes in cellular mRNA expression were assessed by microarray analysis of total RNA isolated from liver and adherent SPC and resident PEC of infected or mock-infected mice 72 h postinfection; all cell samples were processed similarly to samples derived from in vitro sVSG-treated macrophages, and the cDNA was used to probe NimbleGen’s mouse array as above.
Data analysis revealed that >700 genes were up-regulated at least 2-fold over background in wt mice infected with T. b. rhodesiense when compared with mock-infected controls. The pattern of gene expression in vivo closely paralleled the results from our in vitro measurements of sVSG-induced changes in gene expression in macrophages, and included numerous proinflammatory and acute phase genes. The transcription factor IRF-7 and many additional genes downstream of type I IFN signaling were up-regulated in vivo, including IRF-9 and IFN-stimulated gene 15 (ISG15) (Table II). A more comprehensive list of the genes up-regulated during infection is provided in Table III as supplemental data.4
Table II.
Genes associated with type I IFN signaling are up-regulated in vivo during infection: in vivo microarray analysis
| Accession No. | Fold Change | Description |
|---|---|---|
| Inflammatory/stress and immune responses | ||
| Acute phase response | ||
| Inflammation | ||
| M64404 | 2.72 | IL-1R antagonist |
| Other | ||
| NM_011315 | 3.23 | Serum amyloid A3 |
| NM_011314 | 13.5 | Serum amyloid A2 |
| NM_008491 | 4.44 | Lipocalin 2 |
| BC057985 | 2.86 | Orosomucoid 2 |
| M33215 | 2.11 | β-galactoside-binding lectin (L-34) |
| IFN-inducible | ||
| IFN-α/β-associated | ||
| NM_016850 | 11.66 | IFN regulatory factor 7 (Irf7) |
| NM_008390 | 5.78 | IFN regulatory factor 1 (Irf1) |
| NM_008394 | 2.44 | IFN regulatory factor 9 (Isgf3γ) |
| NM_015783 | 14.43 | IFN-stimulated gene 15 (Isg15) |
| NM_029803 | 7.04 | IFN-stimulated gene 12 (Isg12) |
| NM_011909 | 11.35 | Ubiquitin-specific protease 18 (usp18/ubp43) |
| S48937 | 2.60 | p68 kinase (IFN-inducible dsRNA-activated protein kinase) |
| NM_145211 | 2.56 | 2′-5′ oligoadenylate synthetase 1A |
| NM_145227 | 3.70 | 2′-5′ oligoadenylate synthetase 2 |
| NM_145226 | 2.77 | 2′-5′ oligoadenylate synthetase 3 |
| NM_010846 | 10.37 | Myxovirus (influenza virus) resistance 1 (Mx1) |
| IFN-γ-associated | ||
| NM_008390 | 5.78 | IFN regulatory factor 1 (Irf1) |
| NM_008320 | 3.24 | IFN regulatory factor 8 (Irf8) |
| NM_008394 | 2.44 | IFN regulatory factor 9 (Isgf3γ) |
| AK002545 | 9.94 | IFN-inducible protein 1 |
| NM_027320 | 4.03 | IFN-induced protein 35 |
| NM_172648 | 3.81 | IFN-activated gene 205 |
| NM_008329 | 9.30 | IFN-activated gene 204 |
| NM_008330 | 10.42 | IFN-γ -inducible protein 47 |
| NM_018738 | 11.77 | IFN-γ -induced GTPase |
| NM_019440 | 9.30 | IFN-inducible GTPase 2 |
| NM_021792 | 10.78 | IFN-inducible GTPase 1 |
Total RNA was harvested from adherent spleen, liver, and peritoneal cell populations taken from uninfected and infected C57BL/6J mice 72 h postinfection. Total RNA from these three cell populations was pooled, and changes in gene expression were assessed by microarray analysis. Shown are a subset of genes up-regulated 2-fold or more over background in cells from the liver, spleen, and peritoneum of infected mice.
These results reveal for the first time that the pattern of gene expression induced in vivo during infection was similar to the pattern observed in response to sVSG in vitro, demonstrating that the in vitro studies provide an accurate picture of macrophage activation during trypanosome infection and that sVSG is a major component of the innate activation response during early infection. Additionally, these data provided support for our hypothesis that IFN-α/β plays a key role in regulating macrophage activation and cellular function during trypanosome infection.
