

Abstract
Inflammation is a key process in cardiovascular diseases. The extracellular matrix (ECM) of the vasculature is a major target of inflammatory cytokines, and TNFα regulates ECM metabolism by affecting collagen production. In this study, we have examined the pathways mediating TNFα-induced suppression of prolyl-4 hydroxylase alpha1 (P4Hα1), the rate-limiting isoform of P4H responsible for procollagen hydroxylation, maturation, and organization. Using human aortic smooth muscle cells, we found that TNFα activated the MKK4-JNK1 pathway, which induced histone (H) 4 lysine 12 acetylation within the TNFα response element in the P4Hα1 promoter. The acetylated-H4 then recruited a transcription factor, NonO, which, in turn, recruited HDACs and induced H3 lysine 9 deacetylation, thereby inhibiting transcription of the P4Hα1 promoter. Furthermore, we found that TNFα oxidized DJ-1, which may be essential for the NonO-P4Hα1 interaction because treatment with gene specific siRNA to knockout DJ-1 eliminated the TNFα-induced NonO-P4Hα1 interaction and its suppression. Our findings may be relevant to aortic aneurysm and dissection and the stability of the fibrous cap of atherosclerotic plaque in which collagen metabolism is important in arterial remodeling. Defining this cytokine-mediated regulatory pathway may provide novel molecular targets for therapeutic intervention in preventing plaque rupture and acute coronary occlusion.
Keywords: Inflammation, histone, P4Hα1, TNFα, NonO, JNK1
Introduction
Inflammation directly contributes to the pathogenesis of most cardiovascular diseases, including heart failure, cardiomyopathy, atherosclerotic plaque rupture, and aortic aneurysm and dissection [1, 2]. Inflammatory cytokines, such as TNFα, regulate many factors in the pathological processes of cardiovascular diseases; however, one main target for the actions of these cytokines is the extracellular matrix (ECM), which is a primary structural component of the cardiovascular system [3]. Alterations in ECM metabolism, such as abnormal collagen production, result in the initiation and progression of cardiovascular diseases. Reduced collagen synthesis or excessive collagen degradation can lead to aortic aneurysm and atherosclerotic plaque rupture [4].
The ECM, which includes fibrillar (collagen and elastin) and adhesive proteins (laminin and fibronectin), serves as the structural framework for all 3 layers of the arterial wall. Of the more than 39 subtypes of collagens, types I and III are the most common in the arterial wall [5]. Most of the mature collagen fibers comprise 3 identical polypeptide chains called α chains, with at least 1 triple-helical collagenous domain with repeating (Gly-X-Y)n sequences, in which proline and 4-hydroxyproline fill the X and Y positions.
Collagen synthesis and secretion require posttranslational modifications of procollagens, including hydroxylation and glycosylation. Prolyl-4-hydroxylase (P4H), an essential enzyme involved in collagen modification, catalyzes the formation of hydroxyproline from proline residues located in repeating X-Pro-Gly triplets in the procollagens. This step is required for folding the polypeptide chains into stable triple helical molecules [6, 7]; therefore, adequate P4H expression is essential for the formation of functional collagen in the arterial wall and pathogenesis of vascular diseases, e.g. atherosclerosis or aortic aneurysm [8].
P4H is composed of α and β subunits; the rate-limiting α subunit is regulated by various cytokines [9]. Collagen synthesis is actively regulated by cytokines, and TNFα, a cytokine released from active macrophages, is a major participant in the regulation of ECM metabolism. We have previously shown that TNFα suppresses P4Hα1 expression. The TNFα responsive element (TaRE) is located at −32 to +18bp of the P4Hα1 promoter; NonO (human p54nrb) binds as a transcription factor to TaRE and mediates TNFα-mediated P4Hα1 suppression. We have further suggested that the ASK1-JNK pathway may be the downstream pathway for the TNFα effect [10]. In the current study, we have investigated the molecular mechanisms for TNFα-mediated P4Hα1 suppression. In addition to NonO, we have identified another transcription factor, DJ-1[11, 12], which was oxidized by TNFα and required for the NonO-TaRE interaction. Furthermore, we found that the TNFα-MKK4-JNK1 pathway may induce acetylation at lysine 12 of histone 4, which facilitated the NonO-TaRE interaction. Recruitment of histone deacetylases (HDACs) resulted in deacetylation or methylation of lysine 9 at histone 3, which inhibited P4Hα1 transcription. In this study, we have further defined the molecular events mediating the TNFα-induced suppression of P4Hα1. Our findings may identify a new therapeutic target to attenuate inflammation-induced ECM disturbances and consequently cardiovascular diseases.
Materials and Methods
MKK4 and JNK1 adenovirus amplification
Wild type and dominant negative adenovirus for MKK4 and JNK1 were used as previously described [13, 14]. HEK293 cells (ATCC, Manassas, VA) were used to amplify the adenovirus. We infected HEK293 cells, which were cultured in 25ml flasks, with adenovirus by mixing 5 μl adenoviral vectors with 1.5 ml DMEM (Cat# 11965, Invitrogen Corp, Carlsbad, CA) every 15 minutes for 1 hour. The infected cells were continuously cultured for 48 hours before being harvested and stored as MKK4 (1 × 108 PFU/ml) and JNK1 (1.5 × 108 PFU/ml) adenoviral stock solutions.
Cell culture, transfection, and treatments
Human aortic smooth muscle cells (HASMCs) were harvested from fresh human aorta (from organ donors) using an explant culture technique in SMC culture medium (Cat#311–500 Cell Application, San Diego, CA) containing 10% fetal bovine serum (FBS) as described earlier [15]. We cultured cells to the 4th passage before the experiments were carried out. HASMCs were treated with 100 ng/ml TNFα (Cat# T6674, Sigma-Aldrich, St Louis, MO) for 8 hours in 6-well plates.
To study the role of MKK4 and JNK1 in TNFα-mediated P4Hα1 suppression, wild type and dominant negative adenovirus MKK4 and JNK1 were used to infect HASMCs. We mixed 50 μl HASMCs medium with 20 μl adenovirus-infected HEK293 cells, and subjected the cell solution to 3 freeze-thaw cycles for HEK293 cell lysis and adenovirus release. The dose-dependent effects of MKK4 or JNK1 on HASMCS were evaluated by infecting HASMCs with MOI (multiplicity of infection) at 1, 2, 3, 6 and 10 wild type and dominant negative MKK4 or JNK1 adenovirus for 24 hours. At the end of 24 hours, MKK4- and JNK1-treated HASMCs were harvested for mRNA measurements.
