

Abstract
Fructose-1,6-bisphosphate (FBP), an endogenous intracellular metabolite in glycolysis, was found in many preclinical studies to be neuroprotective during hypoxia-ischemia (HI) when administered exogenously. We looked for HI neuroprotection from FBP in a neonatal rat brain slice model, using 14.1 Tesla 1H /31P/13C NMR spectroscopy of perchloric acid slice extracts to ask: 1) if FBP preserves high energy phosphates during HI; and 2) if exogenous [1-13C]FBP enters cells and is glycolytically metabolized to [3-13C]lactate. We also asked: 3) if substantial superoxide production occurs during and after HI, thinking such might be treatable by exogenous FBP's antioxidant effects. Superfused P7 rat cerebrocortical slices (350μm) were treated with 2 mM FBP before and during 30 min of HI, and then given four hours of recovery with an FBP-free oxygenated superfusate. Slices were removed before HI, at the end of HI, and at 1 and 4 hours after HI. FBP did not improve high energy phosphate levels or change 1H metabolite profiles. Large increases in [3-13C]lactate were seen with 13C NMR, but the lactate fractional enrichment was always (1.1±0.5)%, implying that all of lactate's 13C was natural abundance 13C, that none was from metabolism of 13C-FBP. FBP had no effect on the fluorescence of ethidium produced from superoxide oxidation of hydroethidine. Compared to control slices, ethidium fluorescence was 25% higher during HI and 50% higher at the end of recovery. Exogenous FBP did not provide protection or enter glycolysis. Its use as an antioxidant might be worth studying at higher FBP concentrations.
INTRODUCTION
More than 20 years ago Dr. A. K. Markov reported that exogenously administered fructose-1,6-bisphosphate (FBP) could provide impressive hypoxic-ischemic protection[27]. (At that time FBP was often referred to as FDP, ”fructose diphosphate”, which is technically incorrect, as that terminology requires adjacent phosphates.) The original idea behind FBP administration was metabolic rescue: ”Exogenous FBP will restore the activity of glycolysis, which has been inhibited by acidosis, by intervening in the Embden-Meyerhoff pathway both as a metabolic regulator and as a high energy substrate.”[27] Because substrate entry of one exogenous FBP molecule into intracellular glycolysis would spare the expenditure of two ATP's (one by hexokinase, the other by phosphofructokinase), the original belief was that metabolism of exogenous FBP would provide twice the glycolytic energy yield, four ATP's per molecule of substrate.
Ischemic neuroprotection from FBP administration was subsequently demonstrated in animal experiments[6,12,21,35], cell culture studies[11,19,20,31,39], as well as in a brain slice study by us[3]. Our previous investigation of FBP neuroprotection used 31P and 1H ex vivo NMR spectroscopy in a horizontal 4.7 Tesla wide-bore spectrometer. Respiring neonatal (P7) rat cerebrocortical slices superfused in the magnet underwent a 30 minute period of hypoxia-ischemia (10 minutes of hypoxia followed by 20 min stopped-flow, followed by 4 hours of recovery)[3]. In that study as in this one, for one group of slices 2 mM FBP was in the superfusate for an hour before hypoxia-ischemia, and also throughout. Another group was not treated with FBP. At the end of the recovery period, pretreatment with 2 mM FBP resulted in no ATP loss, no lactate increase, and minimal morphologic changes, while no treatment caused a ≈50% ATP loss and an ≈700% increase in lactate.
It is very important to remember, however, that several careful studies found no neuroprotection[2,10,22].
Published conclusions regarding FBP metabolism also differ. In a radioisotope cell culture study of astrocytes where FBP provided protection, [U-14C]FBP was administered prior to hypoxia, but no intracellular 14C-FBP or 14C-lactate was detected, suggesting that FBP did not enter cells[18]. In contrast, NMR studies of superfused porcine vascular smooth muscle given [1,6-13C]FBP found increases in [3-13C]lactate, which were then were taken as evidence for FBP entrance into cells, with subsequent participation in glycolysis[13,14,16].
After completing our 4.7 Tesla study the spectrometer was replaced by a narrow bore 14.1 Tesla NMR system having substantially higher spectral resolution, increased sensitivity, and the capability of doing 13C spectroscopy. This motivated us to use it for better understanding FBP protection. To do so we posed three hypotheses: 1) that as one kind of neuroprotection during hypoxia-ischemia, FBP preserves high energy phosphates; 2) that exogenous [1-13C]FBP will enter cells and glycolysis, and produce [3-13C]lactate; 3) that there is substantial superoxide production during and after hypoxia-ischemia. The third hypothesis was listed because recent FBP studies have emphasized that FBP's antioxidant properties might account for the protection it provides[29,31,39]
RESULTS
1) 31P Data
The quality of our 31P spectra was the same as in recent publications[43,44]. FBP protection of intra- and post-ischemic high energy phosphates was not seen. For each of the two treatment groups there were N=3 experiments; with 5 slices per data point per experiment. The average of 3 experiments means that a final data point represents an average over 15 slices, or 7.5 different animals. The final/initial NTP ratio (taken from signal intensities of the β-NTP resonance) was 0.54±0.14 for the FBP treated group, compared to 0.51±0.14 for the no-treatment group, indicating no large difference between the groups. If one assumes that 100% preservation by FBP is the true reality, (i.e., if the true final/initial NTP ratio were 100%) then the likelihood of our having gotten a 50% ratio, computed using a t-test, is less than 2.5%. Other phosphate ratios for FBP and non-FBP groups, measured 4 hrs after the end of hypoxia-ischemia, were similarly equal within errors. The final ratio of PCr/(β-NTP) after 4 hrs of recovery, compared to the initial ratio at the start of the experiment, was 1.20±.20 for the FBP group, and 0.90±0.10 for the non-FBP group; with 1.13±0.10 and 1.14±0.10 being the corresponding numbers for PCr/(α-NDP).
