

Summary
Protein-protein interaction studies in the S. cerevisiae ergosterol biosynthetic pathway suggest that enzymes in this pathway may act as an integrated multienzyme complex. The yeast sterol 3-ketoreductase (Erg27p) required for C-4 demethylation of sterols has previously been shown to also be required for the function of the upstream oxidosqualene cyclase/lanosterol synthase (Erg7p); thus, erg27 mutants accumulate oxidosqualenes as precursors rather than 3-ketosterones. In the present study, we have created various mutations in the ERG27 gene. These mutations include 5 C-terminal truncations, 6 internal deletions, and 32 point mutants of which 14 were obtained by site directed mutagenesis and 18 by random mutagenesis. We have characterized these ERG27 mutations by determining the following: Erg27 and Erg7 enzyme activities, presence of Erg27p as determined by western immunoblots, ability to grow on various sterol substrates and GC sterol profiles. Mutations of the predicted catalytic residues, Y202F and K206A, resulted in the endogenous accumulation of 3-ketosterones rather than oxidosqualenes suggesting retention of Erg7 enzyme activity. This novel phenotype demonstrated that the catalytic function of Erg27p can be separated from its Erg7p chaperone ability. Other erg27 mutations resulted in proteins that were present, as determined by western immunoblotting, but unable to interact with the Erg7 protein. We also classify Erg27p as belonging to the SDR (short chain dehydrogenase/reductase) family of enzymes and demonstrate the possibility of homo -or hetero-dimerization of the protein. This study provides new insights into the role of Erg27p in sterol biosynthesis.
Keywords: Saccharomyces cerevisiae, ergosterol, Erg7, Erg27, 3-ketoreductase, lanosterol synthase, protein-protein interactions
1. Introduction
ERG27 is one of three enzymes required for the complete demethylation of C-4 methyl groups in sterol biosynthesis. Upon disruption of the S. cerevisiae ERG27 gene, Gachotte et al. observed that not only was 3-ketoreductase activity lost but surprisingly 3-keto intermediates did not accumulate [1]. When the erg27 strain was grown on cholesterol- or ergosterol-supplemented media the endogenous sterol-related intermediates that accumulated were non-cyclic squalene epoxide and squalene diepoxide with little or no accumulation of lanosterol. The accumulation of squalene epoxides is also indicative of a mutation in ERG7 strains [1]. Further analysis by Mo et al. confirmed that erg27 strains were devoid of both Erg27 and Erg7 (lanosterol synthase) enzyme activities [2]. However, both Erg27 and Erg7 enzyme activities could be restored by reintroducing a plasmid containing ERG27 to an erg27 strain. While Milla et al. demonstrated that Erg7 enzyme activity resides mainly in lipid particles [3], Mo et al. demonstrated that Erg27p and Erg7p co-immunoprecipitate and also interact in a yeast membrane two hybrid system [2, 4].
The yeast ERG27 gene belongs to the short chain dehydrogenase/reductase (SDR) family with approximately 3000 primary structures annotated in various sequence databases [5]. Pair-wise combinations of various SDR enzymes indicate that sequence identity is typically only 15–30% but among 30 three-dimensional structures deposited in these databases all possess a highly similar α/β folding pattern with a critical β-sheet typical of a Rossmann fold. The mammalian 17β-hydroxysteroid dehydrogenase type 7 (17β-HSD type 7) is the orthologue of the yeast ERG27, in that it can convert the cholesterol precursor, zymosterone to zymosterol and complements a yeast erg27 mutation [6].
Three-dimensional structures and mutagenesis of SDR enzymes suggest a Rossmann-fold, N- or C- terminal transmembrane domains, and a number of conserved sites. An N-terminal TGX3GXG sequence occurs adjacent to the NADPH binding region; the active site consists of S-Y-K residues where S stabilizes the substrate, Y serves as the catalytic base, and K forms hydrogen bonds with NADPH to promote proton transfer. Other conserved SDR sites are N (also present at the active site which forms a proton relay involving the 2′ OH of the ribose), the side chain of the K residue, and a H20 molecule bound to the carbonyl of N [5, 7]. Further, the functional units of SDRs are with few exceptions either homodimers or homotetramers [8]. Using a fungal 17β-hydroxsteroid dehydrogenase from C. lunatus that is an SDR reductase, Kristan et al. [9] demonstrated that R129 and H111 from the αE-helices interact with D121, E117, and D187 residues from the αE and αF helices of the neighboring subunit and changes R129D and H111L rendered the SDR, 17β-HSDc1, unable to dimerize [9]. While these residues are not conserved in Erg27p, similar residues are likely to be involved in Erg27p multimerization. In the present study, the interaction between Erg7p and Erg27p was explored by determining the effect of various mutations in the ERG27 gene on both the catalytic activity of Erg27p and its possible chaperone-like action toward Erg7p.
2. Materials and Methods
2.1 Strains and growth conditions
The ERG27 gene was deleted in a wild type yeast strain, SCY876 (MATα, upc2-1 hap1Ty ura3 his3 leu2 trp1). The plasmid pRS305 was used as the template for amplification of the LEU2 gene. Forward and reverse PCR primers contained approximately 60 bases of upstream and downstream ERG27 sequence and 20 bases of the leucine gene (listed in supplementary table 1). ERG27 knockouts were confirmed on the basis of sterol profiles as well as PCR reactions using diagnostic primer sequences upstream and downstream of the ERG27 deleted sequence. The ERG7 gene was also disrupted in the SCY876 wild type strain using primers that amplified the histidine selectable marker. The plasmid p423ADH [10] was used as a template for amplification of the HIS3 gene (supplementary table 2). Transformants were grown on complete synthetic medium (CSM) containing 0.67% yeast nitrogen base, 2% glucose, and 0.8 g/l CSM-LEU (amino acid and nitrogen base supplements; Bio101, Vista, CA) or CSM-HIS to select for ERG27 and ERG7 deletants, respectively; in both cases, 20 µg/ml ergosterol was added to ensure growth of sterol auxotrophs.