NonO, DJ-1, and hypoxia-inducible factor-1 (HIF1)–specific siRNA (100 nM) were used for gene-specific inhibition to evaluate the roles of these proteins in TNFα-mediated P4Hα1 suppression. In each experiment, siRNA-mediated gene silencing was allowed for 24 hours before TNFα treatment. The siRNA duplex was chemically synthesized (Integrated DNA Technologies, Houston, TX). The following siRNA sequences for the NonO gene were used: sense, 5′rArArUrCrArUrArCrUrCrCrArArGrGrArArGrCrArUrUrU3′; antisense, 5′rArArArUrGrCrUrUrCrCrUrUrGrGrArGrUrArUrGrArUrUr3. The following siRNA sequences were used for the DJ-1 gene: sense, 5′rArArArCrArGrGrGrArUrGrArCrCrGrUrCrUrCrCrArUrU3′; antisense, 5′rArArUrGrGrArGrArCrGrGrUrCrArUrCrCrCrUrGrUrUrU′. The sequences for siRNA of HIF1 were as follows: sense, 5′rArArCrUrArCrCrArArUrUrCrCrArArCrUrUrGrArArUrU3′; antisense, 5′rArArUrUrCrArArGrUrUrGrGrArArUrUrGrGrUrArGrUrU3′.
To assess the involvement of histone acetylation and deacetylation in TNFα-suppressed P4Hα1 expression, anacardic acid (Cat#270–381-M005, Alexis Corp, Lausen, Switzerland) was used as a histone acetylation transferase inhibitor (HATI), and trichostatin A (TSA) (Cat#T8852-1MG Sigma) was used as an HDAC inhibitor[16–18]. P4Hα1 mRNA expression was determined in HASMCs treated with 20 μM HATI or 500 ng/ml TSA for 24 hours with or without TNFα or JNK1 treatment.
Quantitative realtime RT-PCR
Levels of P4Hα1 mRNA were measured by quantitative realtime RT-PCR. Primers were designed with Beacon Designer 2.0 and chemically synthesized (Integrated DNA Technologies). The primers for P4Hα1 mRNA were sense, 5′CATGACCCTGAG ACTGGAAA3′; and antisense, 5′GCCAGGCACTCTTAGATACT3′. The primers for housekeeping gene human β-actin were sense, 5′CTGGAACGGTGAAGGTGACA3′; and antisense, 5′GGGACTTCCTGTAACAATGCA3′.
RNA Trizol (Cat# 15596-018, Invitrogen) was used to extract total RNA from HASMCs as per manufacturer’s instructions. We tested concentrations of total RNA using spectrophotometry after extracted RNA was dissolved in a final volume of 25 μl RNase free water. Reverse transcription was carried out by adding 1 μg RNA in a final volume of 20 μl. An iScript cDNA synthesis kit (Cat#170-8891, Bio-Rad, Hercules, CA) containing a mixture of oligo(dT) and random primers was used to convert mRNA to cDNA. We used SYBR Green I Supermix kit (cat#170-8882, Bio-Rad) for realtime PCR by adding 1 μl cDNA from the total 20 μl reverse transcription volume. PCR was performed in duplicate using an iCycler iQ realtime PCR detection system for 40 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Estimations of mRNA levels from the value of the threshold cycle (Ct) of the real-time PCR were adjusted by that of β-actin using the formula x-fold expression = 2ΔCt (ΔCt = β-actin Ct – gene of interest Ct).
Nuclear protein extraction and immunoprecipitation
HASMCs were treated with or without TNFα for 8 hours before nuclear protein extraction. Cold PBS was used to wash cell pellets 4 times before cells were lysed on ice and resuspended in NaCl (0.15 M). We added 65% Percoll (Cat#P4937-100ML, Sigma) to 3.5 ml tubes for gradient centrifugation at 30,000g for 15 minutes at 4°C. Then, the cell lysate was added to the top of the gradient solution and centrifuged at 100,000g for 20 minutes at 4°C. We extracted the top gradient layer, which contained the nuclear proteins, and washed and resuspended the proteins in 0.15 M NaCl.
Protein concentrations were measured before immunoprecipitation. Equal amounts of nuclear proteins isolated from HASMCs treated with or without TNFα were resuspended in cold RIPE buffer (Tris-HCl, NaCl SDS, sodium deoxycholate, Triton X-100), and incubated on ice for 20 minutes with proteinase inhibitor. The solution was passed through a 25G1 needle (Cat#305125, Franklin lakes, NJ) to shear the DNA and then centrifuged at 3000g for 10 minutes. The supernatant was mixed with 10 μl (1:100 dilution) nonspecific mouse antibody (Cat#2367, Cell Signaling, Danvers, MA) for 1 hour at 4°C, and then mixed with 45 μl protein A magnetic beads (cat#S1425S, Biolabs) for 1 hour at 4°C for magnetic purification. Finally, the supernatant solution was mixed with mouse anti-NonO antibody (20 μl at 1:100 dilution, Cat#MA3-2024, Affinity Bioreagents, Golden, CO) and incubated overnight at 4°C. Magnetic beads (45 μl) labeled with protein A, which has a high affinity for mouse IgG, were used to pulldown NonO protein complexes, which were washed 3× with RIPE buffer and once with TBE buffer. NonO binding proteins were cleaved from the beads in 50 μl SDS-PAGE buffer by boiling proteins for 5 minutes at 100°C.
1-D electrophoresis and mass spectrometry
Nuclear extracts were subjected to immunoprecipitation using anti-NonO antibody and subsequently subjected to protein separation using 1D SDS-PAGE. Then, the gels were treated with colloidal Coomassie stain to visualize protein bands. Well-resolved protein bands were excised from the gel and subjected to in-gel trypsin digestion, and subsequent isolation of tryptic peptides. Isolated peptides were analyzed by tandem mass spectrometry on an Applied Biosystems/MDS Sciex 4800 MALDI-TOF/TOF instrument. The proteins were identified with the use of GPS Explorer software (v 3.5) with the embedded Mascot® (Matrix Science, London) search engine using the SwissProt database limited to queries against the Homo sapiens sub database (release 51; 16,603 entries).
Chromatin immunoprecipitation (ChIP) assay
A ChIP assay kit (Cat#17-295, Upstate, New York, NY) was used per manufacturer’s protocols. HASMCs treated with or without TNFα were cross-linked with 1% formaldehyde at 37°C for 10 min and rinsed with cold phosphate-buffered saline (PBS). Cell lysis was performed in SDS-lysis buffer by incubation on ice for 10 minutes. The chromatin was fragmented to pieces of 200 to 1000bp by sonication using a Sonifier II 450 (Brason, Danbury, CT) with a 3-mm tip set at a duty cycle 20 and output level 2. Supernatants were collected by centrifuging cell pellets at 15000g for 10 min at 4°C. We pre-cleared supernatants by rotating with 75 μl salmon sperm DNA/protein A agrose-50% slurry for 30 minutes at 4°C. Immunoprecipitation was performed with 2 μg acetyl-histone antibodies from acetyl-Histone antibody sampler kit (Cat#9927, Cell signaling) at 4°C overnight. A salmon sperm DNA/protein A agrose slurry (60 μl) was added to the solution for an additional 1-hour incubation at 4°C. The precipitates of protein A agrose/antibody/chromatin complex were washed twice (5 min each at 4°C) with low-salt buffer, once with high-salt buffer, and once with LiCl buffer. The immune complexes were resuspended in 250 μl elution buffer for 15 minutes. After the elutes were heated at 65°C for 4 hours to reverse cross-linking by addition of 20 μl 5 mol/L NaCl, 10 μl 0.5mol EDTA and proteinase K treatment, DNA was extracted by phenol/chloroform and precipitated with ethanol. The recovered DNA resuspended for use in PCR was analyzed on 1.5% agarose gel. The primers for PCR were 5′-CTCCCTGGCGCTGCCATCGCG-3′ spanning from −60 to −39bp and 5′-CACCTGGAAAGTGGGACGAGAGG-3′ from +83 to +104bp of the P4Hα1 gene. We also used anti-human NonO antibody (Bethyl), anti-human–DJ-1 antibody (Cat#2134, Cell signaling), anti-oxidized–DJ-1 antibody (Cat#HCA024, AbD Serotec, Kidlington, UK), and anti-Di–methyl H3 (lys9) antibody (Cat#9753, Cell Signaling) instead of anti-acetyl–histone antibody for the immunoprecipitation process to perform the transcription factor-specific ChIP assay.