2) 1H metabolite changes
As was the case with 31P NMR, the quality of our 1H spectra was the same as in recent publications[43,44]. Figure 1A shows ratios of metabolite concentrations for lactate, alanine, N-acetyl-aspartate, GABA, glutamate, and succinate relative to initial concentrations (before hypoxia-ischemia). Only six 1H metabolites are quantified for reasons explained in the Methods. Also explained in the Methods is that the no-FBP treatment group was averaged over 3 experiments, while the FBP-treatment group was averaged over 2 experiments. Ratios to initial values are given for each metabolite for three time points (t=0 h, end of hypoxia-ischemia, t=1 h, one hour after ending hypoxia-ischemia, and at t=4 h, the end of the recovery). Bonferroni-Corrected t-tests indicated that the ratios (with standard deviation error bars indicated) were consistent with the null hypothesis: that all metabolite ratio ensembles appear to belong to the same statistical population.
Figure 1A.
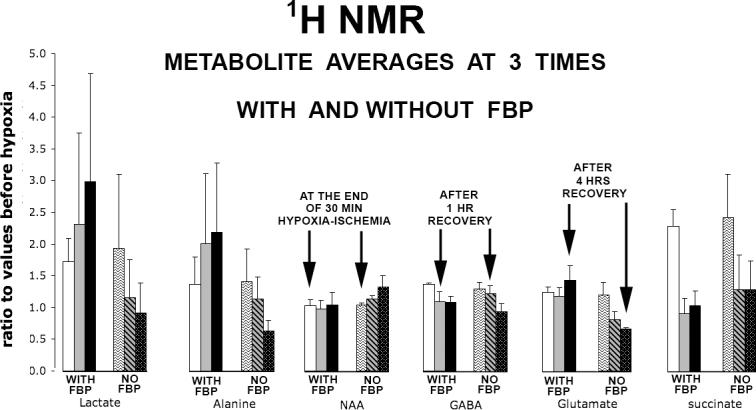
(With FBP)-(Without FBP) bar graph comparisons are shown for three time points for each of six 1H metabolites, with each metabolite being measured relative to its control (value at the start of experiment, t=−0.5 h, before hypoxia-ischemia). Two sets of three vertical bars are shown for each metabolite. Each of the two groups within a set presents metabolite values, going from left to right, for t=0, t=1.0 hrs, and t=4hrs. Each vertical bar represents the average of N separate experiments, with N=2 for the ”With-FBP” group, and N=3 for the ”Without-FBP” group. Twenty brain slices were used for each experiment, with 5 slices being taken at each of four predetermined time points and pooled together for one PCA extraction. Arrows placed over bars illustrate particular time points for different metabolites. Error bars indicate standard deviations. Data from the FBP group are not statistically different from data from the no-FBP group.
3) 1 H metabolomic results
Although time differences in 1H metabolites were generally unimpressive, differences in NTP, NDP, and PCr were obviously much greater at different slice removal times. For example, high energy phosphates decrease to very low values during hypoxia-ischemia. We therefore made four larger sets of variables (”super-spectra”) for each time point by combining β-NTP/PCr and α-NDP/PCr ratios with 1H metabolite ratios. Four classes of ”super-spectra” were defined: one class for each of the four experimental times for removing slices. Figure 1B shows the Scores Plot for a Partial Least Squares multivariate analyses of the ”super-spectra” (Umetrics SIMCA P+ v.11 metabolomics software). Recall that each point in the figure corresponds to a one full set of 1H and 31P metabolite values, i.e., one ”super-spectrum.” The five ”super-spectra” used above to compare 1H metabolite values, (two where FBP was used, three where it was not), are plotted for each of four time points (beginning of the experiment; end of hypoxia-ischemia; after 1 h recovery; after 4 h recovery), giving the plot a total of 5 × 4 = 20 points. Instead of representing each ”super-spectrum” with on the plot with a small symbol, text identifying the spectrum and time point were placed, as described in the Figure legend. The spectra with the highest energy phosphates were from slices prior to hypoxia-ischemia, and these ended up somewhat together in the upper right quadrant. Slices with the lowest energy phosphates unsurprisingly came from the end of hypoxia-ischemia, and these ended up in the lower right quadrant. The points for t=1.0h and 4.0h straddle the x-axis on the left side of the plot. Qualitatively, points clustered close to the origin (x=0, y=0) represent insignificant differences. Significant group differences can be attributed to clusters that are far from each other, and also far from the origin, but still inside Hotelling's Tolerance Ellipse. The Scores Plot confirms the 1H results in Fig 2A: that data ensembles for the end of the recovery period are not significantly different, while data ensembles for the beginning of the experiment are significantly different from data ensembles from the time of greatest insult, the end of hypoxia-ischemia. More generally, the plot shows the potential utility of multivariate analyses for distinguishing important subgroups and identifying outliers.
Figure 1B.