2.2 Construction of ERG27 deletion plasmids
Overlapping C-terminal deletions of ERG27 were generated using a previously described ERG27 containing plasmid ERG27-HA p423ADH [11] as a template. PCR products were generated using a single forward ERG27 primer containing DNA sequence for an N-terminal HA tag and 21 nucleotides corresponding to the beginning of the ERG27 gene in conjunction with reverse primers designed to amplify sequentially shorter regions of the ERG27 C-terminus (primers listed in supplementary table 3). PCR products were digested with EcoRI/SalI and ligated into the p423ADH vector. The QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) was used for constructing deletions within the ERG27 gene. Diverging 5′ phosphorylated primers (Invitrogen) adjacent to the region to be deleted and amplifying in opposite directions were employed to create a linear DNA fragment that when subsequently ligated resulted in a plasmid with the selected deletion (supplementary table 4). An ERG27-containing plasmid (p426GPD) with a C-terminal ERG27-HA was used as the template for the creation of all deletions. Each PCR reaction had a final volume of 50 µl and contained: 5.0 µl 10X reaction buffer, 3.0 µl QuickSolution, 100–200 ng dsDNA template, 125 ng forward primer, 125 ng reverse primer 0.25 mM dATP, 0.25 mM dTTP, 0.25 mM dCTP, 0.25 mM dGTP, and 2.5–3.75U of PfuUltra HF DNA polymerase. The PCR reaction products were digested with Dpn1 at 37°C for 1–2 hrs and separated on a 1% (w/v) agarose gel to purify linear DNA fragments. Blunt end ligation of the linear DNA reconstituted a functional plasmid that was transformed into XL10 Gold E. coli Ultracompetent cells.
All of the resulting plasmids were sequenced to validate each deletion sequence and then used to transform the erg27 deletant yeast strain. Transformants containing either ERG27 C-terminal overlapping deletions or deletions internal to ERG27 were grown on CSM-HIS or CSM-URA dropout media with 20 µg/ml of ergosterol.
2.3 Random and site-directed mutagenesis
Random mutagenesis was used to generate point mutations in the ERG27 gene using the gap-repair protocol of Muhlrad et al. [12]. PCR products containing errors due to Taq DNA polymerase base misincorporation into ERG27 amplicons were co-transformed with a p426ADH vector linearized with EcoRI/SalI such that homologous recombination between vector and PCR product occurred in vivo to reconstitute a functional plasmid (primers listed in supplementary table 5). Yeast cells were transformed and plated on CSM-URA dropout media with 20 µg/ml ergosterol. Sterol auxotrophs were screened by differential growth on CSM-URA without ergosterol. Site directed mutagenesis was performed using the QuickChange II XL Site-Directed Mutagenesis Kit. Primers were constructed such that 12–15 wildtype base pairs bordered each nucleotide change (listed in supplementary table 6). A p426ADH vector with an ERG27 insert was used as the template for all reactions. Each PCR reaction was carried out in a final volume of 50 µl as described above for internal deletions. Upon termination of the PCR reaction, DpnI (1 µl) was added and the reaction mixture was incubated at 37°C for 2 h to ensure complete digestion of the parent vector. The resulting undigested DNA was transformed into XL10 Gold E. coli Ultracompetent cells (Stratagene). All mutations generated were confirmed by DNA sequencing and mutagenized plasmids were transformed into an ERG27 knockout strain. Transformants were grown on CSM-URA dropout media with 20 µg/ml ergosterol.
2.4 Erg7 and Erg27 enzyme assays
The enzyme assay of Erg7p has been previously described [3]. The 3-ketoreductase (Erg27p) enzyme assay was adapted from a protocol by Billheimer et al. with modifications [13]. Each reaction contained 0.1 M potassium phosphate buffer (pH 7.4), 0.2 µmol NADPH and an NADPH generating system of 0.2 µmol NADP+, 5.8 µmol DL-isocitrate, and 0.375U of isocitrate dehydrogenase. 50 nmol of 5α-cholestan-3-one in Tween 80 (1 mg/ml) was used as the substrate. The final volume of the reaction was 0.6 ml. The reaction was initiated by introducing 1.0 mg of protein from a 30,000 × g microsomal fraction followed by a 2 h incubation at 37°C. The reaction was stopped by the addition of 2.5 ml ethanolic KOH (25% w/v). 100 nmol of dihydrolanosterol was added as an internal standard and samples were saponified for 30 min in an 85°C water bath. Lipids were extracted into 3 ml n-heptane, dried under nitrogen, and resuspended in a final volume of 0.2 ml n-heptane. Samples were then analyzed by GC and the formation of 5α-cholestan-3β-ol was quantified. The reaction was found to be linear with time (up to 2 h) and protein (up to 1.25 mg). The amount of product formation was determined by comparison with a cholestan-3-ol standard curve.
2.5 Western blots of Erg27
Yeast cells were usually grown to late log/early stationary phase (OD 0.8–1.2) for all protein preparations. Cells were harvested at 3,000 × g at 4°C, washed once with ice cold H2O, and kept on ice. Cells were broken with an equal volume of acid washed glass beads and breaking buffer (20 mM sodium phosphate buffer, 15 mM NaCl, 1 mM EGTA, 1 mM β-mercaptoethanol, 0.5 mM phenyl methyl sulfonyl fluoride [PMSF], 17 µg/ml Aprotinin, 1.4 µg/ml pepstatin A, 0.5 µg/ml leupeptin, 0.5 mM DTT, 0.35µg/ml Bestatin) was added at 2.0 ml/g wet weight of cells. The cells were then broken at 4°C with eight vortex cycles (1 min vortex: 1 min ice). Cellular debris and glass beads were pelleted with a 10,000 × g clearing spin and the supernatant was then subjected to a 30,000 × g spin. Microsomal pellets were resuspended in breaking buffer with protease inhibitors listed above. Twenty µg of protein was suspended in 1 X SSB (0.0625 M Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.001% bromophenol blue, 50 mM DTT), and boiled for 3 min. Samples were stored at −80°C or used immediately for SDS-PAGE analysis. Protein samples were separated by a 10 % SDS-polyacrylamide gel and transferred to a nitrocellulose membrane for immunological detection. The membrane was then blocked with 5% milk powder (w/v) in 1X PBS (8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4 and 0.24 g/L KH2PO4 [pH 7.4]) with 0.05% Tween-20 (PBST) for 1 h. The membrane was then incubated for 2 h with either HA-Probe (Y11) HRP antibody (Santa Cruz Biotechnology) (1:500) or anti-Erg27 antibody (1:300) both in 5 % milk powder in PBST, washed 3X with 5 % milk powder in PBST and 3X with PBS followed by incubation with goat anti-rabbit IgG-HRP (Santa Cruz biotechnology, 1:5000) for 1 h. Unbound antibody was washed 3 X with 5% milk powder in PBST and 3X with PBS, and Chemiluminescent (BioRad) substrates were added for visualization of protein bands. Anti-Erg27p was prepared from a synthesized 23 amino acid Erg27p epitope (YEGSKRLVDLLHLATYKDLKKLG) (Proteintech Group).
