

Abstract
Down regulation by siRNA or absence of Hypoxic Inducible Factor (HIF-1α) has been shown to lead to increased sensitivity to glycolytic inhibitors in hypoxic tumor cells. In surveying a number of tumor types for differences in intrinsic levels of HIF under hypoxia we find that the reduction of the upstream pathways of HIF, AKT and mammalian target of rapamycin (mTOR) correlate with increased toxic effects of 2-DG in lung cancer cell lines when treated under hypoxia. Since HIF-1α translation is regulated by the mammalian target of rapamycin (mTOR), we examined the effects of blocking mTOR under hypoxia with an analog of rapamycin (CCI-779) in those cell lines that showed increased mTOR and AKT activity and found that HIF-1α down-regulation coincided with increased 2-DG killing. CCI-779 however was ineffective in increasing 2-DG toxicity in cell lines that do not express HIF. This result supports the hypothesis that although mTOR inhibition leads to blockage of numerous downstream targets, CCI-779 increases the toxicity of 2-DG in hypoxic cells through down-regulation of HIF-1α. Overall, our findings show that CCI-779 hyper-sensitizes hypoxic tumor cells to 2-DG and suggest that the intrinsic expression of AKT, mTOR, and HIF in lung cancer, as well as other tumor types, may be important in dictating the decision on how best to use 2-DG alone or in combination with CCI-799 to kill hypoxic tumor cells clinically.
INTRODUCTION
Glucose transporters and glycolytic enzymes are among many of the genes activated by hypoxic inducible factor-1α (HIF-1α), a key regulator of a wide range of cellular responses to lowered O2 tension (1–4). Previously, it was reported that although hypoxic tumor cells are hypersensitive to glycolytic inhibitors such as 2-deoxy-D-glucose (2-DG), HIF-1α appears to confer a level of resistance to this treatment (5). A mechanism offered to explain these results is that HIF-1α increases the expression of hexokinase (HK) which is the enzyme that 2-DG interacts with to block glycolysis, thus greater amounts of this glycolytic inhibitor are required to shut it down. This hypothesis was supported by results which demonstrated that hypoxic cells unable to activate the HIF-response pathway either through siRNA or mutation, showed decreased levels of hexokinase which correlated with increased sensitivity to 2-DG (5). Thus, while tumor cells under hypoxic conditions are compromised in their ability to undergo oxidative phosphorylation and consequently become vulnerable to inhibitors of glycolysis, blocking HIF potentiates the toxicity of this type of treatment.
Since 2-DG is currently in Phase I clinical trials, based on the rational that by killing the hypoxic populations of solid tumors it will raise treatment efficacy when combined with standard chemotherapy, it becomes important to identify mechanisms by which use of this strategy can be maximized. Thus, combining inhibitors of HIF with 2-DG may prove to be one way to increase the effectiveness of this latter agent’s cytotoxic activity in hypoxic tumor cells. In this regard, CCI-779, a soluble analog of rapamycin has been shown to down-regulate the translation of HIF-1α protein by inhibiting the mammalian target of rapamycin, mTOR, which functions as a central modulator of cell growth at the level of mRNA translation (6–9). mTOR controls translation of HIF-1α as well as many other proteins through phosphorylation of two downstream targets, p70S6K and 4E-BP1(10–12). Activation of both of these proteins upregulates the translation of mRNAs that contain polypyrimidine tracts at their 5′ transcriptional (5′TOP) starting sites (13–15). Since HIF-1α mRNA contains such polypyrimidine tracts, its translation is directly regulated by mTOR (14, 16). Upstream of mTOR, the AKT pathway when activated positively stimulates mTOR activity. Thus, tumor cells with intrinsic activation of AKT and or mTOR would be expected to express HIF under hypoxic conditions whereas cell types with low or absent levels of these activated pathways would not. In this regard we report here that human lung cancer cell lines that show intrinsically lower phosphor-AKT (pAKT) and phosphor-mTOR (pmTOR) activity correlates with barely detectable levels of HIF and consequently increased sensitivity to the glycolytic inhibitor 2-DG. Moreover, we find that when mTOR is blocked by CCI-779 in the lung cancer cell lines that express high levels of pAKT and pmTOR, HIF-1α is down-regulated which in turn leads to hypersensitization of hypoxic tumor cells to 2-DG.