IFN-α/β produced during trypanosome infection contributes to early control of parasites
Type I IFN levels were measured indirectly by viral plaque inhibition assays using infected mouse sera as well as culture supernatant fluids from ex vivo SPC and PEC taken early during infection. IFNs were detected in both serum and SPC and PEC cell culture fluids; levels ranged from 52 IU/ml to 176 IU/ml (data not shown). Therefore, type I IFNs are detectable in vivo during trypanosome infection, confirming what we and others have shown previously (39, 40) and providing further validation for our in vitro and in vivo microarray analyses in this biological system.
The contribution of IFN-α/β to host resistance during trypanosome infection was first assessed using Ifnar1−/− mice, which lack the ability to respond to type I IFNs. Parasitemia, IFN-γ levels, and VSG-specific Abs were monitored daily by tail bleed in both wt and Ifnar1−/− mice throughout the course of infection. Clearance of the first peak of parasitemia was consistently delayed by 1 or 2 days in significant numbers of Ifnar1−/− mice (Fig. 3). Likewise, similar levels of IFN-γ were observed in the serum of wt and Ifnar1−/− mice with the exception that there were detectable fluctuations in IFN-γ levels seen in the receptor knockout animals during the first 10 days of infection that were not observed in the wt animals (Fig. 3). Also, unlike the Ubp43−/− mice, below, Th1 cell IFN-γ responses to VSG were not significantly different between wt and Ifnar1−/− mice (data not shown).
FIGURE 3.
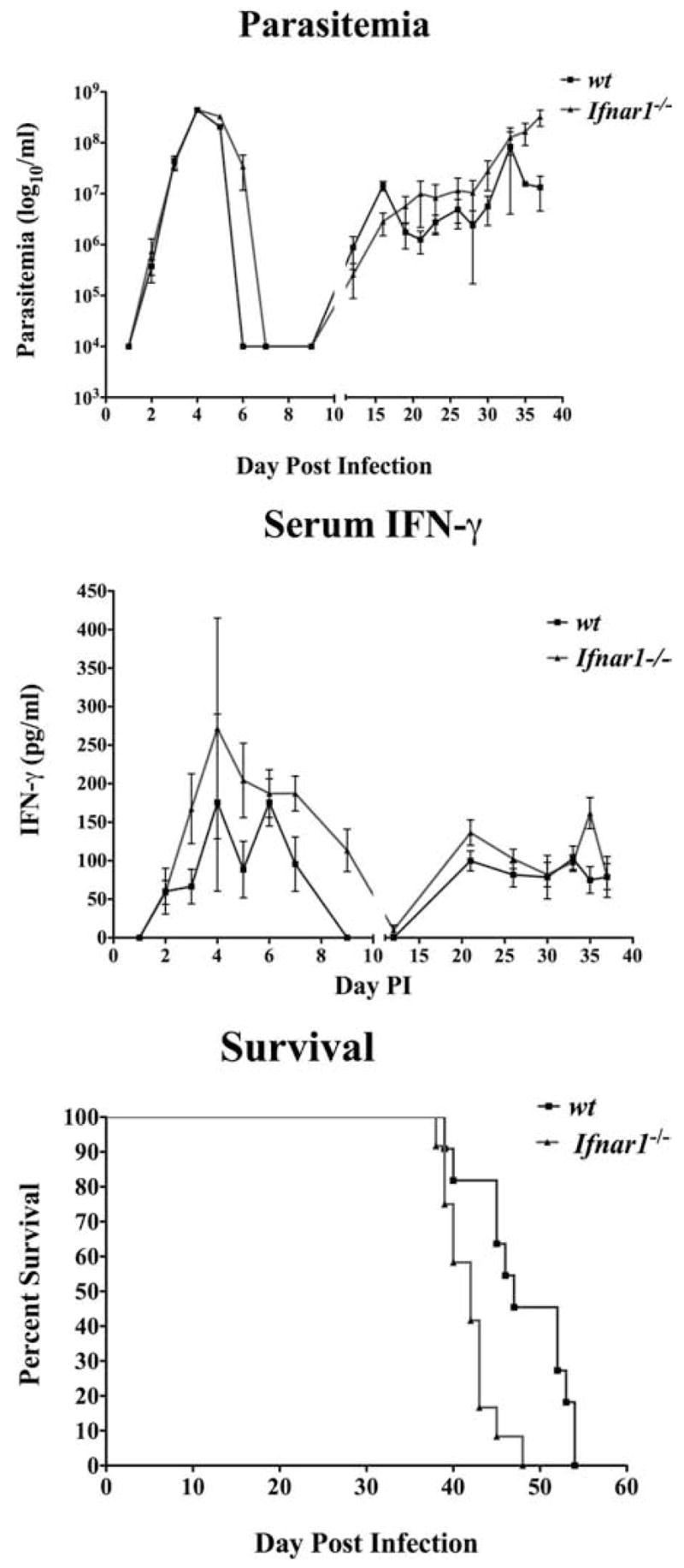
Comparative parasitemias, serum IFN-γ levels, and survival times of trypanosome-infected wt and Ifnar−/− mice. Coordinate measurements are shown of parasitemia (top) and serum IFN-γ (middle) and survival times (bottom) of infected Ifnar1−/− (■) and wild-type C57BL/6 mice (▲) (determinations are from four independent experiments). Mice were injected i.p. with 105 trypanosomes and monitored daily by tail bleed. Parasitemia was assessed at the times shown via enumeration of live parasites. Concentrations of IFN-γ in the serum were determined by sandwich ELISA.