2.6 Erg27p cross-linking
A plasmid containing the wildtype ERG27 gene fused to the C-terminal HA tag was used in all cross-linking experiments. Proteins were cross-linked using the membrane-permeable, homobifunctinal cross-linker EGS [Ethylene glycolbis(succinimidylsuccinate)] in 50 µl of PBS (20 mM sodium phosphate, 0.15M NaCl pH 7.5). Cell homogenates (6,000 × g) or resuspended microsomal pellets (40, 000 × g) containing 125 µg of protein were cross-linked in microfuge tubes containing various final concentrations of EGS (0 mM, 0.6mM, 1.2 mM and 2.5 mM). The reaction mixtures were incubated for 30 min at room temperature, quenched by the addition of 1M Tris, pH 7.5 to a final concentration of 50 mM, and then incubated for 15 min. Immediately, 4x SSB was added to give a final concentration of 1xSSB and boiled for 5 min. Western-blotting was carried out as above using anti-HA and monomers and cross-linked high molecular weight bands were identified.
2.7 Other
Gas chromatographic-mass spectrometric (GC-MS) analyses to determine sterol profiles [14] and spot plate growth assays were carried out as previously described [15]. Quantitation of western blots was carried out using NIH Image J software.
3. Results
3.1 Sterol accumulation in ERG27 and erg7 strains
Mutations in ERG27 and ERG7 accumulate the same sterol intermediates, squalene epoxide and squalene diepoxide (fig. 1A) whereas the wildtype strain accumulates primarily ergosterol and smaller amounts of precursors such as zymosterol, fecosterol, episterol and lanosterol (data not shown) [1]. The inability to synthesize lanosterol, the first sterol molecule in the pathway, from squalene epoxide results in a growth requirement for sterol [16]. Ergosterol or cholesterol can serve as a sterol supplement. However, a feature distinguishing erg27 from erg7 strains is that the erg7 strain can also grow on the 3-keto substrate, ergosta-7,22, diene-3-one, indicating that it retains Erg27 3-ketoreductase activity in the absence of Erg7p (fig.1B). Thus the loss of the ERG27 encoding 3-ketoreductase gene resulting in a concomitant loss of Erg7 enzyme activity is not reciprocal since erg7 strains retain 3-ketoreductase activity. An investigation involving reciprocity between Erg7p and Erg27p using radiolabeled lanosterol and different 3-ketosterone substrates is in progress (S. Taramino, unpublished).
Fig. 1.
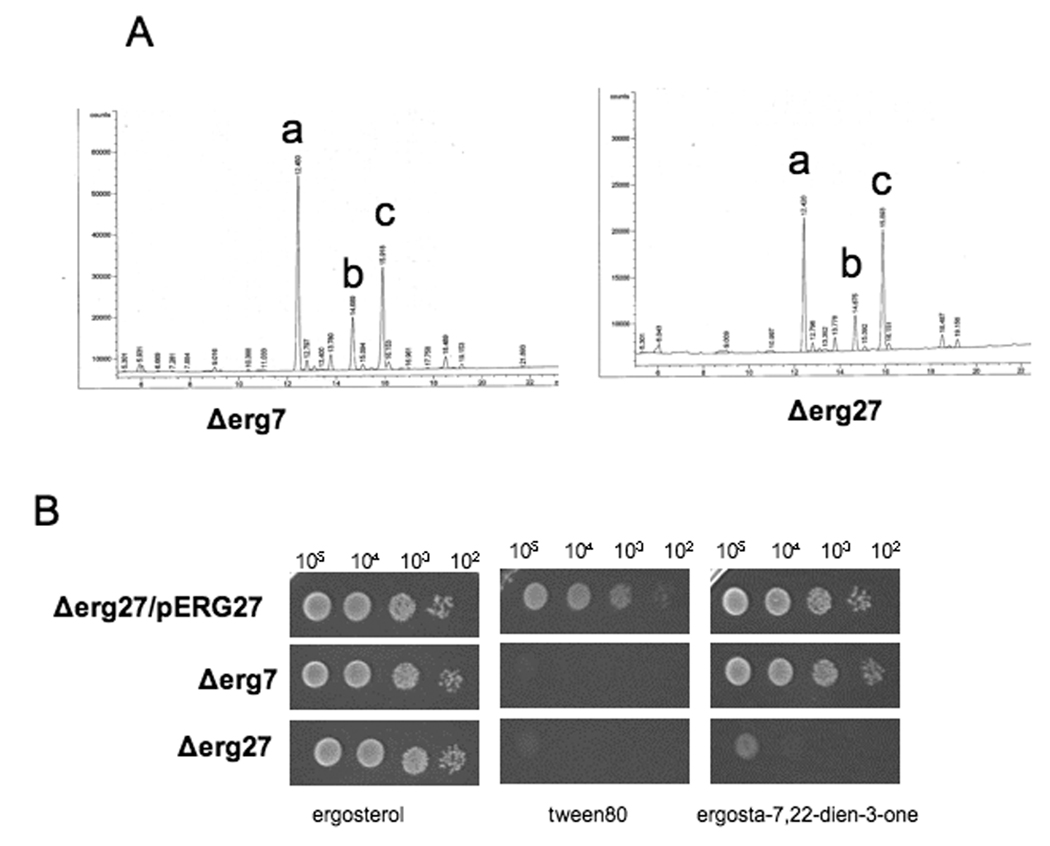
Fig. 1A. GC analyses demonstrates that the sterol profiles of Δerg7 and Δerg27 are the same and the sterol precursors, squalene epoxide (a) and squalene diepoxide (b) accumulate. Cholesterol (c) added to the growth medium is required for growth of Δerg7 and Δerg27 sterol auxotrophs.