Western blot
Nuclear proteins were extracted from HASMCs after various treatments. Proteins were separated in 10% SDS-PAGE gel and transferred to nitrocellulose membranes. Several antibodies, including anti-JNK1 (Cat #9252, Cell Signaling), anti-MKK4 (Cat#9152, Cell Signaling), anti-NonO, anti-DJ-1, anti-oxidized–DJ-1 and anti-HDAC antibodies (Cat#9928, Cell Signaling), were used as primary antibodies in the Western blot. All primary antibodies were diluted 1:1000 for the assay. Anti-mouse IgG-HRP conjugate (1:5000, Cat#7076, Cell Signaling) or anti-rabbit IgG-HRP conjugate (1:5000, Cell signaling) was used as a secondary antibody to detect the bands, which were visualized with ECL plus reagents.
Statistical analyses
All quantitative data are presented as means± SD. Between group differences were compared using the Student’s t-test, and among group differences were analyzed using one-way ANOVA. Two-tailed P < 0.05 was regarded as statistically significant. We used the SPSS v14.0 for Windows (SPSS Inc., Chicago, IL) for statistical analyses.
Results
TNFα suppresses P4Hα1 expression through MKK4 and JNK1 pathways
In HASMCs with overexpressed MKK4, we found that P4Hα1 mRNA levels were reduced to less than 30% by comparing the wildtype with the dominant negative MKK4 overexpression. A dose-dependent effect was noted from 30μl to 100 μl MKK4, whereas the effects at 10 μl or 20 μl were unremarkable (Fig. 1A). The overexpression of MKK4 was confirmed by Western blot (Fig. 1B). Similar suppressive effects were also observed for cells with overexpression of JNK1 (Figs. 1C and 1D). In the experiments described below, infecting HASMCs with 100 μl MKK4 or 100 μl JNK1 adenovirus for 24 hours was used as the standard treatment condition. These data suggest that the MKK4-JNK1 pathway is responsible for TNFα-mediated P4Hα1 suppression. Thus, in the following experiments, we used TNFα and either MKK4 or JNK1 to study the downstream molecular mechanisms.
Fig. 1. Wild-type MKK4 and JNK1 adenovirus suppress P4Hα1 expression in human aortic smooth muscle cells (HASMCs).



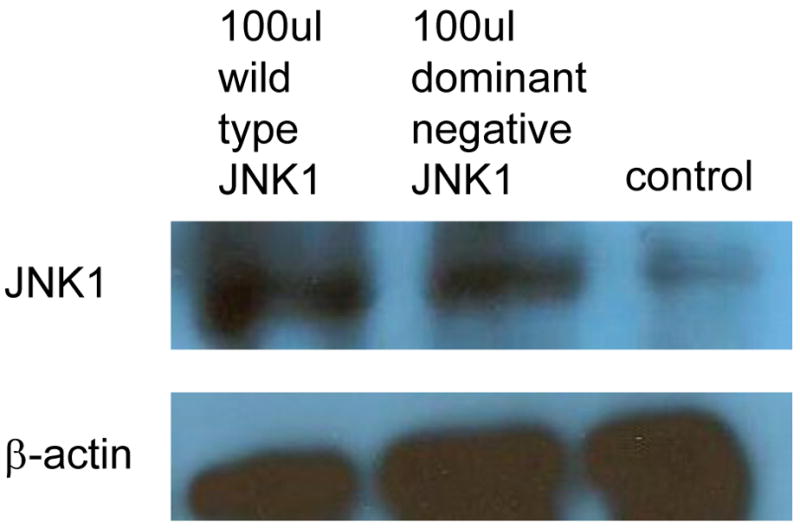
A. Levels of P4Hα1 mRNA were measured by quantitative realtime RT-PCR from HASMCs infected with 30, 60, and 100 μl wild-type and dominant negative MKK4 adenovirus. **P<0.01, by Student’s t test for comparisons between cells treated with different doses of MKK4 and control cells. House-keeping control β-actin was regarded as 1, i.e. 100% expression. B. Western blot results with anti-MKK4 antibody showing overexpression of MKK4 in HASMCs infected with MKK4 adenovirus. β-actin was used as a control. C. P4Hα1 mRNA levels were determined by RT-PCR from HASMCs infected with 30, 60, and 100 μl wild-type and dominant negative JNK1 adenovirus. **P<0.01, by Student’s t test for comparisons between cells treated with different doses of JNK1 and control cells. Housekeeping control β-actin was regarded as 1, i.e. 100% expression. D. Western blot result with anti-JNK1 antibody showing overexpression of JNK1 in HASMCs infected with JNK1 adenovirus. β-actin was used as a control.
NonO is a transcription factor responsible for TNFα-MKK4-JNK1–mediated P4Hα1 suppression
To evaluate the role of NonO in the TNFα-MKK4-JNK1–mediated suppression of P4Hα1, we used NonO siRNA to suppress NonO expression before MKK4 and JNK1 overexpression. RT-PCR was performed to measure P4Hα1 mRNA levels at 24 hours. NonO silencing abolished almost 60%–70% of the MKK4- (Fig. 2A) or JNK1-mediated (Fig. 2B) P4Hα1 suppression. In contrast, the dominant negative forms of MKK4 or JNK1 or NonO siRNA alone did not affect the expression of P4Hα1.
Fig. 2. Silencing NonO abolishes the MKK4-JNK1–mediated P4Hα1 suppression.