Both wt and Ifnar1−/− mice exhibited significant levels of serum IFN-γ after 2 wk of infection, but this did not correlate with distinct reductions in parasite burden. One notable difference between wt and Ifnar1−/− animals was in the levels of VSG-specific IgM and IgG3, which were decreased in Ifnar1−/− mice through day 5 postinfection (Fig. 4). However, levels of IgG1 and IgG2a were nearly identical among the two groups of mice during the same period of infection before clearance of the first peak of parasitemia. These observations may explain the 1 or 2 day delay in parasite clearance observed during the first peak of parasitemia in Ifnar1−/− mice. Overall, Ifnar1−/− mice were more susceptible to infection (mean survival time of 43 days) when compared with their wt counterparts (mean survival time of 54 days; Fig. 3).
FIGURE 4.

Comparative VSG-specific Ab responses of infected IFNAR1 KO mice. Ab titers measured in the plasma of infected Ifnar1−/− (▲) and wt C57BL/6 mice (■). Mice were infected i.p. with 105 parasites, blood was collected by tail bleed at the time points shown, and plasma Ab titers for IgM, IgG1, IgG2a, and IgG3 were measured by sandwich ELISA.
Type I IFNs may regulate IFN-γ production during trypanosome infection
A second approach to assess the impact of type I IFNs on host resistance during infection was to use Ubp43−/− mice, which are hypersensitive to type I IFNs. UBP43 binds to IFNAR2 and inhibits the interaction between JAK1 and IFNAR2; this is independent of its isopeptidase activity. Therefore, the absence of UBP43 leads to enhanced and prolonged STAT1 phosphorylation via type I IFN signaling (41). Parasitemia, serum IFN-γ, VSG-specific Ab levels, and VSG-specific Th1 cell responses (IFN-γ-secreting cells) were monitored in both wt and Ubp43−/− mice during the course of infection. Most Ubp43−/− animals did not completely clear the first wave of parasitemia (Fig. 5); these mice maintained higher levels of parasites in the blood throughout infection compared with the wt controls. No significant difference in the levels of VSG-specific Abs was observed between the two groups of mice during the first week of infection, with the notable exception of IgG3 levels, which were much lower in Ubp43−/− mice by day 10 postinfection (Fig. 6). Levels of IFN-γ in serum, however, were either below the limit of detection or were very low in Ubp43−/− mice when compared with wt controls, and VSG-specific Th1 cell responses and IFN-γ production were significantly depressed in Ubp43−/− mice (Fig. 5). Thus, the VSG-specific Th1 cell IFN-γ response, which is the cornerstone of host resistance to African trypanosomes, mirrors the serum IFN-γ level reductions seen in the Ubp43−/− mice. Additionally, Ubp43−/− mice were significantly more susceptible to infection in general when compared with the wt controls (Fig. 5).