Materials and Methods
Cell lines
SCLC 1 & SCLC SR-2 cell lines were derived from the bone marrow, and SCLC B & SCLC BC came from the lymph nodes as described previously (17, 18). NSCLC A and NSCLC ALC lines were established from metastatic adenocarcinomas to the brain(19). HEPA-1, and C4 cell lines(20) were purchased from ATCC (Rockville, MD).
Compounds and antibodies
CCI-779 was kindly provided by Wyeth-Ayrest (Madison, NJ). 2-deoxy-D-Glucose (2-DG) was purchased from Sigma Chemicals (St. Louis, MO). Hexokinase type II, AKT, phosphor-AKT(Ser473), mTOR, phosphor-mTOR(Ser2448), and phosphor-P70S6K(Thr389) were purchased from Cell Signaling Inc (Danvers, MA). HIF-1α was purchased from BD Bioscience (San Jose, CA).
Cytotoxicity Assay
Cells were seeded in 24-well dishes at the following numbers per well: 8 × 104 transfected SCLC, 4 × 104 HEPA-1 or C4, 1.5× 104 SCLC, and 4 × 104 NSCLC. Transfected SCLC cells were incubated and 95% air under 5%CO2 at 37°C for 6 to 8 hr to allow attachment and transferred to either normoxic or hypoxic conditions. All and 95% air at 37°C for 24hr other cell lines were cultured under 5%CO2 before the various treatment conditions. Cells were treated under hypoxia for 48hr with different combinations of 2-DG and CCI-779 as described in the figure legends. The culture mediums as well as the trypsinized cells were collected and this mixture was centrifuged at 400 × g for 5 min, The supernatant was discarded, and the cells were resuspended in 1 mL Hank’s buffer and assayed for live and dead cells using a Vi-Cell cell viability analyzer (Beckman Coulter, Inc., Fullerton, CA).
Hypoxia
Cells were exposed to hypoxia (0.5% O2) by incubation in a hypoxia glove box (Coy Laboratory Products, Inc., Grass Lake, MI). After an initial exposure to low oxygen, all subsequent treatments were given within the glove box to prevent cellular damage due to re-oxygenation. Additionally, if the procedure required a change of medium after hypoxic exposure, the replacement medium was equilibrated to the low-oxygen environment 24 h before use. Normoxia-equilibrated medium was used for the normoxic control cells.
siRNA
8×105 cells were seeded in a 60mm petri dish and incubated under normal culture conditions for 24hr. Dharmafect 1 transfection reagent was then used to transfect 100nM of HIF-directed SMARTpool® siRNA or siCONTROL® (Dharmacon Co., Denver, CO). Cells were incubated in the presence of transfection medium for 24h under 5%CO2 and 95% air at 37°C. The transfection medium was then removed, cells were gently rinsed with PBS 3 times, and fresh culture medium was replaced. Cells were then allowed to recover for 24 hr under normal culture conditions, and then trypsinized and reseeded either in 24 well plates or 100mm Petri-dishes depending on the proceeding experimental protocols as described in the appropriate figure legends.
ATP
Cells were seeded at 1.5 × 104 and grown under normal culture conditions for 24 h. The cells were either maintained under normoxia or transferred to hypoxia for 24 h. The medium was then replaced with 100 μL of fresh medium conditioned in either normoxic or hypoxic conditions, and the appropriate doses of 2-DG with or with out combination of CCI-779 were given. Following 6 h of treatment, the Cell Titer-Glo kit (Promega) was used to quantify ATP by luminescence as measured on a FLUOstar OPTIMA (BMG Lab Tech, Durham, NC). Each data point was normalized to an average of three tandem cell counts.