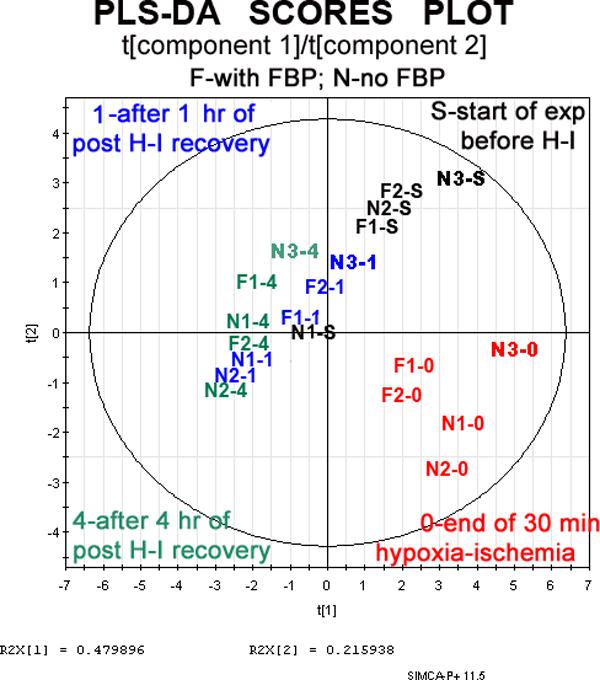
A 2D scatter plot is shown for a Partial Least Squares Discriminant Analysis (PLS-DA) done with the multivariate analysis program SIMCA Plus v.11. Biomarker datasets for each experiment consisted of that time point's 1H NMR metabolites measured relative to starting values, along with two ratios from that time point's 31P spectra: β-NTP/PCr and α-NDP/PCr. Each biomarker dataset provides coordinates for a single point in the plot. However, instead of plotting a point, the name of the data set is placed at the point's coordinates. ”F” means FBP was used; ”N” means FBP was not used. Times when slices were removed are represented by: ”S”, the start of the experiment; ”0”, end of hypoxia; ”1.0” and ”4”, after 1.0 hrs and 4 hrs of recovery. Coordinates in a PLS-DA multivariate analysis are obtained from a new set of axes in multidimensional space is formed after scaling and centering each biomarker, and after a Principal Component Analysis finds the axes of largest and second largest variance. The outer circle is this plot's Hotelling's Tolerance Ellipse. Points outside the circle, and there are none in this case, would be ”outliers”, indicating that the statistical model is poor.
Figure 2.

Three representative 13C NMR spectra are shown for Perchloric Acid (PCA) Extracts from slices taken from the superfusion chamber at different times: (A) before hypoxia-ischemia; (B) at the end of hypoxia-ischemia; (C) after four hours of recovery following hypoxia-ischemia. Resonances for particular metabolites are numbered: 1) lactate C3, which is substantially increased in B and C relative to A ; 2) β−hydroxybutyrate C4; 3) GABA C3; 4) glutamate C3; 5) glutamate C4; 6) succinate C2, C3; 7) GABA C2; 8) taurine. (D) a different portion of the same spectrum shown in C, showing an increased resonance peak for C2 of lactate, which is barely discernable in (E). (F) A representative portion of a 1H spectrum at the end of hypoxia-ischemia. The large, narrow doublet at 1.33 ppm is from the methyl protons on the C3 of unlabeled lactate. When the C3 of lactate is labeled with 13C, two doublets occur as indicated by arrows instead of the narrow doublet at 1.33 ppm. In all spectra where 13C-glucose is was not used, the combined area of the two small doublets was 1.1% of the large central doublet, which is what occurs from natural abundance 13C.
4) 13 C Data
[1-13C]FBP was administered prior to and during hypoxia-ischemia, and subsequent increases in [3-13C]lactate were detected. However, it was not possible to say that FBP metabolism was observed, because increases seen in 13C spectra were those expected from natural abundance 13C, which is approximately 1.1% that of 12C. Portions of three representative 13C spectra arranged vertically at the left of Figure 2 compare spectra from the beginning and end of a hypoxia-ischemia experiment in which 2 mM [1-13C]FBP administration was begun before the insult and continued until reoxygenation. After 30 minutes of hypoxia-ischemia, a 3 to 4 fold increase can be seen in [3-13C]lactate, the spectral peak labeled as ”1”. Indeed, looking at this comparison alone initially suggested to us that [1-13C]FBP was metabolized. However, as explained below, one cannot draw that conclusion. All 1H spectra in the 13C-FBP experiment had the increase in labeled lactate being proportional to the increase in unlabeled lactate. The [3-13C]lactate fractional enrichment was always 1.1% of the total lactate. Thus the observed increase in [3-13C]lactate was that expected from an increase in total lactate, without prior administration of 13C enriched substrates.
In the 1H NMR spectrum of Figure 2F, the large doublet resonance peak at 1.33 ppm comes from the methyl protons of [3-12C]lactate. Unlike 12C, which has no nuclear spin, 13C has a nuclear spin of ½, which causes a very small additional ”up” or ”down” magnetic field in the vicinity of C3's methyl protons. This small additional field produces two 1H doublet resonance peaks: one on either side of the central 1.33 ppm peak, ≈0.1 ppm away, as shown in Figure 2F. All three lactate peaks increased equally during hypoxia-ischemia. Indeed, except in experiments where 13C enriched substrates were administered, such happened in all hypoxia data ever taken by us, not just in this FBP study. Figures 2D and 2E were taken from the same 13C NMR spectrum as Figures 2A-2C, but are at a larger scale. They show a hypoxia-induced increase in [2-13C]lactate that is also attributable to naturally abundant 13C. (Similar information about the C1 of lactate was not attainable, as it does not have an associated 1H resonance, and the intensity of its 13C NMR resonance is too small to be observed in our experiments.) If we estimate the error in our quantification of the satellite peaks to be ±0.5% of the total lactate pool, then a 3-standard deviation upper limit for [3-13C]lactate production from [1-13C]FBP can be estimated as ≈1.5%.
5) Increases in intracellular superoxide
Ethidium, the red fluorescent product of superoxide and hydroethidine, was histologically detected intracellularly in slices incubated in hydroethidine for 30 minutes before, during, and after hypoxia-ischemia. Figure 3A shows that superoxide was almost totally absent in control slices, those whose incubation period ended just before beginning hypoxia-ischemia. A small scattering of bright cells that converted hydroethidine to ethidium is easily recognized on the right in the 50 μm ”injury layer” (where the blade cut the slice from the brain.) Figure 3B, which has the slice in the same orientation, was exposed during hypoxia-ischemia and removed prior to reoxygenation. It shows a substantial number of ethidium stained cells throughout the parenchyma, but not as many as in slices that were removed after 4 hours of recovery, shown in Figures 3C and 3D. No FBP was involved for the slice shown in Figure 3C. The slices shown in Figures 3C and 3D were from the same litter of animals in an extra experiment that had parallel paths for FBP and no-FBP superfusion. Image intensities relative to background intensities, quantified using ”NIH Image” (http://rsb.info.nih.gov/nih-image), were 1.10±0.06 for control, 1.36±0.14 for end of hypoxia-ischemia; 1.72±0.21 and 1.61±0.13 for end of recovery with and without FBP. Bonferroni-corrected t-tests found p-values less than .05 for the control group relative to each of the other 3 groups, but not for the FBP group relative to the non-FBP group. In any event, the images suggest that the ex vivo slice model is appropriate for investigating ROS mediated injuries.