FIGURE 5.

Comparative parasitemias, serum and Th1 cell IFN-γ responses, and survival times in trypanosome-infected wt and Ubp43−/− mice. Coordinate measurements are shown of parasitemia (top), IFN-γ in the serum and in VSG-stimulated T cell cultures (middle), and survival times of infected Ubp43−/− (■) and wt mice (▲) (all determinations are from four independent experiments). Mice were infected i.p. with 105 trypanosomes and monitored daily by tail bleed. Parasitemia was assessed at the times shown via enumeration of live parasites on a hemacytometer. Concentrations of IFN-γ in the serum were determined by sandwich ELISA, and IFN-γ secretion by VSG-specific CD4 T cells was enumerated by ELISPOT analysis as detailed in Materials and Methods.
FIGURE 6.

Comparative VSG-specific Ab responses of infected UBP43 KO mice. Ab titers in the blood of infected Ubp43−/− (■) and wt C57BL/6 mice (▲) were determined. Mice were injected i.p. with 105 parasites, blood was collected by tail bleed at the time points shown, and Ab titers for IgM, IgG1, IgG2a, and IgG3 were measured in the plasma by sandwich ELISA.
Discussion
It is clear from in vitro experiments that the trypanosome sVSG molecule possesses significant immunoregulatory capabilities. Early work established that sVSG activates cells of the innate immune system and that such activation events may be enhanced by exposure of the cells to IFN-γ (20, 34). What is not known is which aspects of the immune response regulated by IFN-γ play a role in host resistance and which genes are the critical activation targets during the time between parasite recognition and activation of host-acquired immunity. Recent work using cells from infected, genetically defined mice has demonstrated that sVSG-induced production of TNF-α by macrophages is linked to MyD88-dependent signaling events (20). However, little is known about the specific signaling events initiated by sVSG in macrophages or which genes in addition to IFN-γ play a central role in conferring host resistance to the parasite.
The results presented herein provide two novel observations with respect to innate immune system activation in this disease. Our assessment of NF-κB and MAPK pathway activation confirmed that significant levels of sVSG, known to activate macrophage gene expression, do so at least in part through activating NF-κB and MAPK signaling intermediates. This is consistent with our recent observation that sVSG directly engages a scavenger receptor on macrophages and induces signaling events that are at least partly dependent on MyD88 signaling but are TRAF6 independent (20, 24). While ideally it would be useful to have signaling data on macrophages directly activated in situ during infection, these types of studies are almost impossible to carry out for technical reasons and because of small differences in the timing or progression of infection in individual animals. However, as a means to mimic events occurring in vivo, the levels of sVSG used in cultures here were similar to those detected in blood of infected mice at the peak of parasitemia based on measurements of parasite burden and the known mean number of VSG molecules per trypanosome (23, 42, 43).
The first important finding regarding events initiated by sVSG-mediated activation is that type I IFNs, which are produced by macrophages in vitro in response to sVSG and in vivo during trypanosome infection, play a role in the early control of parasite burden in infected mice. The demonstration of type I IFN induction in response to sVSG stimulation in vitro and in vivo during infection is concordant with previous work demonstrating the presence of IFN-α/β detectable in the sera of infected mice during T. b. rhodesiense infection (39, 40). These results are also consistent with earlier unpublished findings discussed by Bancroft and colleagues (39), which demonstrated that a significant reduction in the first wave of parasitemia occurred in T. b. brucei-infected mice treated with IFN-α/β or poly(I:C). However, the authors also observed that overall survival was not enhanced in these mice despite the induced decrease in parasitemia. This early observation is consistent with our second novel finding, that excessive reactivity to type I IFNs can lead to detrimental effects for the host by modulating IFN-γ production and overall resistance.