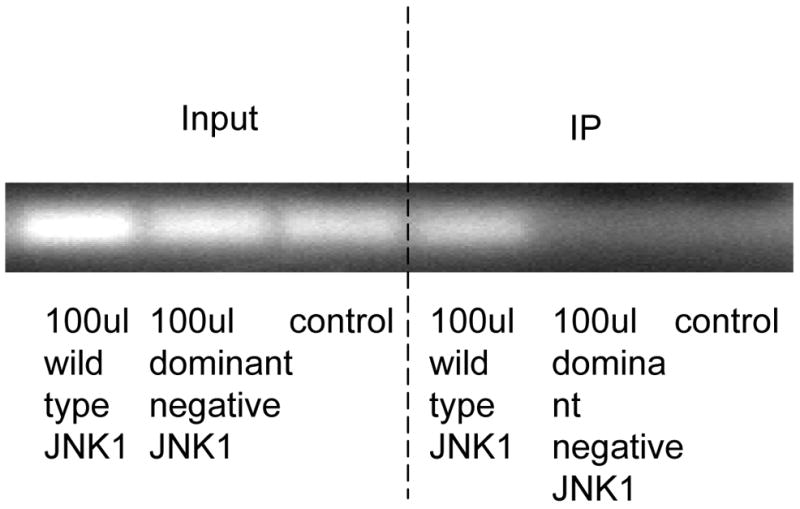
A. Expression of NonO was silenced by transfection of NonO-specific siRNA. P4Hα1 mRNA levels were measured by RT-PCR after HASMCs were infected with 100 μl wild-type and dominant negative MKK4 adenovirus for 24 hours. House-keeping control β-actin was regarded as 1, i.e. 100% expression. B. Effects of NonO silence on HASMCs infected with 100 μl wild-type and dominant negative JNK1 adenovirus. The levels of P4Hα1 mRNA were measured and compared among different treatment conditions. *P<0.05, **P<0.01, by Student’s t test for comparisons between cells with different treatments and control cells. House-keeping control βactin was regarded as 1, i.e. 100% expression. C. NonO expression in HASMCs treated with TNFα, MKK4, and JNK1 was measured by Western blot. HASMCs were infected with 100 μl wild-type or dominant negative MKK4 or JNK1 adenovirus for 24 hours, and 100ng/ml TNFα was added to cells for 8 hours. Anti-NonO antibody was used as primary antibody; β-actin was used as a control. D. HASMCs were infected with 100 μl wild-type and dominant negative JNK1 adenovirus. The ChIP assay was performed, and anti-NonO antibody was used for the immunoprecipitation. PCR primers covering the −60 to +104bp region of the P4Hα1 promoter were used to amplify recovered protein-bound DNA.
We evaluated whether overexpression of MKK4 or JNK1 or TNFα in HASMCs regulated NonO expression. None of these factors changed the protein levels of NonO in HASMCs (Fig. 2C). Using the ChIP assay in which anti-NonO antibody was used for immunoprecipitation, we showed that JNK1 overexpression significantly increased the binding between TaRE within the P4Hα1 promoter region and the NonO protein (Fig. 2D), which was the same as the effects by TNFα as shown previously [10]. In contrast, overexpression of the dominant negative JNK1 had no effect. This finding suggests that NonO is responsible for the TNFα-MKK4-JNK1–induced pathway of P4Hα1 suppression.
DJ-1 is involved in TNFα-MKK4-JNK1-NonO–mediated P4Hα1 suppression
Although NonO was found to be involved in TNFα-MKK4-JNK1–mediated P4Hα1 suppression, we studied other nuclear proteins that may be involved in the regulation. By subjecting the anti-NonO antibody–precipitated nuclear protein complex to 1–D electrophoresis and LC-MS/MS investigation, we identified a list of nuclear proteins in binding with NonO protein (data not shown). Among them, DJ-1 was the only protein found in TNFα-treated HASMCs that could potentially be involved in NonO-mediated gene regulation. After DJ-1 expression was silenced by DJ-1–specific siRNA in HASMCS, we observed a 50% recovery of P4Hα1 expression in HASMCs treated with TNFα or JNK1 (Fig. 3A).
Fig. 3. Effects of silencing DJ-1 on TNFα-JNK1–induced suppression of P4Hα1.

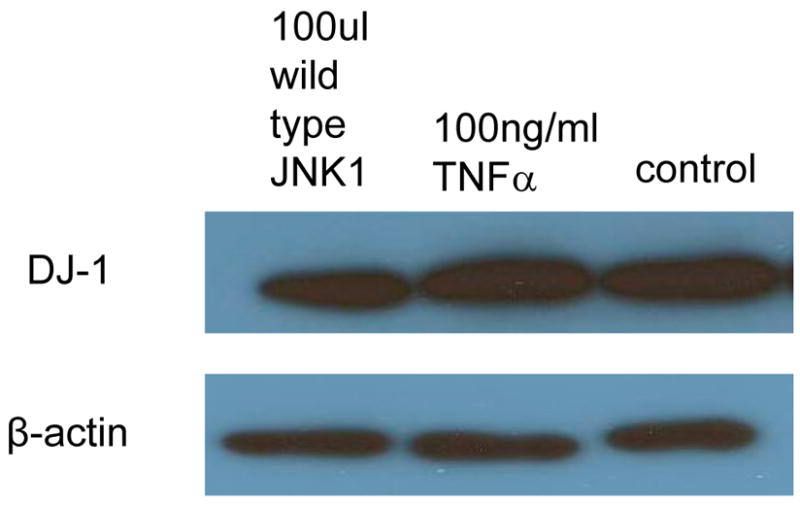

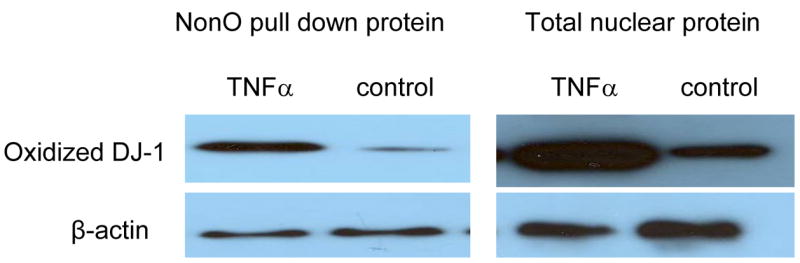

A. DJ-1-specific siRNA was applied to suppress DJ-1 expression. HASMCs were infected with 100 μl wild-type and dominant negative JNK1 adenovirus or treated with 100 ng/ml TNFα. P4Hα1 mRNA levels were measured by quantitative RT-PCR. *P<0.05, **P<0.01, by Student’s t test for comparisons between cells treated with JNK1 or TNFα and control cells. House-keeping control β-actin was regarded as 1, i.e. 100% expression. B. Western blot results with anti-DJ-1 antibody showing expression of DJ-1 in HASMCs treated with or without 100 μl wild-type JNK1 adenovirus or with100 ng/ml TNFα. C. ChIP assay examining the binding of NonO protein with TaRE element (−32bp - +18bp) in the P4Hα1 promoter. Anti-NonO antibody was used in the ChIP assay in HASMCs treated with or without 100ng/ml TNFα or with 100 μl wild-type JNK1 adenovirus with or without prior DJ-1 siRNA silencing. PCR was used to amplify protein-bound TaRE P4Hα1 promoter. D. Detection of oxidized DJ-1 in anti-NonO antibody pulldown proteins or total proteins from HASMCs treated with or without 100ng/ml TNFα. E. ChIP assays were performed using anti-DJ-1 antibody or anti-oxidized DJ-1 antibody to study DJ-1-TaRE or oxidized DJ-1-TaRE interactions in HASMCs treated with or without 100ng/ml TNFα or with 100 μl wild-type JNK1 adenovirus. NonO specific siRNA was applied to silence NonO protein expression.
Like the effects on NonO protein, neither JNK1 nor TNFα affected the protein levels of DJ-1 (Fig. 3B). To examine the effect of DJ-1 on the NonO-TaRE interaction, HASMCs were treated with TNFα and JNK1 with or without DJ-1 siRNA. Using the ChIP assay with the anti-NonO antibody for immunoprecipitation, we showed that TNFα- or JNK1-induced NonO-TaRE binding was abolished when the expression of DJ-1 was suppressed by gene specific siRNA (Fig. 3C). This finding suggests that DJ-1 is involved in the NonO-P4Hα1 promoter interaction.