Figure 3.

Representative ethidium fluorescence in slices removed at times indicated in the figure. Slices were incubated for 30 minutes with hydroethidine, which is converted to ethidium by superoxide radicals. Higher magnification images (200×) are overlaid at the lower left corners of lower magnification images (100×). The pial layer of the slice is at the left, and scale bars are 100 μm and 30 μm respectively, in low and high magnification images. Qualitatively, the least oxidation is before hypoxia-ischemia, and the most is at the end of 4 hrs recovery (p<0.05).
DISCUSSION
NON-PRESERVATION OF HIGH ENERGY PHOSPHATE LEVELS BY EXOGENOUS FBP
Our finding that exogenous FBP could not prevent falling NTP levels contrasts with our earlier brain slice study done in the previous laboratory, where with pretreatment but not post-treatment NTP preservation was 100%[3]. We attribute this to two things: 1) the conclusion in previous studies with FBP failures[10,22,23,34] that FBP is not protective if hypoxia-ischemia is sufficiently severe; and 2) that our new laboratory's superfusion systems provided less oxygen availability. Our previous studies in which NTP preservation was found were performed inside a 34 cm wide-bore, 4.7 Tesla magnet, using a 30 mm diameter slice chamber containing 80 superfused brain slices. The superfusate arrived by large diameter tubing from a nearby ACSF reservoir. After moving to the new laboratory and its 14.1 Tesla system, it was no longer easy to do ex vivo NMR spectroscopy of slices superfused in the magnet. The narrow magnet bore was restricted to either 5 mm or 8 mm NMR tubes. When we conducted preliminary studies using slices that were superfused and respiring in an 8 mm NMR tube, we learned that achieving a 37°C temperature in the magnet bore was more difficult, because of the need for longer tubing and the ACSF's reservoir being further from the magnet[25]. We learned to carefully regulate slice temperatures in the magnet by using the temperature-dependent 1H chemical shift of water relative to lactate[25], and we did complete one ex vivo hypoxia experiment[15]. However, even with active warming of the longer tubing, a 41°C temperature was needed for the ACSF as it left the remotely located reservoir, which meant that some oxygen was driven out of solution. When FBP preservation did not occur in slices superfused in the new magnet, we turned to the benchtop experiments in this study, and made NMR measurements of metabolites extracted from quickly frozen brain slices. The hypoxia PO2 in the new laboratory's slice chamber was measured with an Ocean Optics SF2000 Fiber Optic Spectrophotometer to be ≈7 mm Hg, which we believe was lower than than the hypoxic PO2 of the larger, less crowded, no longer available old chamber, where hypoxic PO2 was measured in ACSF aliquots taken to a clinical blood gas machine.
NON-OBSERVATION OF FBP METABOLISM
Our non-observation of FBP metabolism agrees with the radioisotope cell culture study that was cited earlier[18]. Our conclusion that observed 13C lactate increases come from natural abundance 13C does not agree with the already cited ex vivo and extract 13C NMR studies involving exogenously administered [1,6-13C]FBP[13,14,16]. However, none of the three publications addressed whether or not their observed increases in 13C metabolites might contain contributions from natural abundance 13C, nor did any publication show or cite relevant 1H spectra, such as those presented by us in Figure 1F, which made clear that natural abundance 13C could account for all of our 13C-lactate increases. In any event, our findings do not necessarily contradict their conclusions, as non-observation of FBP metabolism in brain tissue need not imply that its metabolism cannot occur in the types of muscle tissue that they studied.
SUPEROXIDE INCREASES DURING HYPOXIA-ISCHEMIA AND RECOVERY
We remarked in earlier publications that ”alternative hypotheses of FBP protection have included calcium chelation, free-radical scavenging, and protease inhibition.”[3] In recent years three studies of free radical mechanisms in FBP protection have pointed to antioxidant effects being a primary protective mechanism[29,31,39]. A nonspecific antioxidant effect would be consistent with FBP being successful in so many widely differing circumstances. Other recent work has reminded us that the keto carbonyl group of many metabolic intermediates confers strong antioxidant properties to α-keto acids (R-CO-COO−) for H2O2 radical scavenging via peroxidative decarboxylation[1,9]. For example, in the case of pyruvate, R is CH3, and the products of the reaction are acetate and the formate radical, with the latter easily converting to H2O + CO2. Such scavenging is preferable, for example, to superoxide removal by superoxide dismutase, which consumes two H+ ions that could be used by ATPase to generate ATP[5]. Interesting experiments in which DNA injury has lead to PARP-1 activation and depletion of NAD+, an intermediate essential for glycolysis, have been hypothesized and then found true, verifying that PARP-1 related cellular injury can be ameliorated by exogenous administration of TCA Cycle intermediates such as pyruvate and α-ketoglutarate[42,44]. In those studies the motivation for administering pyruvate and α-ketoglutarate was to provide the TCA Cycle with substrates distal to glycolysis, thereby boosting ATP production in a manner reminiscent of Dr. Markov's original motivation. However, others were publishing papers at the same time of those studies, advocating the use of α-keto TCA substrates as antioxidants[7,8,36-38]. We recently compared two post oxidative stress rescue protocols in a study aimed at distinguishing the relative importance of ethyl pyruvate's antioxidant and nutrient mechanisms[26,43]. In the first regimen ethyl pyruvate, which efficiently delivers to cells pyruvate that is metabolized[43], was added to ACSF glucose. In the second regimen the additive to ACSF glucose was a nonmetabolizable radical scavenger, PBN. Addition of the antioxidant provided the same ATP protection as addition of the metabolic substrate[26], suggesting that a similar result might occur if FBP were the additive instead of ethyl pyruvate. Strong neuroprotection by edaravone, a radical scavenger developed for clinical use in humans[33], has also been demonstrated in a P7 brain slice model very similar to ours[30]. Should future studies of FBP's power to rescue be planned, it would seem prudent to consider having a treatment group with one or more nonmetabolizable antioxidants.