We demonstrate that mice that are hyperresponsive to the presence of type I IFN fail to release significant amounts of IFN-γ into the serum or directly by VSG-stimulated Th1 cells. The IFN-γ response of activated Th1 cells is a cornerstone of host resistance to trypanosomes, and the depressed response of the T cells mirrors the depressed IFN-γ serum response observed. Thus, the observation that Ubp43−/− mice are more susceptible to infection than are their wild-type counterparts is novel and likely fully based on the lower IFN-γ responses seen. Overall, these studies reveal that a host innate immune response that is prominent during early infection appears to have both beneficial and detrimental effects on long-term host resistance. Modest production of type I IFN appears to aid in efficient parasite clearance by host Ab early during infection and can therefore be said to contribute to host resistance. However, an overly robust or “uncontrolled” type I IFN response, as is the case with Upb43−/− mice, is associated with a defect in host IFN-γ production that is detrimental to host resistance and may be due to antagonistic effects of IFN-α/β on the production of IFN-γ by Th1 cells later during infection. Note that there are differences in the survival times and other infection parameters of the wt mice used for the Infar1−/− and Ubp43−/− experiments. Dr. Dong-Er Zhang at the Scripps Research Institute provided the matched wt and knockout founder strains used here, and the animals were further crossed and selected at the University of Wisconsin–Madison. Although biological differences are inherent in infectious disease model systems, especially with African trypanosomes, this may have been amplified because the wt mouse strains used for the two gene knockout groups have been bred and maintained independently and thus may be somewhat different due to genetic drift. Regardless, the observations and trends are clear and unequivocal within each experimental group of mice, revealing centrally important effects of type I IFNs on infection.
Our results are consistent with related observations in the trypanosome system. Recent findings by Kierstein et al. (37) demonstrated that expression of the IFNR α gene (Ifnar1) was associated with heightened susceptibility in mice infected with Trypanosoma congolense. As noted above, an earlier study by Mansfield and colleagues (40) also demonstrated that the early and secondary peaks of IFN produced in relatively susceptible animals were composed entirely of IFN-α/β. This was in contrast to relatively resistant animals in which the early release of IFN was composed predominantly of IFN-α/β, but in which the second release of IFN was composed almost entirely of IFN-γ. Collectively, these results suggest that relative susceptibility may be defined in part by an inability to make a critical switch to IFN-γ production by Th1 cells, due to differences in the regulation of IFN-α/β production and/or signaling. In fact, studies in viral disease systems demonstrate that IFN-γ may not be present at times when IFN-α/β is being produced systemically (44, 45). These studies showed that IFN-α/β actually inhibited IFN-γ production by NK and T cells and that this inhibition was dependent on both the IFN-α/β receptor and signal transducer and activator of transcription 1 (STAT1). In contrast, in the absence of STAT1, these cytokines promoted IFN-γ production via STAT4 signaling in a manner reminiscent of IL-12 signaling (45). Thus, type I IFN signaling was found to occur via both STAT1-dependent and -independent mechanisms and to exert distinct effects on IFN-γ production. Our previous in vitro work demonstrating that sVSG has dramatic effects on STAT1 activation is consistent with this idea and suggests that modulating the activity of individual STAT transcription factors during infection ultimately determines how IFN-α/β, together with other innate cytokines such as IL-12, will shape subsequent adaptive immune responses.
Thus, IFN-α/β is produced early during infection in both relatively susceptible and relatively resistant animals and this is associated with an enhancement of early host resistance that may be mediated in part by enhanced Ab responses. However, a robust response to IFN-α/β does not appear to be beneficial for overall or long-term host resistance and may actually down-regulate host IFN-γ production by Th1 cells, which is known to be critical for host resistance to trypanosome infection. Future studies in our laboratory will be aimed at determining whether in vivo neutralization of IFN-α/β in Ubp43−/− mice can restore IFN-γ production in these animals, whether treatment of Ubp43−/− mice with exogenous IFN-γ can increase overall resistance, and whether mechanisms similar to those observed in viral disease systems also regulate the production of IFN-γ in response to type I IFN in relatively susceptible and resistant animals during trypanosome infection. These studies will be key to understanding whether differences in regulation of IFN-α/β production and signaling are related to differences in STAT recruitment among strains of mice that differ in their ability to produce IFN-γ and thus differ in their susceptibility to trypanosome infection. Regardless of the outcome of our future studies, it remains both intriguing and surprising that an extracellular parasite such as T. b. rhodesiense invokes such a highly polarized Th1 cell adaptive immune response that is modulated in part by an innate immune IFN-α/β response.