Westernblot analysis
Cells were seeded at 1 × 105/ml onto 100 mm dishes, and allowed to attach overnight, then treated with various doses of 2-DG, with and without CCI-779 under either normoxic or hypoxic conditions. At 48 hrs, cells were harvested by scraping in RIPA buffer which contains 10 mM Tris pH7.4 100 mM NaCl, 1 mM EDTA, 20 mM Na4P2O7, 2 mM Na3VO4 10%, 0.5% deoxycholate, 1 mM PMSF) and a phosphatase and protease inhibitor cocktail (purchased from Sigma). Cell lysis was completed by sonication and centrifugation. The total protein was separated on an 8% SDS-PAGE, transferred onto a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ) and immunoblotted with the indicated antibodies. Bands were visualized using an enhanced chemiluminescence reagent (Pierce Biotechnology, Inc., Rockford, IL). The membrane was stripped using a stripping buffer (Pierce Biotechnology) for successive re-blotting with additional antibodies. Equal protein loading was verified using the Micro BCA Protein Assay (Pierce Biotechnology) as well as using actin as a loading control (actin antibody was purchased from Sigma). Band Density (intensity × mm2) was measured for each protein using a molecular imager Chemidoc system with Quality One software (Bio-Rad, Richmond, CA).
RESULTS
Reduced AKT and mTOR activity correlates with low levels of HIF and increased sensitivity to 2-DG in lung cancer cell lines under hypoxia
Since down-regulation or absence of HIF renders hypoxic cells more sensitive to glycolytic inhibitors we surveyed a number of cell lines for their intrinsic ability to express this transcription factor as well as its upstream regulators AKT and mTOR. Under hypoxia, low pAKT and pmTOR levels (which are the active forms) were found in 2 cell lines (NSCLC) correlating with undetectable levels of HIF-1α (Fig. 1 A, B) and to increased sensitivity to 2-DG, as compared to 4 cell lines (SCLC) that express higher levels of these proteins (Fig. 1C). The average of 6 fold higher mTOR activity and 10 fold higher AKT activity detected in the SCLC lines (Quantification