Because TNFα activation can cause oxidative stress and oxidatively modify DJ-1, we used Western blot analysis to detect oxidized DJ-1 in both total nuclear proteins and anti-NonO–immunoprecipitated nuclear proteins. TNFα treatment resulted in an increase in oxidized DJ-1, which was also seen in NonO-bound DJ-1 (Fig. 3D). Furthermore, the ChIP assay (with anti-oxidized DJ-1 antibody) showed DJ-1–TaRE binding, which was abrogated by NonO siRNA (Fig. 3E). In contrast, no direct binding was seen between native DJ-1 and TaRE. These findings indicate TNFα-induced oxidized DJ-1 rather than native DJ-1 could play a role in bringing the NonO protein to the TaRE element.
Role of histone acetylation in TNFα-MKK4-JNK1-NonO–mediated P4Hα1 suppression
To understand how TNFα-MKK4-JNK1-NonO regulates P4Hα1 promoter activity, we studied the histone (H3 and H4) phosphorylation and acetylation status in HASMCs treated with TNFα or JNK1. Anti-H3 or H4 antibodies were used for immunoprecipitation, and primers for the TaRE region within the P4Hα1 promoter were used for PCR amplification in the ChIP assay. As shown in Fig. 4A, the H3 serine 10 residue (H3S10) was phosphorylated in HASMCs; TNFα treatment did not change the phosphorylation status. H3 lysine 23 (H3L23) and H4 lysine 8 (H4L8) were acetylated and not modified by TNFα or JNK1. Furthermore, H3 lysine 18 (H4L18) was not deacetylated, and further treatment with TNFα or JNK1 did not change the deacetylation status (Fig. 4A). In contrast, although TNFα or JNK1 induced H4 lysine 12 (H4L12) acetylation, the treatments induced deacetylation at H3 lysine 9 (H3L9) (Fig. 4B).
Fig. 4. TNFα-JNK1 induced modification.
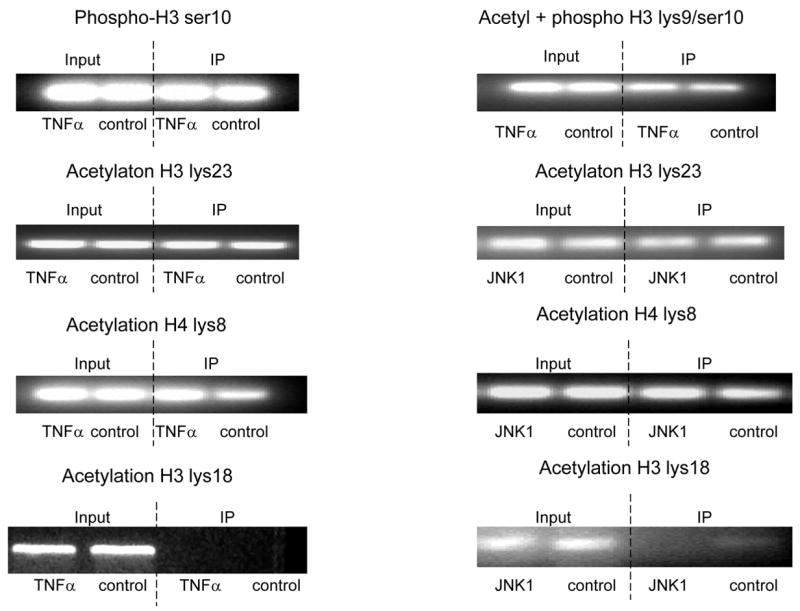
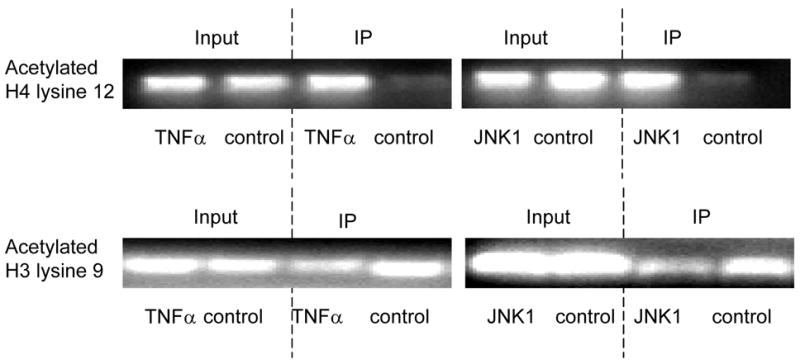

ChIP assays were performed with specific histone (H) antibodies in HAMSCs treated with or without TNFα or JNK1. Primers covering the −60 to +104bp region of the P4Hα1 promoter were used to amplify histone-bound DNA in PCR. A. ChIP assays using anti-phospho-H3 serine10, acetyl/phospho-H3 lysine9/serine10, acetyl-H3 lysine23, acetyl-H4 lysine8 and acetyl-H3 lysine18 antibodies. Experiments show no change in the acetylation or phosphorylation in H3 and H4 at the listed amino acid sites. B. ChIP assay using anti-acetyl-H4 lysine 12 or anti-acetyl-H3 lysine 9 in cells treated with or without TNFα or JNK1. TNFα or JNK1 treatment induced H4-lysine 12 acetylation and H3-lysine 9 deacetylation. C. HASMCs treated with or without TNFα were further treated with 20 μM HATI or 500ng/ml TSA (HDAC inhibitor). ChIP assays were performed using anti-acetyl-H4 lysine 12 or anti-acetyl-H3 lysine 9 antibodies. HATI treatment inhibited the TNFα-induced H4 lysine12 acetylation; TSA blocked the TNFα-induced deacetylation of H3 lysine. Exposure to HATI or TSA alone did not change the acetylation status at lysine 9 of H3 and lysine 12 of H4.
To further study TNFα- or JNK1-induced histone modifications within the TaRE region of the P4Hα1 promoter, we treated HASMCs with either 20 μM HATI (anacardic acid) or 500ng/ml HDAC inhibitor (TSA). HATI abolished the TNFα-induced H4L12 acetylation, and the HDAC inhibitor abrogated TNFα-induced H3L9 deacetylation (Fig. 4C). These data suggest that H4L12 acetylation and H3L9 deacetylation may be involved in TNFα-MKK4-JNK1–induced P4Hα1 suppression.
Effects of NonO on TNFα-JNK1–mediated histone modification
NonO is essential for TNFα-induced P4Hα1 suppression, and TNFα alters histone modification in the TaRE region of the P4Hα1 promoter; therefore, studying the involvement of NonO in P4Hα-TaRE histone modification is a logical approach. Fig. 5A shows that silencing NonO by gene specific siRNA had no effect on TNFα-induced H4L12 acetylation. However, NonO suppression abolished TNFα-induced H3L9 deacetylation. Furthermore, silencing NonO eliminated TNFα-induced methlyation of H3L9 (Fig. 5B).
Fig. 5. Effects of NonO on TNFα-induced histone acetylation/deacetylation and methylation.