Acknowledgments
We thank Vicki Leatherberry and Jim Schrader for help with animal infections, and we acknowledge Tajie Harris, Brian Leppert, and Bailey Freeman for helpful discussions during the completion of these studies.
Footnotes
Supported by funds from U.S. Public Health Service Grants AI048242 and AI051421 (to D.M.P.) and AI22441 and AI073346 (to J.M.M.), as well as by a predoctoral fellowship to R.L. from the Ford Foundation.
Abbreviations used in this paper: VSG, variant surface glycoprotein; GIP, glycosylinositolphosphate; IRF, IFN regulatory factor; mfVSG, GPI-anchored membrane form of the VSG molecule; ODN, oligodeoxynucleotide; PBSG, PBS containing 1 mM EDTA and 1% glucose; PEC, peritoneal cells; SPC, spleen cells; sVSG, soluble VSG; TRAF, TNFR-associated factor; wt, wild type.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.De Gee AL, Mansfield JM. Genetics of resistance to the African trypanosomes: IV. Resistance of radiation chimeras to Trypanosoma rhodesiense infection. Cell Immunol. 1984;87:85–91. doi: 10.1016/0008-8749(84)90132-1. [DOI] [PubMed] [Google Scholar]
- 2.De Gee AL, Levine RF, Mansfield JM. Genetics of resistance to the African trypanosomes: VI Heredity of resistance and variable surface glycoprotein-specific immune responses. J Immunol. 1988;140:283–288. [PubMed] [Google Scholar]
- 3.Greenblatt HC, Diggs CL, Rosenstreich DL. Trypanosoma rhodesiense: analysis of the genetic control of resistance among mice. Infect Immun. 1984;44:107–110. doi: 10.1128/iai.44.1.107-111.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine RF, Mansfield JM. Genetics of resistance to the African trypanosomes: III. Variant-specific antibody responses of H-2-compatible resistant and susceptible mice. J Immunol. 1984;133:1564–1569. [PubMed] [Google Scholar]
- 5.Seed JR, Sechelski JB. African trypanosomes: inheritance of factors involved in resistance. Exp Parasitol. 1989;69:1–8. doi: 10.1016/0014-4894(89)90164-1. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg JM. Human African trypanosomiasis: clinical presentation and immune response. Parasite Immunol. 2004;26:469–476. doi: 10.1111/j.0141-9838.2004.00731.x. [DOI] [PubMed] [Google Scholar]
- 7.Hertz CJ, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-γ dependent. J Immunol. 1998;161:6775–6783. [PubMed] [Google Scholar]
- 8.Schleifer KW, Filutowicz H, Schopf LR, Mansfield JM. Characterization of T helper cell responses to the trypanosome variant surface glycoprotein. J Immunol. 1993;150:2910–2919. [PubMed] [Google Scholar]
- 9.Schopf LR, Filutowicz H, Bi XJ, Mansfield JM. Interleukin-4-dependent immunoglobulin G1 isotype switch in the presence of a polarized antigen-specific Th1-cell response to the trypanosome variant surface glycoprotein. Infect Immun. 1998;66:451–461. doi: 10.1128/iai.66.2.451-461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Namangala B, De Baetselier P, Noel W, Brys L, Beschin A. Alternative versus classical macrophage activation during experimental African trypanosomosis. J Leukocyte Biol. 2001;69:387–396. [PubMed] [Google Scholar]
- 11.Magez S, Geuskens M, Beschin A, Delfavero H, Verschueren H, Lucas R, Pays E, Debaetselier P. Specific uptake of tumor necrosis factor-α is involved in growth control of Trypanosoma brucei. J Cell Biol. 1997;137:715–727. doi: 10.1083/jcb.137.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincendeau P, Daulouede S. Macrophage cytostatic effect on Trypanosoma musculi involves an L-arginine-dependent mechanism. J Immunol. 1991;146:4338–4343. [PubMed] [Google Scholar]