CONNECTION WITH PREVIOUS STUDIES OF EXOGENOUS FBP
During the past twenty years of brain research post-ischemic outcome improvements by FBP have been found by some to be dramatic, by others to be simply modest[6,11,12,19-21,31,35,39], and by a smaller number to be completely nonexistent[10,22,23]. FBP studies showing neuroprotection pose a formidable intellectual challenge: do improved outcomes come from important, already known injurious mechanisms that are typically focused upon, such as glutamate excitotoxicity, calcium overload, acidosis, oxygen radicals, PARP-1 activation, apoptosis, etc? Or, are there new, important, undiscovered neuroprotective mechanisms that successful FBP treatments are trying to reveal? We initially hoped for the latter, but we reversed our views after not finding substantial protection, if any, in the experiments reported in this paper. Although each year fewer publications appear in support of hypoxic/ischemic protection by exogenous FBP[17,28,31,32,41], there has not been a total cessation. We therefore feel it is important to share data from our negative experiments, which support earlier impressions that FBP is not metabolized in brain tissue, most likely because it does not enter cells. It is tempting, but not possible, to conclude that when FBP administration is helpful, its benefits can be attributed to known mechanisms, with further studies being unlikely to produce high-impact mechanistic findings. Conclusions from our study are limited because cell damage was assessed with a limited set of outcome measures. We have not ruled out FBP modulation of other important protective mechanisms, for example activation of Phospholipase C and intracellular survival pathways[4]. It is possible that FBP was initiating protective steps that we did not look for or detect.
MATERIALS AND METHODS
ANIMAL PREPARATION, ARTIFICIAL CEREBROSPINAL FLUID (ACSF)
The protocol was approved by the UCSF's Institutional Animal Care and Use Committee (IACUC), and very close to earlier protocols[43,44]. Briefly, in each experiment 20 cerebrocortical slices (350 μm thick) were obtained from ten 7-day-old (P7) Sprague-Dawley rats and superfused with fresh, oxygenated artificial cerebrospinal fluid (oxy-ACSF) that consisted of a modified Krebs balanced salt solution. The superfusion chamber was immersed in a water bath that was kept at 37°C and the oxy-ACSF flow rate was 10 ml/min. ACSF was maintained at constant PCO2 (40 mm Hg), PO2 (600 to 650 mm Hg), and pH (7.4). Thirty minutes of hypoxiaischemia (10 minutes of hypoxia followed by 20 min stopped-flow) began after 3 h of metabolic recovery from decapitation. Recovery began at t=0, and continued until t=4 hrs. Slice sampling was done at t= −0.5 h, 0h, 1h, and 4h, with slices immediately being either frozen in liquid nitrogen immediately or placed in a fixation solution. Chemicals were obtained from Sigma-Aldrich (St. Louis, MO), with FBP as D-fructose-1,6-bisphosphate trisodium salt octahydrate (>98% pure, Fluka Chemical Corporation, Ronkonkoma, NY). 13C-FBP was obtained as D-[1-13C]fructose-1,6-bisphosphate sodium salt, 99% 13C, from Omicron Biochemicals Inc., South Bend, IN.
In the three experiments without FBP and in two of three experiments with FBP, [U-13C]glucose was used as the ACSF glucose. This was because FBP effects were optimistically expected, and we wanted in that case to see if glucose metabolism was altered. It has been proposed that exogenous FBP could increase activity of the Pentose Phosphate Pathway[24,40] (PPP). An increase in reduced glutathione (GSH/GSSG) can result from enhanced glucose-6-phosphate delivery to the PPP for NADPH regeneration[40].
Unlabeled FBP was used in two of the three experiments with FBP, while [1-13C]FBP was used in the third, which also used unlabeled glucose. Thus [U-13C]glucose was used in 5 experiments, while unlabeled glucose was used in one. Because 13C satellite peaks distort the 1H spectrum in many places, only 6 metabolites were found where it was clear that there was no interference. In principle one could avoid overlapping satellites by applying a broadband 13C decoupling pulse during 1H acquisitions, (eliminating 13C magnetization by continuously flipping it). However, this presents practical problems that include heating the sample and possibly damaging the radiofrequency probe.) Thus it was not done. Using similar reasoning, the 1H spectrum from the [1-13C]FBP, while used to assess FBP metabolism, was not used for quantifying the 6 metabolites.