Fig. 1B. Spot plate analysis of deletion strain erg27 transformed with an ERG27 plasmid (Δerg27/pERG27), or deletion strains erg7, and erg27. Strains were grown on CSM-Ura media supplemented with 20 µg/ml indicated sterol (ergosterol or ergosta-7, 22,-3-one) or tween 80 (without sterol). Serial dilutions of 105-102 cells were spotted onto indicated plates.
3.2 ERG27 deletion analyses
Two types of deletions were generated to discern whether all or parts of the Erg27 protein were required for Erg7 enzyme activity. The first set of deletions involved overlapping truncations at the ERG27 C-terminus. Wildtype and deletion constructs were transformed into an erg27 strain. Only the full-length Erg27 protein containing 347 amino acids resulted in a wildtype sterol profile, an Erg7 enzyme specific activity of 0.45 nmol/h/mg and an Erg27p that could be detected by western blots (using anti-HA which targets the N-terminus HA epitope). Overlapping ERG27 plasmids containing 282, 211, 144, or 74 N-terminal amino acids resulted in mutant sterol profiles (only squalene epoxides) and no detectable Erg27 protein (data not shown). These truncated ERG27 constructs behaved as erg27 auxotrophs. It is likely that these mutants gave rise to unstable proteins that were proteolytically degraded.
The second set of deletions encompasses internal deleted regions of Erg27p (indicated in fig. 2A as numbered boxes). Fig. 2A is an alignment plot showing Erg27p in four different yeast species (S. cerevisiae, C. glabrata, K. lactis and C. albicans), mouse, and humans. The amino acid regions encompassed by regions 1, 2, and 5 represent amino acid sequences conserved among the four fungi; amino acid regions 3 and 6 are sequences conserved in fungi and mammals; region 4 is also fairly conserved and encompasses the active site. However, deletion of any of these six regions resulted in erg27 auxotrophy, the accumulation of squalene epoxides, loss of oxidosqualene cyclase (erg7p) activity and an inability to grow on 3-keto sterol intermediates. However, unlike the previous set of deletions, all internal deletions with the exception of deletion 4 resulted in an Erg27 protein as detected by western blots (fig. 2B).
Fig. 2.

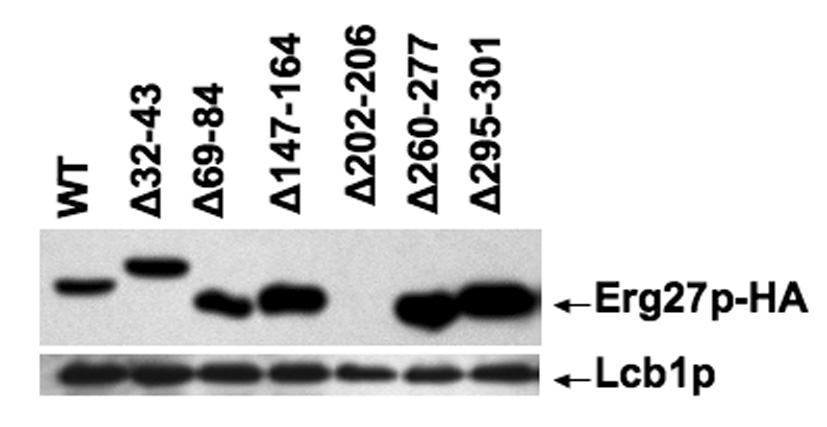
Fig. 2A. 3-ketoreductase sequence alignment with Erg27p internal deletions indicated as boxed areas. Homology plot of from Saccharomyces cerevisiae (top), Candida glabrata, Kluyvermyces lactis, Candida albicans, Mus musculus, Homo sapiens (bottom). Regions selected for internal deletions are outlined in red. Region 1 deleted amino acids 32–43 (Δ32–43); region 2, Δ69–84; region 3, Δ147–164; region 4, Δ202–206; region 5, Δ260–277; region 6, Δ295–301.
Fig. 2B. Western immunoblot from wildtype (wt) Erg27p and internal deletion strains. Erg27p was detected using HA-Probe (Y11) HRP antibody (1:500). Lcb1p, involved in sphingolipid biosynthesis, was used as a loading control and detected with anti-Lcb1p (1:500),
3.3 Random and site directed mutagenesis analyses of ERG27 mutants
In an attempt to isolate ERG27 mutants that would retain Erg27 activity but not Erg7 activity and reciprocally mutants that would retain Erg7activity and not Erg27 activity, we performed both random and site-directed mutagenesis. We also anticipated obtaining mutants that would retain both activities (wildtype or nearly-wildtype) and those that would retain neither activity. The random mutagenesis protocol generated 520 transformant colonies that were screened on media lacking ergosterol; 26 failed to grow and 35 grew poorly resulting in 61 possible ERG27 mutant genes. DNA sequencing of each ERG27 containing plasmid resulted in 19 plasmids containing alterations in the ERG27 gene. Plasmids were again transformed back into an erg27 deletion strain to insure repeatability of the phenotype. Ten plasmids had ERG27 alterations that resulted in a single amino acid substitution, seven resulted in two amino acid substitutions, one plasmid with three substitutions, and one with four amino acid substitutions. Mutant plasmids are listed in table 1 as belonging to group I, high 3-ketosterone accumulation as determined by GC analyses; group II, high squalene epoxide accumulation; and group III, intermediate sterol accumulation in which sterol profiles were somewhere intermediate between wildtype and groups I and II. Group I mutants are novel in that they represent mutations that retain Erg7 activity but lack Erg27 activity. Group IV mutants are similar to wildtype. For site-directed mutagenesis, all amino acid changes were substitutions to alanine with the exception of the active site amino acids, tyrosine (Y202) that was changed to F to remove the catalytic hydroxyl group, and asparagine (N153) changed to L to minimize steric effects and eliminate the polar carbonyl group. Of the 14 mutants obtained by site-directed mutagenesis, four were mutations at the active site, three involved substitutions at the NADPH binding site and seven represented substitutions at sites conserved in short chain dehydrogenases/ reductases. Of the latter group, two mutants had two substitutions in sequential amino acids. These mutants are also listed in table 1 according to category. Whereas the erg27 deletion strain transformed with a wildtype ERG27 plasmid accumulates ergosterol and ergosterol precursors (zymosterol, fecosterol, episterol, ergosta-5,7,22,24(28)-tetraene-ol, and lanosterol) when grown in cholesterol-supplemented medium, both erg27 and erg7 deletion strains (transformed with control vector) accumulate only squalene epoxides (table 2). Group I mutants accumulate 3-ketosterones as these erg27 alleles lack Erg27 but retain Erg7 enzyme activities. In this group, 4-methylcholesta-8,24-diene-3-one, 4,4 dimethylcholesta-8,24-diene-3-one and ergosta-7,24(28)-diene-3-one accumulate. This group contains mutations in the active site-Y202F and K206A as well as a mutation in the NADPH binding site-G10A and additionally two other mutations in a highly conserved site R40A (obtained by site-directed mutagenesis) and the double mutation, S183P/F235S. Group II mutants accumulate high levels of squalene epoxides with sterol profiles very similar to the erg27 deletion mutant. These mutants include L23P, L212P, and S180C. Group III mutants are interesting in that they represent a mixture of sterol profiles in which some mutants will accumulate both squalene epoxides and 3-keto sterones. An example is the P166L/S179L ERG27 allele obtained by random mutagenesis in which 27% squalene epoxides and 16% 3-keto sterones accumulate. Fig. 3 correlates the growth responses of each category with regard to growth with and without various sterols. Whereas group I mutants generally appear to be unable to grow without sterol supplement, the mutant bearing the G10A mutation (which affects the NADPH binding site) grows without sterol supplementation; even though the G10A mutant accumulates large amounts of 3-keto sterones, it also accumulates ergosterol and ergosterol precursors.