- 13.Vincendeau P, Daulouede S, Veyret B, Darde ML, Bouteille B, Lemesre JL. Nitric oxide-mediated cytostatic activity on Trypanosoma brucei gambiense and Trypanosoma brucei brucei. Exp Parasitol. 1992;75:353–360. doi: 10.1016/0014-4894(92)90220-5. [DOI] [PubMed] [Google Scholar]
- 14.Mansfield JM, Paulnock DM. Regulation of innate and acquired immunity in African trypanosomiasis. Parasite Immunol. 2005;27:361–371. doi: 10.1111/j.1365-3024.2005.00791.x. [DOI] [PubMed] [Google Scholar]
- 15.Magez S, Radwanska M, Beschin A, Sekikawa K, De Baetselier P. Tumor necrosis factor α is a key mediator in the regulation of experimental Trypanosoma brucei infections. Infect Immun. 1999;67:3128–3132. doi: 10.1128/iai.67.6.3128-3132.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sternberg JM. Elevated serum nitrate in Trypanosoma brucei rhodesiense infections: evidence for inducible nitric oxide synthesis in trypanosomiasis. Trans R Soc Trop Med Hyg. 1996;90:395. doi: 10.1016/s0035-9203(96)90519-2. [DOI] [PubMed] [Google Scholar]
- 17.Schleifer KW, Mansfield JM. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J Immunol. 1993;151:5492–5503. [PubMed] [Google Scholar]
- 18.Askonas BA. Macrophages as mediators of immunosuppression in murine African trypanosomiasis. Curr Top Microbiol Immunol. 1985;117:119–127. doi: 10.1007/978-3-642-70538-0_6. [DOI] [PubMed] [Google Scholar]
- 19.Vickerman K. Antigenic variation in trypanosomes. Nature. 1978;273:613–617. doi: 10.1038/273613a0. [DOI] [PubMed] [Google Scholar]
- 20.Drennan MB, Stijlemans B, Van Den Abbeele J, Quesniaux VJ, Barkhuizen M, Brombacher F, De Baetselier P, Ryffel B, Magez S. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol. 2005;175:2501–2509. doi: 10.4049/jimmunol.175.4.2501. [DOI] [PubMed] [Google Scholar]
- 21.Harris TH, Cooney NM, Mansfield JM, Paulnock DM. Signal transduction, gene transcription, and cytokine production triggered in macrophages by exposure to trypanosome DNA. Infect Immun. 2006;74:4530–4537. doi: 10.1128/IAI.01938-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magez S, Stijlemans B, Radwanska M, Pays E, Ferguson MA, De Baetselier P. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J Immunol. 1998;160:1949–1956. [PubMed] [Google Scholar]
- 23.Paulnock DM, Coller SP. Analysis of macrophage activation in African trypanosomiasis. J Leukocyte Biol. 2001;69:685–690. [PubMed] [Google Scholar]
- 24.Leppert BJ, Mansfield JM, Paulnock DM. The soluble variant surface glycoprotein of African trypanosomes is recognized by a macrophage scavenger receptor and induces IκBα degradation independently of TRAF6-mediated TLR signaling. J Immunol. 2007;179:548–556. doi: 10.4049/jimmunol.179.1.548. [DOI] [PubMed] [Google Scholar]
- 25.Mansfield JM, Bagasra O. Lymphocyte function in experimental African trypanosomiasis: I. B cell responses to helper T cell-independent and -dependent antigens. J Immunol. 1978;120:759–765. [PubMed] [Google Scholar]
- 26.Mansfield JM. Immunobiology of African trypanosomiasis. Cell Immunol. 1978;39:204–210. doi: 10.1016/0008-8749(78)90094-1. [DOI] [PubMed] [Google Scholar]
- 27.Moulton JE, Coleman JL. Immunosuppression in deer mice with experimentally induced trypanosomiasis. Am J Vet Res. 1977;38:573–579. [PubMed] [Google Scholar]
- 28.Mathias S, Perez R, Diffley P. The kinetics of gene expression and maturation of IL-1 alpha after induction with the surface coat of Trypanosoma brucei rhodesiense or lipopolysaccharide. J Immunol. 1990;145:3450–3455. [PubMed] [Google Scholar]
- 29.Mansfield JM. Immunobiology of African trypanosomiasis: a revisionist view. In: Boothroyd JC, Komuniecki R, editors. Molecular Approaches to Parasitology. Wiley-Liss; New York: 1995. pp. 477–496. [Google Scholar]
- 30.Wellhausen SR, Mansfield JM. Lymphocyte function in experimental African trypanosomiasis: II. Splenic suppressor cell activity. J Immunol. 1979;122:818–824. [PubMed] [Google Scholar]