A. NonO-specific siRNA was transfected into HASMCs treated with or without TNFα. ChIP assay was performed using anti-acetyl-H4 lysine 12 or anti-acetyl-H3 lysine 9 antibodies. Histone-bound DNA was amplified by primers covering −60 to +104bp region of the P4Hα1 promoter. Silencing NonO did not affect TNFα-induced H4 lysine 12 acetylation but did partially restore the H3 lysine 9 acetylation induced by TNFα treatment. B. Anti-methyl-H3 lysine 9 antibody was used in the ChIP assay in HASMCs treated with or without 100ng/ml TNFα or NonO-specific siRNA. NonO silence by gene specific siRNA blocked TNFα-induced H3 lysine 9 methylation.
We analyzed the effects of histone modification on the binding affinity between NonO protein and the TaRE element in the P4Hα1 promoter by treating HASMCs with HATI or TSA. Fig. 6A shows that NonO-TaRE binding was significantly reduced when histone acetylation was inhibited by HATI (20 μM). In addition, HATI treatment reversed the TNFα-JNK1-induced suppression of P4Hα1 expression (Fig. 6B). In contrast, deacetylation inhibition by the HDAC inhibitor TSA (0.5 μM) did not affect NonO-TaRE binding (Fig. 6A). However, TSA treatment recovered the TNFα-JNK1-mediated suppression of P4Hα (Fig. 6C). Although TSA and HATI had opposite effects on histone acetylation, both appeared to reverse TNFα-JNK1-induced P4Hα1 suppression. This effect was accompanied by the reduced NonO-TaRE interaction induced by TNFα-JNK1 in cells treated with HATI (Fig. 6A) when TSA had no effect on the TNFα-JNK1–induced NonO-TaRE interaction. These findings suggest that TNFα-JNK1 treatment may induce H4L12 acetylation, which promotes the binding of NonO to TaRE. This binding then causes H3L9 deacetylation and methylation, which is responsible for reduced P4Hα1 promoter activity.
Fig. 6. Effects of histone acetylation on the NonO-P4Hα1-TaRE interaction.



A. HASMCs were treated with TNFα and either HATI (20 μM) or TSA (500ng/ml). Anti-NonO antibody was used in the ChIP assay. NonO-bound DNA was amplified in PCR using primers covering the region of −60 to +104bp in the P4Hα1 promoter. HATI treatment attenuated the TNF-induced NonO-P4Hα1 promoter interaction, whereas TSA treatment had no effect on the interaction. However, TSA treatment of control cells did slightly increase the interaction when compared with baseline level. B. and C. Expression of P4Hα1 in HASMCs treated with TNFα (100ng/ml) or JNK1 (100 μl) with or without HATI (20 μM) or TSA (500 ng/ml). Both HATI and TSA reversed the suppressive effects on P4Hα1 expression by the TNFα-JNK1 activation, although TSA-induced restoration was more complete than that induced by HATI. House-keeping control β-actin was regarded as 1, i.e. 100% expression.
To study how NonO is involved in histone modification, we conducted a co-immunoprecipitation assay. Nuclear proteins were extracted from HASMCs treated with or without TNFα. NonO antibody was used to pull down NonO-binding proteins, which were then subjected to Western blot analysis with anti-HDAC antibodies. TNFα treatment significantly increased the NonO-HDAC1 and NonO-HDAC2 interaction (Fig. 7). We noted binding of HDAC3 to NonO, but this interaction was not affected by TNFα treatment (Fig. 7). No interaction was detected between NonO and HDAC4, HDAC5, HDAC6, and HDAC7, either with or without TNFα treatment.
Fig. 7. Interaction between NonO and HDACs.

Anti-NonO antibody was used to immunoprecipitate nuclear proteins extracted from HASMCs treated with or without TNFα. The pulldown NonO-binding proteins were then subjected to Western blot analysis with antibodies against HDAC1, 2, 3, 4, 5, 6 and 7. HDAC1 and HDAC2 binding with NonO was increased by TNFα treatment. Minimal change was noted in HDAC3, with no binding with HDAC 4, 5, 6, and 7.
Discussion
Inflammation is a double-edged sword. Controlled inflammation is essential for remodeling of the myocardium and vascular wall, whereas uncontrolled inflammation leads to excessive tissue destruction and the eventual development of cardiovascular disease. Of the plethora of inflammatory cytokines, TNFα is one of the most potent ones implicated in the development of cardiovascular diseases, such as atherosclerosis [19]. Previously, we have identified the TNFα response element (TaRE) at −32 to +18bp in the P4Hα1 promoter, which binds NonO as a transcription factor in response to TNFα-mediated P4Hα1 suppression. In addition, we have suggested that the ASK1-JNK pathway may be activated by TNFα. In the current study, we have shown that the activation of TNFα-ASK1-MKK4-JNK1-NonO is essential for P4Hα1 suppression; knock-down of any of the molecules in the signaling pathway relieves the suppression of P4Hα1. In addition, oxidized DJ-1 induced by TNFα treatment appears to be essential for the NonO-TaRE interaction, and NonO could also be phosphorylated for the interaction with P4Hα1 promoter. Furthermore, our findings showed that TNFα-JNK1 could induce H4L12 acetylation. Acetylated H4L12 recruited NonO to the TaRE region, which resulted in the deacetylation of H3L9. These consecutive reactions inactivate transcription of P4Hα1.
Several intracellular pathways could be triggered following TNFα activation during inflammation. The importance of JNK in the TNFα downstream pathway appears to correlate with transcriptional activity [20]. To activate JNK, TNFα binds to TNFR1 on the cell surface and activates downstream cascades [21, 22]. The finding that JNK-specific (SP600125) and ASK1-specific inhibitors (thioredoxin) prevent TNFα-mediated P4Hα1 suppression suggests the involvement of the ASK1-JNK1 pathway [10]. Using the wild-type and dominant negative MMK4 and JNK1 overexpression vectors in this study, we validated the requirement of the ASK1-MKK4-JNK1 pathway in TNFα-mediated P4Hα1 suppression. Our results support previous findings that ASK1-MKK4-JNK1 is a classical inflammatory pathway [23, 24].
Using 2-D gel electrophoresis and MALDI-TOF, we identified another transcriptional factor, DJ-1, involved in the NonO-TaRE complex. A 20KD conserved protein, DJ-1 binds to RNA-binding proteins to regulate RNA-protein interaction and indirectly regulates the androgen receptor (AR) [11, 25, 26]. DJ-1 expression may be stimulated by several cytokines, including TNF-related-apoptosis-inducing-ligand/Apo-2L (TRAIL) [27]. Other studies have also reported that DJ-1, together with NonO, regulates the neuroprotective genetic program in the pathogenesis of Parkinson’s disease [28, 29]. DJ-1 is specially responsive to oxidative damage which usually leads to oxidative modifications of DJ-1 [30]. In our study, DJ-1 was constitutively expressed in HASMCs. Treatment with TNFα, however, induced oxidative modification of DJ-1, which facilitated the NonO-TaRE interaction. The role of DJ-1 in the NonO-TaRE interaction was further supported by our studies using DJ-1– specific siRNA and the ChIP assay, in which oxidized but not native DJ-1 interacted with the NonO-TaRE complex.