Table 1.
ERG27 mutations obtained as a result of site-directed mutagenesis (SDM) or random mutagenesis (RM) correlated with sterol profiles and predicted site of lesion
| Type of sterol profile | SDM or RM | Predicted site of lesion in Erg27p | |
|---|---|---|---|
| Group I | High 3-keto sterone accumulation | ||
| Y202F | SDM | Active site | |
| K206A | SDM | Active site | |
| N153L | SDM | Active site | |
| R40A | SDM | SDR conserved site | |
| G10A | SDM | NADPH binding site | |
| D75G/N153D | RM | ||
| S183P/F235S | RM | ||
| S205P/S237G | RM | ||
| Group II | High squalene epoxide accumulation | ||
| L212P | RM | ||
| S180C | RM | ||
| L23P | RM | ||
| S205P/S237G | RM | ||
| L42P/D68G/A152T | RM | ||
| Group III | Intermediate sterol accumulation | ||
| Q311*1 | RM | ||
| P232A | SDM | SDR conserved site | |
| R32K/S239P | RM | ||
| I135V/L148P | RM | ||
| P166L/S179L | RM | ||
| Q46R/D54G/N146V/K206E | RM | ||
| Wildtype-like sterol accumulation | |||
| Group IV | T9A | ||
| S13A | SDM | NADPH binding site | |
| D75A | SDM | NADPH binding site | |
| N84S | SDM | SDR conserved site | |
| I111M | RM | ||
| V140A | RM | ||
| F150L | RM | ||
| M182V | RM | ||
| S189A | RM | ||
| D314A | SDM | Active site | |
| K337E | SDM | SDR conserved site | |
| T26I/F160L | RM | ||
| F246A/F247A | RM | ||
| SDM | SDR conserved site |
Q311* results from a stop codon at amino acid 311. Slashes indicate multiple amino acid substitutions
Table 2.
GC profiles of erg27 strains transformed with plasmids containing wildtype ERG27 or various ERG27 alleles derived from mutagenesis
| Accumulating Sterols | |||||
|---|---|---|---|---|---|
| Strain | Squalene epoxides | 3-ketosterones | Ergosterol | Other sterol intermediates | Cholesterol |
| Controls | |||||
| ERG27 (wt) | - | 4.8 | 33.1 | 49.1 | 12.9 |
| erg27 | 75.4 | - | - | - | 24.6 |
| erg71 | 62.3 | - | - | - | 37.7 |
| Group I | |||||
| Y202F | 3.0 | 38.3 | 4.5 | 14.9 | 39.2 |
| K206A | - | 27.7 | 10.9 | 37.3 | 24.1 |
| R40A | - | 36.4 | 8.2 | 30.4 | 25.1 |
| S183P/F235S | 4.9 | 33.6 | 1.8 | 31.5 | 28.1 |
| G10A | - | 21.8 | 21.5 | 35.6 | 21.1 |
| Group II | |||||
| L212P | 57.2 | - | - | - | 42.8 |
| S180C | 73.0 | - | - | - | 27.0 |
| L23P | 63.6 | - | - | - | 36.4 |
| Group III | |||||
| P166L/S179L | 27.1 | 15.5 | 1.8 | 22.1 | 33.5 |
| Q46R/D54G/N146V/K206E | 8.8 | 17.2 | 10.2 | 25.3 | 38.5 |
| Q311* | 14.3 | 4.0 | 14.7 | 40.6 | 26.5 |
erg7 was not transformed and its sterol profile is listed for comparison.
ERG27 and erg27 controls are erg27 strains containing a wildtype ERG27 insert or the empty vector, respectively.
Group I–III strains are erg27 strains containing plasmids transformed with various ERG27 mutated alleles.
Numbers represent percent of total sterol. Other sterol intermediates are ergosterol precursors that accumulate in wildtype strains and are listed in text.
Fig. 3.