PCA EXTRACTION AND 1H/31P/13C SPECTROSCOPY
PCA extraction and NMR tube loading were also done as in earlier studies[43,44]. In brief, five frozen slices from each time point were pulverized in liquid nitrogen. The resulting fine powder was placed in 7 ml of 12% perchloric acid (PCA) at 4°C. Final extracts were lyophilized (BenchTop 2K lyophilizer, Virtis, Gardiner, NY, USA) and the weight of dry powder was measured. Each lyophilized sample was dissolved in 99.9% D2O and neutralized with NaOD or DCl. After 1H and 13C NMR spectroscopy was completed, EDTA was added to chelate and remove line-broadening cations. 1H, 31P, and 13C chemical shifts were referenced respectively to 3-trimethylsilyl-tetradeuterosodium propionate (TMSP), methylene diphosphonate (MDP), and TMSP. NMR studies of the PCA extracts were performed in the UCSF Magnetic Resonance Laboratory using a 14.1 Tesla (600-MHz) Varian UNITY spectrometer with an INOVA console and a customized, multinuclear Z-SPECT radiofrequency probe that was optimized for this project (3NG600−8, Nalorac Division of Varian, Martinez, CA, USA). As in recent experiments, basic one-pulse, 90° tip-angle RF sequences were used for obtaining 1H spectra at 599.92 MHz, 31P spectra at 242.86 MHz, and 13C spectra at 126 MHz. Proton spectra were composed from 64 transients, with the interpulse delay being 5 seconds. 13C acquisitions consisted of 4,096 complex data points, a spectral width of 30,000 Hz, and a WALTZ-16 scheme for broadband proton decoupling.
IN SITU DETECTION OF SUPEROXIDE ANIONS (O2−)
Hydroethidine (HEt, Invitrogen, D1168), which is selectively oxidized by the superoxide anion to red fluorescing ethidium bromide (Et), was prepared as a 1 mg/ml stock solution in dry, N2-sparged dimethylsulfoxide (DMSO), and stored in the dark at −80°C. Working solutions were made freshly using ACSF in a 1:1000 dilution, with final concentrations being 3.3 μM HEt and 0.1% DMSO. In experiments done with and without FBP treatments, slices were transferred from the original superfusion chamber into a parallel chamber containing the HEt working solution. These slices were exposed to HEt for 30 minutes that began prior to three sampling time points: t = −0.5 h, t = 0 h (no FBP only), and t = 4 h. After incubation for 30 minutes, slices were taken out and then fixed overnight in freshly prepared 4% formaldehyde at 4°C. Thereafter the fixed slices were washed twice in PBS, and then immediately embedded in optimum cutting temperature (OCT) compound (Sakura Fineteck), and cut into 10-μm-thick sections on a cryostat (Leica, CM1900, Solms, Germany). Sections were then studied with fluorescence microscopy, with excitation wavelengths being 510−550 nm, and emission wavelengths >580 nm for Et detection. Photomicrographs of the cerebral cortex were taken at 100× and 200× magnification with a Zeiss Axioskop Fluorescence Microscope. Image intensities were analyzed on a Macintosh computer using the public domain program ”NIH Image” (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
PRINCIPAL COMPONENT ANALYSIS
NMR intensities were analyzed with the SIMCA P+ v.11 multivariate analysis software by Umetrics, Inc. of Umea, Sweden. The SIMCA P+ approach begins mathematically by creating an N dimensional space, where N is the number of metabolites quantified per time point from NMR spectra, which is 9 in our case: 6 from a 1H spectrum and 3 from a 31P spectrum. The numerical values for each variable then undergo Pareto scaling, which establishes the same standard deviation in each variable's distribution. For each NMR spectrum the six scaled metabolite values define a single point in the six dimensional space. After each NMR spectra is represented by a point, a Principal Component Analysis in 6 dimensions finds the principal axis, which is the best straight line fit to all points, and then the orthogonal axis that defines the plane having maximum cluster separations when data points are projected onto it. After performing a Principal Component Analysis (PCA), the software, which considered the data points as independent (X) variables, uses a Partial Least Squares Discriminant Analysis (PLS-DA) to redefine all of the variables, defining some that are independent (X) and others that are dependent (Y). These are then projected onto a two dimensional plane, as in the PCA.
Acknowledgments
Financial support fo this research came from NIH Award R01 GM34767 and the UCSF Department of Anesthesia. Dr. Vladimir Basus provided substantial help with NMR issues during the initial period of the study. We are grateful to Dr. Mark S.J. Kelly and Davide Tonelli for technical advice and assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bhattacharya R, Tulsawani R. In vitro and in vivo evaluation of various carbonyl compounds against cyanide toxicity with particular reference to alpha-ketoglutaric acid. Drug Chem Toxicol. 2008;31:149–61. doi: 10.1080/01480540701688865. [DOI] [PubMed] [Google Scholar]
- 2.Cannella DM, Kapp JP, Munschauer FE, Markov AK, Shucard DW. Cerebral resuscitation with succinate and fructose-1, 6-diphosphate. Surg Neurol. 1989;31:177–82. doi: 10.1016/0090-3019(89)90113-4. [DOI] [PubMed] [Google Scholar]