Chromatin/histone modification is a critical process in transcriptional regulation. Several types of histone modifications, including acetylation, phosphorylation, and methylation, alter the tertiary structure of chromatins to make them accessible to transcriptional factors or, if tightly packed, inaccessible to all factors [31, 32]. Many inflammatory cytokines trigger histone modification at different sites [33, 34]. Several pathways, including JNK and NFκB, participate in TNFα-induced histone acetylation in which histone 4 could be hyper-acetylated by JNK [35, 36]. Our experiments show that TNFα-JNK induce acetylation at H4L12, resulting in the formation of an inactive chromatin state, which is consistent with the previous hypothesis [37]. The additional H3L9 methylation may further shut down transcription [38, 39]. Previous studies showing that H3L9 methylation is usually followed by its deacetylation support our findings [40]. In fact, H4L12 acetylation and H3L9 methylation as combinatorial histone modifications represent an “imprint” for inactive chromatin [41]. This effect is consistent with our findings. Synchronized H4L12 acetylation and H3L9 deacetylation would reinforce the inactive chromatin status within the P4Hα1 promoter region. Thus, TNFα-JNK1-induced acetylation at H4L12 and deacetylation and methylation at H3L9 would be sufficient to silence P4Hα1 expression. The sufficiency of HATI (anacardic acid) as an acetylation inhibitor and TSA as a deacetylation inhibitor in histone modification was identified by the fact that HATI abolished the TNFα-induced H4L12 acetylation and HDAC inhibitor abrogated TNFα-induced H3L9 deacetylation. The elimination of TNFα-JNK1–mediated P4Hα1 suppression by treatment with HATI or TSA suggests that histone modification is crucial in TNFα-mediated P4Hα1 suppression.
According to our findings, HATI inhibited H4L12 acetylation, which led to the disruption of NonO-TaRE binding. However, silencing of NonO did not interfere with H4L12 acetylation caused by TNFα. These findings suggest that acetylation at H4L12 is essential for NonO-TaRE binding. Although TSA-induced acetylation of H3L9 had no effect on NonO-TaRE binding, NonO silencing significantly reversed TNFα-induced H3L9 deacetylation. This finding suggests that NonO-TaRE binding is necessary for deacetylation at H3L9. In studies of the mechanisms of NonO-associated histone acetylation/deacetylation, we showed that the binding of HDAC1/HDAC2 with NonO was significantly increased in HASMCs treated with TNFα. The binding between HDAC1/2 and NonO-TaRE may contribute to the deacetylation of H3L9. Other studies have shown that NonO could work together with PTB-associated splicing factor (PSF) to enhance recruitment of HDAC1 to target genes [42].
One limitation of our study is that the post-translational modification of NonO has not been fully defined. The NonO protein is constitutively expressed in HASMCs treated with or without TNFα; however, binding between the NonO protein and the P4Hα1 promoter is seen only in TNFα-treated cells. Two possibilities can explain this binding: posttranslational modification of the NonO protein or additional protein(s) that facilitate the binding of NonO with TaRE. Our findings suggest that DJ-1, oxidized by TNFα, could act as a bridging protein that brings NonO to TaRE in the P4Hα promoter. However, our initial experiments using 2-D electrophoresis, 2-D Western blot, and specific phosphorylated protein stains show that TNFα treatment may result in NonO phosphorylation, which could facilitate the direct binding of NonO to the P4Hα1 promoter. Indeed, our preliminary experiments using 2-D gel electrophoresis and phospho-protein specific staining suggest possible phosphorylation of NonO by TNFα treatment (online supplemental data). Furthermore, a global scanning of site-specific phosphorylation by Olsen and colleagues has shown the possible phosphorylation of the NonO protein at the threonine 450 site, which is the phosphorylation target of the JNK family [43]. Further study is necessary to identify the amino acid residue(s) in the NonO protein that is phosphorylated by TNFα, and the functional consequences of NonO phosphorylation.
In summary, our study has established a tentative signaling pathway for TNFα-induced suppression of P4Hα1. First, TNFα activates the ASK1-MKK4-JNK1 pathway, which oxidizes DJ-1, possibly phosphorylates NonO, and induces H4L12 acetylation. This cascade of events leads to the interaction of NonO with TaRE, which is followed by recruitment of HDACs and H3L9 deacetylation and methylation. Then finally, P4Hα1 transcription is inhibited (Fig 8). Our study is the first to establish this chain of molecular events, which may provide novel therapeutic targets for modulating pathologic processes induced by inflammation. Specifically, this inflammation-induced suppression of collagen may be important to aortic aneurysm and dissection and the stability of the fibrous cap of atherosclerotic plaques.
Fig. 8. Diagram of mechanisms in TNFα-mediated P4Hα1 suppression.
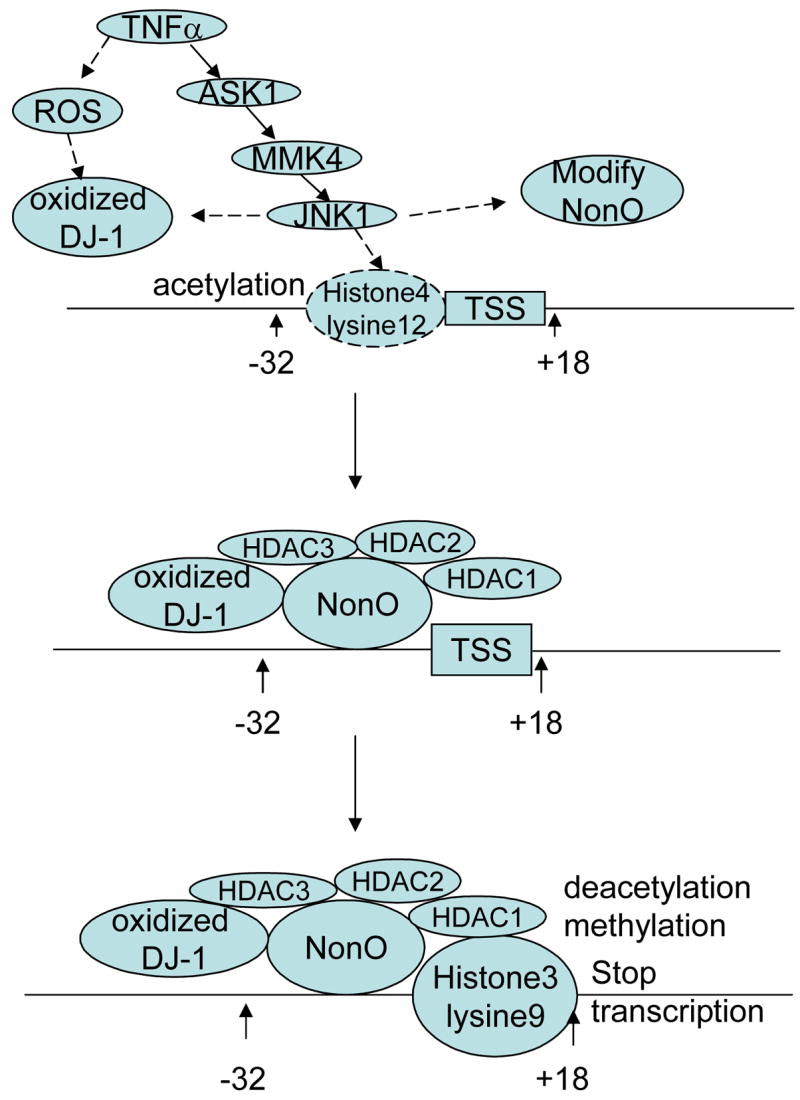
Potential pathways for TNFα-induced P4Hα1 suppression. It appears that TNFα first oxidizes DJ-1, phosphorylates NonO and acetylates H4 lysine 12. This effect is followed by NonO-P4Hα1 promoter interaction, which is facilitated by the oxidized DJ-1. This interaction leads to HDACs binding to the NonO-TaRE complex, which, in turn, induces deacetylation and methylation of the H3 lysine 9; the joint actions suppress P4Hα1 transcription.