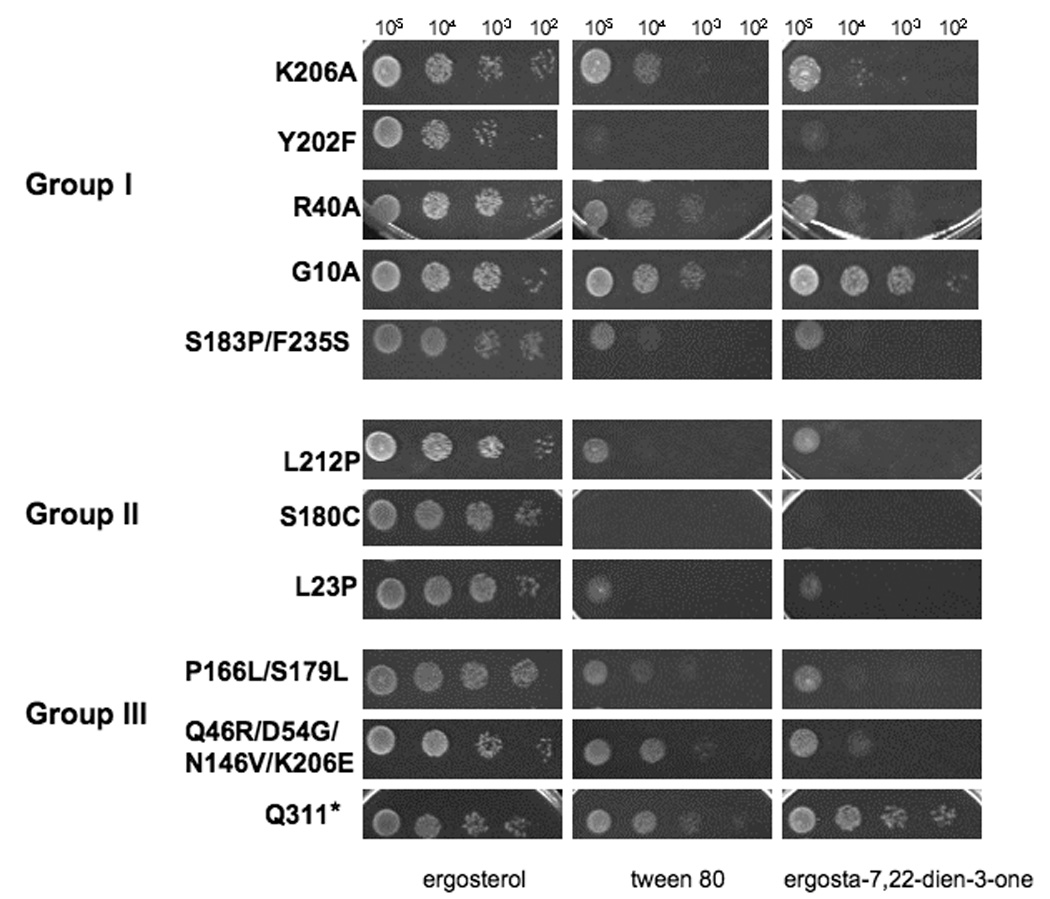
Spot plate assays of ERG27, erg27, and various ERG27 mutated alleles (Groups I–III) grown on medium containing ergosterol, tween 80 (no sterol supplement) and ergosta-7,22-diene-3-one. Serial dilutions of 105-102 cells were spotted onto indicated plates. Growth responses of erg27 and erg27/pERG27 controls are given in fig.1B
3.4 Erg27 and Erg7 enzyme activities and western immunoblots
Table 3 is a compilation of Erg27 and Erg7 enzyme assays together with western immunoblots of the Erg27 protein. Group I mutants which accumulate high levels of 3-ketosterones show marked reductions in Erg27 activity and near wildtype levels of Erg7 activity. Group II mutants generally demonstrate marked decreases in both enzyme activities. Interestingly, all mutants with the exception of the control erg27 deletion strain, L23P, L212P, and Q311*(stop codon at amino acid 311), produce Erg27 protein as demonstrated by western immunoblots. The small amounts of activity in erg27 deletion strains may result from other 3-ketoreductase activities such as the 3-ketoreductase involved in the fatty acid elongase system required for the synthesis of very long-chain fatty acids [17].
Table 3.
Erg27 and Erg7 enzyme assays and Erg27p western immunoblots derived from ERG27, erg27 and plasmids containing group I,II,III ERG27 alleles.
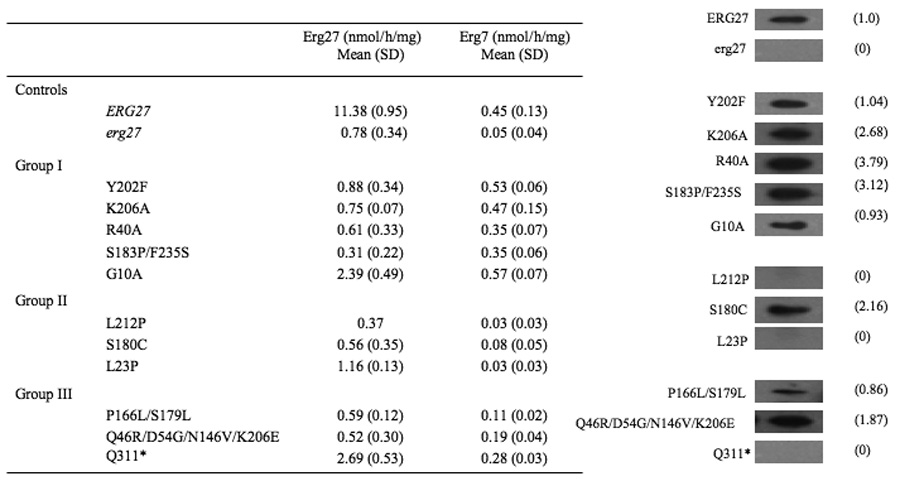 |
All Erg27 and Erg7 specific activities are averages of three independent experiments with the exception of L212P which is an average of two experiments; numbers in parenthesis for specific activities are standard deviations. Numbers in parenthesis adjacent to western immunoblots indicate amount of Erg27p as quantified using NIH Image J software.
3.5 Cross-linking of the Erg27 enzyme
In an attempt to demonstrate whether Erg27p might functionally exist as a dimer or higher order multimer, total homogenate and 32,000 × g resuspended microsomal fractions were cross-linked with the homobifunctinal cross-linker EGS. Fig. 4 demonstrates that as increasing concentrations of EGS are added, the 33–34 kDa monomer (as detected with HA antibody) disappears and a higher molecular weight dimer (which migrates at twice the size of the monomer) appears suggesting that Erg27p may exist as a dimer consistent with other SDR proteins that are dimers or tetramers. Less likely is the formation of an Erg27/Erg7 heterodimer in which the expected mass would have a molecular weight exceeding 115 kDa.