- 3.Espanol MT, Litt L, Hasegawa K, Chang LH, Macdonald JM, Gregory G, James TL, Chan PH. Fructose-1,6-bisphosphate preserves adenosine triphosphate but not intracellular pH during hypoxia in respiring neonatal rat brain slices. Anesthesiology. 1998;88:461–72. doi: 10.1097/00000542-199802000-00025. [DOI] [PubMed] [Google Scholar]
- 4.Fahlman CS, Bickler PE, Sullivan B, Gregory GA. Activation of the neuroprotective ERK signaling pathway by fructose-1,6-bisphosphate during hypoxia involves intracellular Ca2+ and phospholipase C. Brain Res. 2002;958:43–51. doi: 10.1016/s0006-8993(02)03433-9. [DOI] [PubMed] [Google Scholar]
- 5.Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson's disease: evidence supporting it. Ann Neurol. 1992;32:804–12. doi: 10.1002/ana.410320616. [DOI] [PubMed] [Google Scholar]
- 6.Farias LA, Willis M, Gregory GA. Effects of fructose-1,6-diphosphate, glucose, and saline on cardiac resuscitation. Anesthesiology. 1986;65:595–601. doi: 10.1097/00000542-198612000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Fink MP. Ringer's ethyl pyruvate solution: a novel resuscitation fluid. Minerva Anestesiol. 2001;67:190–2. [PubMed] [Google Scholar]
- 8.Fink MP. Reactive oxygen species as mediators of organ dysfunction caused by sepsis, acute respiratory distress syndrome, or hemorrhagic shock: potential benefits of resuscitation with Ringer's ethyl pyruvate solution. Curr Opin Clin Nutr Metab Care. 2002;5:167–74. doi: 10.1097/00075197-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Fink MP. Ethyl pyruvate: a novel anti-inflammatory agent. Crit Care Med. 2003;31:S51–6. doi: 10.1097/00003246-200301001-00008. [DOI] [PubMed] [Google Scholar]
- 10.Fujii E, Kodama Y, Takahashi N, Roman C, Ferriero D, Gregory G, Parer JT. Fructose- 1,6-bisphosphate did not affect hippocampal neuronal damage caused by 10 min of complete umbilical cord occlusion in fetal sheep. Neurosci Lett. 2001;309:49–52. doi: 10.1016/s0304-3940(01)02026-2. [DOI] [PubMed] [Google Scholar]
- 11.Gobbel GT, Chan TY, Gregory GA, Chan PH. Response of cerebral endothelial cells to hypoxia: modification by fructose-1,6-bisphosphate but not glutamate receptor antagonists. Brain Res. 1994;653:23–30. doi: 10.1016/0006-8993(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 12.Gregory GA, Yu AC, Chan PH. Fructose-1,6-bisphosphate protects astrocytes from hypoxic damage. J Cereb Blood Flow Metab. 1989;9:29–34. doi: 10.1038/jcbfm.1989.4. [DOI] [PubMed] [Google Scholar]
- 13.Hardin CD, Roberts TM. Metabolism of exogenously applied fructose 1,6-bisphosphate in hypoxic vascular smooth muscle. Am J Physiol. 1994;267:H2325–32. doi: 10.1152/ajpheart.1994.267.6.H2325. [DOI] [PubMed] [Google Scholar]
- 14.Hardin CD, Roberts TM. Compartmentation of glucose and fructose 1,6-bisphosphate metabolism in vascular smooth muscle. Biochemistry. 1995;34:1323–31. doi: 10.1021/bi00004a027. [DOI] [PubMed] [Google Scholar]
- 15.Hirai K, Sugawara T, Chan PH, Basus VJ, James TL, Litt L. Cytochrome c associated apoptosis during ATP recovery after hypoxia in neonatal rat cerebrocortical slices. J Neurochem. 2002;83:309–19. doi: 10.1046/j.1471-4159.2002.01130.x. [DOI] [PubMed] [Google Scholar]
- 16.Juergens TM, Hardin CD. Fructose-1,6-bisphosphate as a metabolic substrate in hog ileum smooth muscle during hypoxia. Mol Cell Biochem. 1996;154:83–93. doi: 10.1007/BF00248465. [DOI] [PubMed] [Google Scholar]
- 17.Kaakinen T, Heikkinen J, Dahlbacka S, Alaoja H, Laurila P, Kiviluoma K, Salomaki T, Romsi P, Tuominen H, Biancari F, Lepola P, Nuutinen M, Juvonen T. Fructose-1,6-bisphosphate supports cerebral energy metabolism in pigs after ischemic brain injury caused by experimental particle embolization. Heart Surg Forum. 2006;9:E828–35. doi: 10.1532/HSF98.20061079. [DOI] [PubMed] [Google Scholar]
- 18.Kelleher JA, Chan PH, Chan TY, Gregory GA. Energy metabolism in hypoxic astrocytes: protective mechanism of fructose-1,6-bisphosphate. Neurochem Res. 1995;20:785–92. doi: 10.1007/BF00969690. [DOI] [PubMed] [Google Scholar]
- 19.Kelleher JA, Chan TY, Chan PH, Gregory GA. Protection of astrocytes by fructose 1,6-bisphosphate and citrate ameliorates neuronal injury under hypoxic conditions. Brain Res. 1996;726:167–73. [PubMed] [Google Scholar]
- 20.Kelleher JA, Gregory GA, Chan PH. Effect of fructose-1,6-bisphosphate on glutamate uptake and glutamine synthetase activity in hypoxic astrocyte cultures. Neurochem Res. 1994;19:209–15. doi: 10.1007/BF00966818. [DOI] [PubMed] [Google Scholar]
- 21.Kuluz JW, Gregory GA, Han Y, Dietrich WD, Schleien CL. Fructose-1,6-bisphosphate reduces infarct volume after reversible middle cerebral artery occlusion in rats. Stroke. 1993;24:1576–83. doi: 10.1161/01.str.24.10.1576. [DOI] [PubMed] [Google Scholar]
- 22.LeBlanc MH, Farias LA, Evans OB, Vig V, Smith EE, Markov AK. Fructose-1,6-bisphosphate, when given immediately before reoxygenation, or before injury, does not ameliorate hypoxic ischemic injury to the central nervous system in the newborn pig. Crit Care Med. 1991;19:75–83. doi: 10.1097/00003246-199101000-00018. [DOI] [PubMed] [Google Scholar]
- 23.LeBlanc MH, Farias LA, Markov AK, Evans OB, Smith B, Smith EE, Brown EG. Fructose-1,6-diphosphate, when given five minutes after injury, does not ameliorate hypoxic ischemic injury to the central nervous system in the newborn pig. Biol Neonate. 1991;59:98–108. doi: 10.1159/000243329. [DOI] [PubMed] [Google Scholar]