Acknowledgments
This work is supported by an Established Investigator Award (AHA-0440001N) from the American Heart Association and NHLBI P50HL083794 to Dr. Xing Li Wang, and a National 973 Research Project (No.2006CB503803) grant awarded to Dr. Yun Zhang. We are most grateful to Dr. Rebecca Bartow, medical editor at the Texas Heart Institute, for her editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Libby P. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Chamorro A, Hallenbeck J. Stroke. 2006;37:291–3. doi: 10.1161/01.STR.0000200561.69611.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li YY, Feng YQ, Kadokami T, McTiernan CF, Draviam R, Watkins SC, Feldman AM. Proc Natl Acad Sci U S A. 2000;97:12746–51. doi: 10.1073/pnas.97.23.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahkonen O, Su M, Hakovirta H, Koskivirta I, Hormuzdi SG, Vuorio E, Bornstein P, Penttinen R. Circ Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 5.Plenz GA, Deng MC, Robenek H, Volker W. Atherosclerosis. 2003;166:1–11. doi: 10.1016/s0021-9150(01)00766-3. [DOI] [PubMed] [Google Scholar]
- 6.Kivirikko KI, Helaakoski T, Tasanen K, Vuori K, Myllyla R, Parkkonen T, Pihlajaniemi T. Ann N Y Acad Sci. 1990;580:132–42. doi: 10.1111/j.1749-6632.1990.tb17925.x. [DOI] [PubMed] [Google Scholar]
- 7.Kivirikko KI, Pihlajaniemi T. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–98. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 8.Rocnik EF, Chan BM, Pickering JG. J Clin Invest. 1998;101:1889–98. doi: 10.1172/JCI1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Annunen P, Autio-Harmainen H, Kivirikko KI. J Biol Chem. 1998;273:5989–92. doi: 10.1074/jbc.273.11.5989. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, Zhang MX, Shen YH, Burks JK, Zhang Y, Wang J, Lemaire SA, Yoshimura K, Aoki H, Coselli JS, Wang XL. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.144881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi K, Taira T, Niki T, Seino C, Iguchi-Ariga SM, Ariga H. J Biol Chem. 2001;276:37556–63. doi: 10.1074/jbc.M101730200. [DOI] [PubMed] [Google Scholar]
- 12.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. Proc Natl Acad Sci U S A. 2006;103:15091–6. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S. J Biol Chem. 2002;277:10244–50. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Nat Med. 2005;11:1330–8. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, LeMaire SA, Chen L, Shen YH, Gan Y, Bartsch H, Carter SA, Utama B, Ou H, Coselli JS, Wang XL. Circulation. 2006;114:I200–5. doi: 10.1161/CIRCULATIONAHA.105.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. J Biol Chem. 2003;278:19134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 17.Davidson SM, Townsend PA, Carroll C, Yurek-George A, Balasubramanyam K, Kundu TK, Stephanou A, Packham G, Ganesan A, Latchman DS. Chembiochem. 2005;6:162–70. doi: 10.1002/cbic.200400246. [DOI] [PubMed] [Google Scholar]
- 18.Young DA, Billingham O, Sampieri CL, Edwards DR, Clark IM. Febs J. 2005;272:1912–26. doi: 10.1111/j.1742-4658.2005.04622.x. [DOI] [PubMed] [Google Scholar]
- 19.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Arterioscler Thromb Vasc Biol. 2004;24:2137–42. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 20.Davis RJ. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 21.Micheau O, Tschopp J. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 22.Pantano C, Shrivastava P, McElhinney B, Janssen-Heininger Y. J Biol Chem. 2003;278:44091–6. doi: 10.1074/jbc.M308487200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou G, Golden T, Aragon IV, Honkanen RE. J Biol Chem. 2004;279:46595–605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]
- 24.Mittler RS, Foell J, McCausland M, Strahotin S, Niu L, Bapat A, Hewes LB. Immunol Res. 2004;29:197–208. doi: 10.1385/IR:29:1-3:197. [DOI] [PubMed] [Google Scholar]
- 25.Hod Y, Pentyala SN, Whyard TC, El-Maghrabi MR. J Cell Biochem. 1999;72:435–44. [PubMed] [Google Scholar]
- 26.Nachaliel N, Jain D, Hod Y. J Biol Chem. 1993;268:24203–9. [PubMed] [Google Scholar]
- 27.Hod Y. J Cell Biochem. 2004;92:1221–33. doi: 10.1002/jcb.20159. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA. Hum Mol Genet. 2005;14:1231–41. doi: 10.1093/hmg/ddi134. [DOI] [PubMed] [Google Scholar]
- 29.Zhong N, Kim CY, Rizzu P, Geula C, Porter DR, Pothos EN, Squitieri F, Heutink P, Xu J. J Biol Chem. 2006;281:20940–8. doi: 10.1074/jbc.M601935200. [DOI] [PubMed] [Google Scholar]
- 30.Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L. J Biol Chem. 2006;281:10816–24. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger SL. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 32.McDonald OG, Owens GK. Circ Res. 2007;100:1428–41. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- 33.Akimzhanov AM, Yang XO, Dong C. J Biol Chem. 2007;282:5969–72. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 34.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Faseb J. 2004;18:1897–9. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 35.Alberts AS, Geneste O, Treisman R. Cell. 1998;92:475–87. doi: 10.1016/s0092-8674(00)80941-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee CW, Lin WN, Lin CC, Luo SF, Wang JS, Pouyssegur J, Yang CM. J Cell Physiol. 2006;207:174–86. doi: 10.1002/jcp.20549. [DOI] [PubMed] [Google Scholar]
- 37.Turner BM. Bioessays. 2000;22:836–45. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 38.Delaval K, Govin J, Cerqueira F, Rousseaux S, Khochbin S, Feil R. Embo J. 2007;26:720–9. doi: 10.1038/sj.emboj.7601513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowak SJ, Corces VG. Trends Genet. 2004;20:214–20. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 40.Fuks F. Curr Opin Genet Dev. 2005;15:490–5. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Jenuwein T, Allis CD. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 42.Dong X, Sweet J, Challis JR, Brown T, Lye SJ. Mol Cell Biol. 2007;27:4863–75. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell. 2006;127:635–48. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]