Fig. 4.
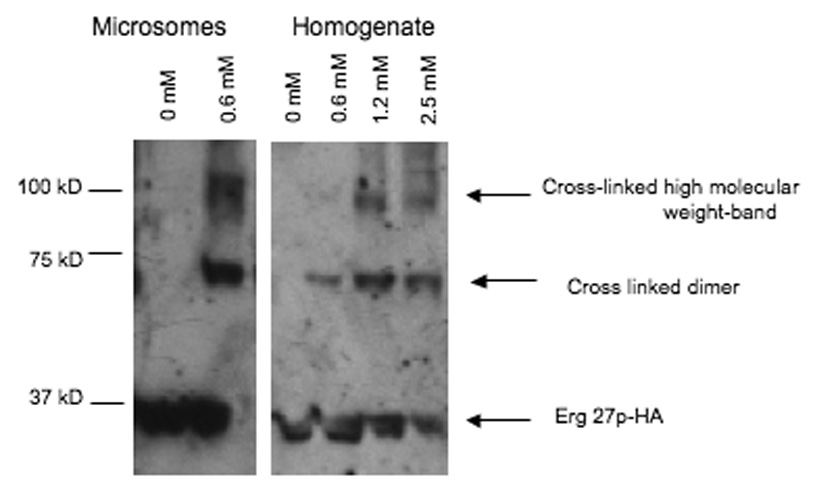
Erg27p from resuspended microsomal pellets (40, 000 × g, lanes 1–2) or cell homogenates (6000 × g, lanes 3–6) migrates as a dimer and even higher molecular weight band after treatment with the EGS cross-linker. The concentrations of cross-linker are: lane 1, 0. 0mM; lane 2, 0.6mM; lane 3, 0.0mM; lane 4, 0.6mM, lane 5, 1.2mM; lane 6, 2.5mM. Erg27p was detected using HA-Probe (Y11) HRP antibody (1:500).
3.6 Erg27 enzyme and lipid particles
The Erg7 enzyme has been demonstrated to localize primarily to lipid particles [3, 18, 19]. We previously demonstrated that Erg7p and Erg27p interact by co-immunoprecipitation and two-hybrid analyses in the ER [4, 11]. Our original hypothesis was that Erg27p escorted Erg7p to lipid particles where it was primarily localized. We thus tested the hypothesis that if Erg7p were retained in the ER then Erg27p would not be required. A yeast strain, H1246, devoid of lipid particles due to the deletion of four genes required for sterol and triacylglycerol formation (ARE1, ARE2, DGA1, and LRO1) was examined. H1246 has no discernible lipid particles as determined by fluorescence microscopy [20]. Sorger et al. demonstrated that in the quadruple deletion strain Erg7p now localized to the 30,000 and 40,000 × g microsomal fractions [21]. We deleted ERG27 in an H1246 background and demonstrated that H1246 erg27 had an average Erg7 specific activity of 0.05 nmol/h/mg protein, whereas the non-disrupted H1246 ERG27 had an average specific activity of 0.99 nmol/h/mg protein. This suggests that even in the absence of lipid particles when Erg7p localizes to the ER, Erg27p is still required for Erg7 enzyme activity.
4. Discussion
The observation that Erg27p, an enzyme required for C-4 demethylation, is required for Erg7p activity [2] suggests a possible regulatory mechanism whereby loss of a downstream enzyme results in the elimination of all sterol synthesis. Hitchcock et al. [22] has suggested that the Erg27 enzyme is a candidate for ubiquitination. If Erg27p is indeed ubiquitinated and degraded, a chain reaction would occur in which Erg7p would become unstable and shut down all sterol synthesis. Other possibilities exist in that Erg27p is merely required for the proper folding and conformation of the lanosterol synthase (Erg7p) enzyme. It may be that multimerization of Erg27p is required for Erg7p stability. Mo et al. have demonstrated by two-hybrid analysis that these two enzymes interact [4]. The observation that erg27 mutants in Candida albicans [23] also accumulate squalene epoxides suggest that this interaction may be evolutionarily conserved, at least in fungal pathways.
While Erg27p is required for Erg7p activity, it is clear that erg7 mutants still retain the 3-ketoreductase activity (Erg27) since erg7 mutants can grow on 3-ketosterone intermediates and convert these to the alcohol (data not shown). In this study we have succeeded in separating the two activities by obtaining ERG27 mutants that retain Erg7 activity but not Erg27 activity. These mutants (Y202F and K206A) are likely at the active site of Erg27p which we now demonstrate are critical amino acid residues for reducing the 3-keto group to the 3-hydroxyl. Both mutations reduced Erg27 activity by more than 93%. We additionally demonstrated that only the G10A conversion that lies in a putative NADPH binding region had a dramatic effect in reducing Erg27 catalytic activity by 79%. Two other mutant strains also resulted in significant accumulation of 3-keto sterones (loss of Erg27 activity) but still significant Erg7 activity. R40 is a highly conserved amino acid residue and is present in all the above cited four fungal species as well as in mouse and human. Site-directed mutagenesis of R40 to A resulted in a phenotype similar to Y202F as did the double mutant, obtained by random mutagenesis-S183P/F235S. However, while we were able to generate mutants that had Erg7 but not Erg27 activity, we were not able to generate the reciprocal type (Erg27 activity but no Erg7 activity). It is possible that these mutants can not be obtained. The internal deletion strains that we created were informative and basically behaved as group II mutants accumulating squalene epoxides. Interestingly, with the exception of the deletion 4 which eliminated the active site, all internal deletions resulted in the accumulation of the Erg27 protein as seen in western blots. Thus in our study, group II mutations in which high levels of squalene epoxides accumulate can result from the complete loss or retention of the Erg27 protein. Finally, the Q311*(stop) mutation was particularly interesting as no protein could be observed by western immunoblots but a small amount of ergosterol was nevertheless present as was Erg7 and Erg27 enzyme activities. We suggest that an unstable Erg27 protein was degraded during our extraction procedure. A similar situation was observed in the C-terminal truncations where no protein could be observed by western blot analysis but this time the cells were unable to grow without sterol. These observations suggest that the C-terminus of Erg27p may be important for enzyme stability.