- 31.Grosskinsky CM, Askonas BA. Macrophages as primary target cells and mediators of immune dysfunction in African trypanosomiasis. Infect Immun. 1981;33:149–155. doi: 10.1128/iai.33.1.149-155.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mabbott NA, Sutherland IA, Sternberg JM. Suppressor macrophages in Trypanosoma brucei infection: nitric oxide is related to both suppressive activity and lifespan in vivo. Parasite Immunol. 1995;17:143–150. doi: 10.1111/j.1365-3024.1995.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 33.Sternberg JM, Mabbott NA. Nitric oxide-mediated suppression of T cell responses during Trypanosoma brucei infection: soluble trypanosome products and interferon-gamma are synergistic inducers of nitric oxide synthase. Eur J Immunol. 1996;26:539–543. doi: 10.1002/eji.1830260306. [DOI] [PubMed] [Google Scholar]
- 34.Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-γ-induced nitric oxide production via reduction in STAT1 phosphorylation in African trypanosomiasis. J Immunol. 2003;171:1466–1472. doi: 10.4049/jimmunol.171.3.1466. [DOI] [PubMed] [Google Scholar]
- 35.Cross GA. Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei. Parasitology. 1975;71:393–417. doi: 10.1017/s003118200004717x. [DOI] [PubMed] [Google Scholar]
- 36.Hereld D, Krakow JL, Bangs JD, Hart GW, Englund PT. A phospholipase C from Trypanosoma brucei which selectively cleaves the glyco-lipid on the variant surface glycoprotein. J Biol Chem. 1986;261:13813–13819. [PubMed] [Google Scholar]
- 37.Kierstein S, Noyes H, Naessens J, Nakamura Y, Pritchard C, Gibson J, Kemp S, Brass A. Gene expression profiling in a mouse model for African trypanosomiasis. Genes Immun. 2006;7:667–679. doi: 10.1038/sj.gene.6364345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pottathil R, Chandrabose KA, Cuatrecasas P, Lang DJ. Establishment of the interferon-mediated antiviral state: possible role of superoxide dismutase. Proc Natl Acad Sci USA. 1981;78:3343–3347. doi: 10.1073/pnas.78.6.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Askonas BA, Bancroft GJ. Interaction of African trypanosomes with the immune system. Philos Trans R Soc Lond Biol. 1984;307:41–49. doi: 10.1098/rstb.1984.0107. [DOI] [PubMed] [Google Scholar]
- 40.De Gee AL, Sonnenfeld G, Mansfield JM. Genetics of resistance to the African trypanosomes: V. Qualitative and quantitative differences in interferon production among susceptible and resistant mouse strains. J Immunol. 1985;134:2723–2726. [PubMed] [Google Scholar]
- 41.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diffley P, Strickler JE, Patton CL, Waksman BH. Detection and quantification of variant specific antigen in the plasma of rats and mice infected with Trypanosoma brucei brucei. J Parasitol. 1980;66:185–191. [PubMed] [Google Scholar]
- 43.Radwanska M, Magez S, Michel A, Stijlemans B, Geuskens M, Pays E. Comparative analysis of antibody responses against HSP60, invariant surface glycoprotein 70, and variant surface glycoprotein reveals a complex antigen-specific pattern of immunoglobulin isotype switching during infection by Trypanosoma brucei. Infect Immun. 2000;68:848–860. doi: 10.1128/iai.68.2.848-860.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen L, Knipe DM, Finberg RW. Mechanism of virus-induced Ig subclass shifts. J Immunol. 1994;152:478–484. [PubMed] [Google Scholar]
- 45.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA. Critical role for STAT4 activation by type 1 interferons in the interferon-γ response to viral infection. Science. 2002;297:2063–2066. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]