of Fig. 1A) corresponds with robust amounts of HIF-1α (Fig. 1B). Interestingly, the levels of the inactive forms of mTOR and AKT (unphosphorylated) under normoxia and hypoxia are the same for both lung cancer cell types (Fig. 1A). Overall these results demonstrate that decreased levels of HIF-1α in cells growing under hypoxia correlate with decreased AKT and mTOR activity and increased sensitivity to 2-DG.
Figure 1.

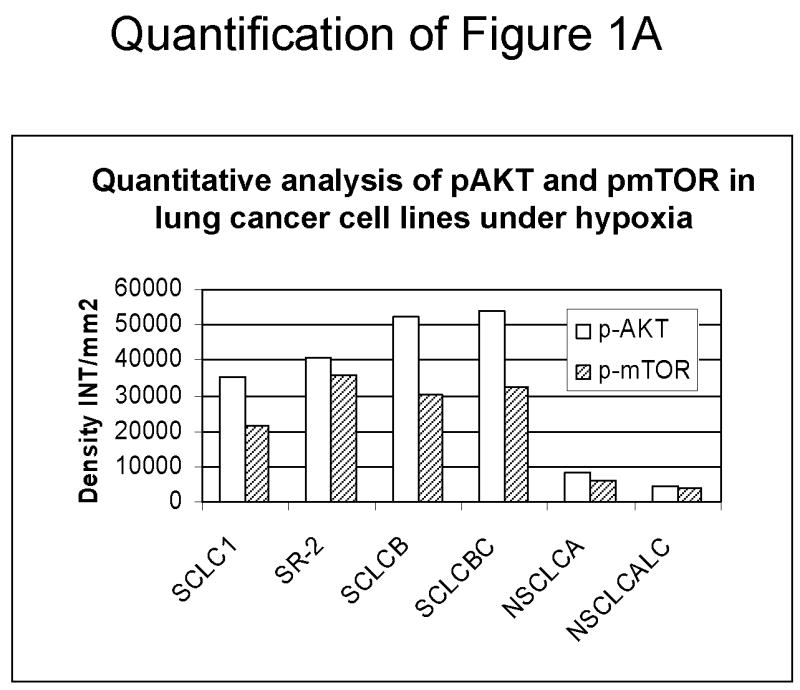
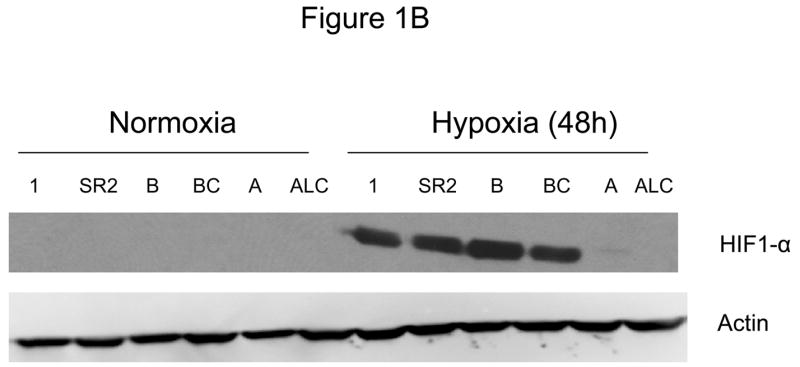

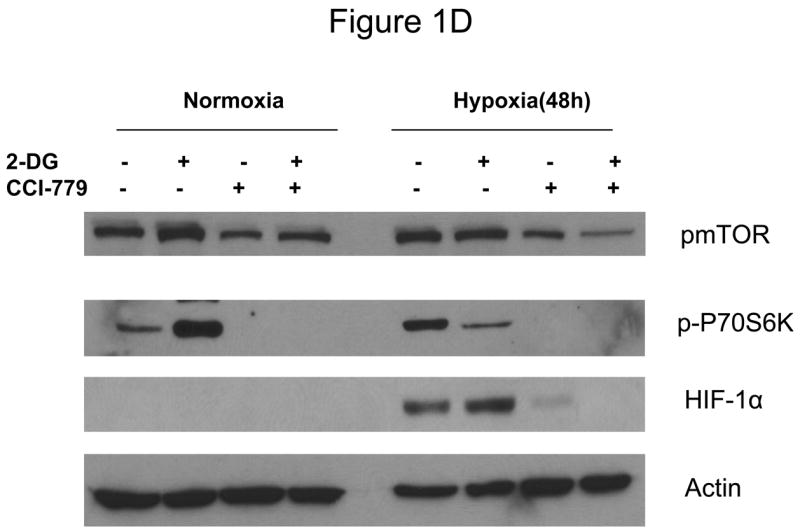

Decreased AKT and mTOR activity in lung cancer cell lines correlates with reduced levels of HIF-1α and increased sensitivity to 2-DG under hypoxia. (A) Western blots and quantitative analysis of pAKT and pmTOR levels in lung cancer cell lines grown under normoxia or hypoxia for 48hrs. Under normoxia as well as hypoxia, 2 NSCLC lines displayed intrinsically low levels of pAKT and pmTOR as compared to 4 SCLC lines. (B) Immunoblots of cells grown under normoxic or hypoxic conditions for 48hr showed increased HIF-1α in those cell lines with expressing high levels of pAKT and pmTOR (SCLC lines) whereas in the NSCLC lines which expressed lower levels of pAKT and pmTOR, HIF-1α was barely detectable. (C) Low levels of HIF-1α in NSCLC lines correlated with increased sensitivity to 2-DG under hypoxia (48h). Each data point is the average of triplicate samples with S. D.; P<0.05. (D) Immunoblots of pmTOR, p-P70S6K, and HIF-1α in SCLC line B treated with 4mM of 2-DG or 0.1μg/ml of CCI-779 alone and in-combination under both normoxic and hypoxic conditions for 48hr. Note that under hypoxic conditions, CCI-779 down-regulates HIF-1α and when 2-DG is added, HIF-1α is completely eliminated. (E) Immunoblot of HKII expression in SCLC line B when treated with 0.1μg/ml of CCI-779 alone or in combination with 4mM of 2-DG under 48h of normoxia and hypoxia. Quantitative analysis indicates that HKII is decreased by 30% when SCLC line B under hypoxia is treated with CCI-779 alone and further reduced when co-treated with 2-DG (4mM).