Supplementary Material
Acknowledgements
This investigation was supported by a National Institutes of Health Grant GM62104 to M.B and by the University of Turin (Italy) to G.B.
Abbreviations
- CSM
complete synthetic medium
- EGS
ethylene glycolbis(succinimidylsuccinate) cross linker
- HA
peptide from human hemagglutinin
- SDR
short-chain dehydrogenase/reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gachotte D, Sen SE, Eckstein J, Barbuch R, Krieger M, Ray BD, Bard M. Characterization of the Saccharomyces cerevisiae ERG27 gene encoding the 3-keto reductase involved in C-4 sterol demethylation. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12655–12660. doi: 10.1073/pnas.96.22.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mo C, Milla P, Athenstaedt K, Ott R, Balliano G, Daum G, Bard M. In yeast sterol biosynthesis the 3-keto reductase protein (Erg27p) is required for oxidosqualene cyclase (Erg7p) activity. Biochim. Biophys. Acta. 2003;1633:68–74. doi: 10.1016/s1388-1981(03)00088-x. [DOI] [PubMed] [Google Scholar]
- 3.Milla P, Athenstaedt K, Viola F, Oliaro-Bosso S, Kohlwein SD, Daum G, Balliano G. Yeast oxidosqualene cyclase (Erg7p) is a major component of lipid particles. J. Biol. Chem. 2002;277:2406–2412. doi: 10.1074/jbc.M104195200. [DOI] [PubMed] [Google Scholar]
- 4.Mo C, Bard M. A systematic study of yeast sterol biosynthetic protein-protein interactions using the split-ubiquitin system. Biochim. Biophys. Acta. 2005;1737:152–160. doi: 10.1016/j.bbalip.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem. Biol. Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 6.Marijanovic Z, Laubner D, Moller G, Gege C, Husen B, Adamski J, Breitling R. Closing the gap: identification of human 3-ketosteroid reductase, the last unknown enzyme of mammalian cholesterol biosynthesis. Mol. Endocrinol. 2003;17:1715–1725. doi: 10.1210/me.2002-0436. [DOI] [PubMed] [Google Scholar]
- 7.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh D, Sawicki M, Pletnev V, Erman M, Ohno S, Nakajin S, Duax WL. Porcine carbonyl reductase. structural basis for a functional monomer in short chain dehydrogenases/reductases. J. Biol. Chem. 2001;276:18457–18463. doi: 10.1074/jbc.M100538200. [DOI] [PubMed] [Google Scholar]
- 9.Kristan K, Deluca D, Adamski J, Stojan J, Lanisnik Rizner T. Dimerization and enzymatic activity of fungal 17beta-hydroxysteroid dehydrogenase from the short-chain dehydrogenase/reductase superfamily. BMC Biochem. 2005;6:28. doi: 10.1186/1471-2091-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 11.Mo C, Valachovic M, Randall SK, Nickels JT, Bard M. Protein-protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9739–9744. doi: 10.1073/pnas.112202799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- 13.Billheimer JT, Chamoun D, Esfahani M. Defective 3-ketosteroid reductase activity in a human monocyte-like cell line. J. Lipid Res. 1987;28:704–709. [PubMed] [Google Scholar]
- 14.Gachotte D, Eckstein J, Barbuch R, Hughes T, Roberts C, Bard M. A novel gene conserved from yeast to humans is involved in sterol biosynthesis. J Lipid Res. 2001;42:150–154. [PubMed] [Google Scholar]
- 15.Wilcox LJ, Balderes DA, Wharton B, Tinkelenberg AH, Rao G, Sturley SL. Transcriptional profiling identifies two members of the ATP-binding cassette transporter superfamily required for sterol uptake in yeast. J. Biol. Chem. 2002;277:32466–32472. doi: 10.1074/jbc.M204707200. [DOI] [PubMed] [Google Scholar]
- 16.Kalb VF, Woods CW, Turi TG, Dey CR, Sutter TR, Loper JC. Primary structure of the P450 lanosterol demethylase gene from Saccharomyces cerevisiae. DNA. 1987;6:529–537. doi: 10.1089/dna.1987.6.529. [DOI] [PubMed] [Google Scholar]
- 17.Han G, Gable K, Kohlwein SD, Beaudoin F, Napier JA, Dunn TM. The Saccharomyces cerevisiae YBR159w gene encodes the 3-ketoreductase of the microsomal fatty acid elongase. J. Biol Chem. 2002;277:35440–35449. doi: 10.1074/jbc.M205620200. [DOI] [PubMed] [Google Scholar]
- 18.Mullner H, Zweytick D, Leber R, Turnowsky F, Daum G. Targeting of proteins involved in sterol biosynthesis to lipid particles of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1663:9–13. doi: 10.1016/j.bbamem.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Leber R, Landl K, Zinser E, Ahorn H, Spok A, Kohlwein SD, Turnowsky F, Daum G. Dual localization of squalene epoxidase, Erg1p, in yeast reflects a relationship between the endoplasmic reticulum and lipid particles. Mol. Biol. Cell. 1998;9:375–386. doi: 10.1091/mbc.9.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandager L, Gustavsson MH, Stahl U, Dahlqvist A, Wiberg E, Banas A, Lenman M, Ronne H, Stymne S. Storage lipid synthesis is non-essential in yeast. J. Biol Chem. 2002;277:6478–6482. doi: 10.1074/jbc.M109109200. [DOI] [PubMed] [Google Scholar]
- 21.Sorger D, Athenstaedt K, Hrastnik C, Daum G. A yeast strain lacking lipid particles bears a defect in ergosterol formation. J. Biol Chem. 2004;279:31190–31196. doi: 10.1074/jbc.M403251200. [DOI] [PubMed] [Google Scholar]
- 22.Hitchcock AL, Auld K, Gygi SP, Silver PA. A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc. Natl. Acad. Sci. U.S.A. 2003;100:12735–12740. doi: 10.1073/pnas.2135500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierson CA, Jia N, Mo C, Lees ND, Sturm AM, Eckstein J, Barbuct R, Bard M. Isolation, characterization, and regulation of the Candida albicans ERG27 gene encoding the sterol 3-keto reductase. Med Mycol. 2004;42:461–473. doi: 10.1080/1369378032000141471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.