- 24.Lian XY, Khan FA, Stringer JL. Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci. 2007;27:12007–11. doi: 10.1523/JNEUROSCI.3163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litt L, Hirai K, Basus VJ, James TL. Temperature control of respiring rat brain slices during high field NMR spectroscopy. Brain Res Brain Res Protoc. 2003;10:191–8. doi: 10.1016/s1385-299x(02)00218-0. [DOI] [PubMed] [Google Scholar]
- 26.Litt L, Liu J, M S, S Y, G-Y Y, M K, TL J. Similar ATP protection but different metabolomic profiles during brain slice recovery from H2O2 stress with ethyl pyruvate and the nonmetabolizable antioxidant PBN under review. 2008 [Google Scholar]
- 27.Markov AK. Hemodynamics and metabolic effects of fructose 1−6 diphosphate in ischemia and shock--experimental and clinical observations. Ann Emerg Med. 1986;15:1470–7. doi: 10.1016/s0196-0644(86)80946-5. [DOI] [PubMed] [Google Scholar]
- 28.Markov AK, Warren ET, Cohly HH, Sauls DJ, Skelton TN. Influence of fructose-1,6-diphosphate on endotoxin-induced lung injuries in sheep. J Surg Res. 2007;138:45–50. doi: 10.1016/j.jss.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 29.Mazzio EA, Soliman KF. Cytoprotection of pyruvic acid and reduced beta-nicotinamide adenine dinucleotide against hydrogen peroxide toxicity in neuroblastoma cells. Neurochem Res. 2003;28:733–41. doi: 10.1023/a:1022813817743. [DOI] [PubMed] [Google Scholar]
- 30.Nakano-Okuda Y, Hasegawa K, Hirai K, Kanai-Ochiai R, Morimoto M, Sugimoto T. Effects of edaravone on N-methyl-D-aspartate (NMDA)-mediated cytochrome c release and apoptosis in neonatal rat cerebrocortical slices. Int J Dev Neurosci. 2006;24:349–56. doi: 10.1016/j.ijdevneu.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Park JY, Kim EJ, Kwon KJ, Jung YS, Moon CH, Lee SH, Baik EJ. Neuroprotection by fructose-1,6-bisphosphate involves ROS alterations via p38 MAPK/ERK. Brain Res. 2004;1026:295–301. doi: 10.1016/j.brainres.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 32.Song X, Wu B, Takata T, Wang X, Oizumi XS, Akisaki T, Yokono K, Sakurai T. Neuroprotective effect of D-fructose-1,6-bisphosphate against beta-amyloid induced neurotoxicity in rat hippocampal organotypic slice culture: involvement of PLC and MEK/ERK signaling pathways. Kobe J Med Sci. 2005;51:73–83. [PubMed] [Google Scholar]
- 33.Tabrizchi R. Edaravone Mitsubishi-Tokyo. Curr Opin Investig Drugs. 2000;1:347–54. [PubMed] [Google Scholar]
- 34.Tortosa A, Rivera R, Ambrosio S, Bartrons R, Ferrer I. Fructose-1,6-bisphosphate fails to ameliorate delayed neuronal death in the CA1 area after transient forebrain ischaemia in gerbils. Neuropharmacology. 1993;32:1367–71. doi: 10.1016/0028-3908(93)90032-x. [DOI] [PubMed] [Google Scholar]
- 35.Trimarchi GR, Arcadi FA, De Luca R, Imperatore C, Santoro G, Trimarchi F, Costa G. Neuroprotective activity of fructose-1,6-bisphosphate following transient forebrain ischemia in the Mongolian gerbil. Jpn J Pharmacol. 1993;62:215–22. doi: 10.1254/jjp.62.215. [DOI] [PubMed] [Google Scholar]
- 36.Varma SD, Devamanoharan PS, Ali AH. Formation of advanced glycation end (AGE) products in diabetes: prevention by pyruvate and alpha-keto glutarate. Mol Cell Biochem. 1997;171:23–8. doi: 10.1023/a:1006846501081. [DOI] [PubMed] [Google Scholar]
- 37.Varma SD, Devamanoharan PS, Ali AH. Prevention of intracellular oxidative stress to lens by pyruvate and its ester. Free Radic Res. 1998;28:131–5. doi: 10.3109/10715769809065799. [DOI] [PubMed] [Google Scholar]
- 38.Varma SD, Hegde KR, Kovtun S. Oxidative damage to lens in culture: reversibility by pyruvate and ethyl pyruvate. Ophthalmologica. 2006;220:52–7. doi: 10.1159/000089275. [DOI] [PubMed] [Google Scholar]
- 39.Vexler ZS, Wong A, Francisco C, Manabat C, Christen S, Tauber M, Ferriero DM, Gregory G. Fructose-1,6-bisphosphate preserves intracellular glutathione and protects cortical neurons against oxidative stress. Brain Res. 2003;960:90–8. doi: 10.1016/s0006-8993(02)03777-0. [DOI] [PubMed] [Google Scholar]
- 40.Winiarska K, Drozak J, Wegrzynowicz M, Jagielski AK, Bryla J. Relationship between gluconeogenesis and glutathione redox state in rabbit kidney-cortex tubules. Metabolism. 2003;52:739–46. doi: 10.1016/s0026-0495(03)00035-0. [DOI] [PubMed] [Google Scholar]
- 41.Xu K, Stringer JL. Pharmacokinetics of fructose-1,6-diphosphate after intraperitoneal and oral administration to adult rats. Pharmacol Res. 2008 doi: 10.1016/j.phrs.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ying W, Chen Y, Alano CC, Swanson RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab. 2002;22:774–9. doi: 10.1097/00004647-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Zeng J, Liu J, Yang GY, Kelly MJ, James TL, Litt L. Exogenous ethyl pyruvate versus pyruvate during metabolic recovery after oxidative stress in neonatal rat cerebrocortical slices. Anesthesiology. 2007;107:630–40. doi: 10.1097/01.anes.0000281898.01966.1e. [DOI] [PubMed] [Google Scholar]
- 44.Zeng J, Yang GY, Ying W, Kelly M, Hirai K, James TL, Swanson RA, Litt L. Pyruvate improves recovery after PARP-1-associated energy failure induced by oxidative stress in neonatal rat cerebrocortical slices. J Cereb Blood Flow Metab. 2007;27:304–15. doi: 10.1038/sj.jcbfm.9600335. [DOI] [PubMed] [Google Scholar]