Blocking mTOR with CCI-779 correlates with down-regulation of HIF-1α and increased 2-DG toxicity in hypoxic tumor cells
In order to determine whether blockage of mTOR results in down-regulation of HIF and thereby increases the sensitivity of hypoxic cells to 2-DG, SCLC line B was treated with the water soluble analog of Rapamycin, CCI-779. At a non-toxic dose, CCI-779 is found to completely block mTOR activity as measured by assaying its direct down-stream target phosphor-P70S6K (p-P70S6K). This in turn coincides with the lowering of HIF-1α as well as hexokinase type II (HKII) levels by CCI-779 (Fig. 1D, E).
Co-treatment of SCLC B with 2-DG and CCI-779 results in increased toxicity as compared to cells treated with 2-DG alone (Fig. 2A). Interestingly, the percent of dead cells elicited by this treatment in SCLC B is strikingly similar to that in NSCLC cells treated with 2-DG alone where HIF is not expressed; 78% and 88% compared with 73% and 82% respectively. However, when NSCLC line A, which has low mTOR activity, was treated with CCI-779 under hypoxia, 2-DG toxicity did not increase and in fact a slight protective effect was observed (Fig. 2B). Overall, these data demonstrate that inhibiting mTOR with CCI-779 in lung cancer cell lines that intrinsically express high levels of this protein, leads to blockage of HIF-1α translation culminating in increased sensitivity of hypoxic tumor cells to 2-DG.
Figure 2.

Blocking mTOR with CCI-779 correlates with down-regulation of HIF-1α and increased sensitivity to 2-DG. (A, B) Under hypoxia CCI-779 increased the cytotoxicity of 2-DG in SCLC line B which expresses high levels of pmTOR but not in NSCLC line A which expresses low levels of pmTOR. Each data point is the average of triplicate samples with S.D. *P=0.002, **P=0.004
CCI-779 does not increase 2-DG toxicity in hypoxic cells when HIF-1α is downregulated by siRNA or is non-functional in HIF-1β mutant cells
Inhibition of mTOR is known to affect the translation of numerous proteins. To address the question of whether the increased sensitivity to 2-DG by CCI-779 was specific to its effects on HIF under hypoxia, siRNA directed against HIF-1α or mutant HIF-1β cells that cannot express functional HIF were used. In SCLC cells where HIF-1α is significantly decreased with siRNA (Fig. 3A) addition of CCI-779 did not augment 2-DG cytotoxicity (Fig. 3B). As expected, in SCLC cells transfected with scrambled siRNA where HIF is expressed under hypoxia, treatment with CCI-779 down-regulated HIF which correlated with increased sensitivity to 2-DG (Fig. 3C). Similarly, CCI-779 did not increase the sensitivity to 2-DG in a hepatoma mutant cell line deficient in HIF-1β (c4), while 2-DG sensitivity was augmented with CCI-779 treatment under hypoxia in its wild type cell counterpart (Hepa-1) (Fig. 4A&B). Moreover, in the two NSCLC lines studied, CCI-779 did not increase sensitivity to 2-DG under hypoxia (where HIF-1α is barely detectable) while in the four SCLC lines (where HIF is expressed) it did (Fig. 4C). Interestingly, in all three models where HIF was non-functional under hypoxia, CCI-779 not only failed to increase sensitivity to 2-DG but displayed a slight protective effect. Overall, the results in the HIF+ and HIF− models presented above support the interpretation that CCI-779 increases sensitivity to 2-DG in hypoxic tumor cells by down-regulating HIF-1α.
Figure 3.
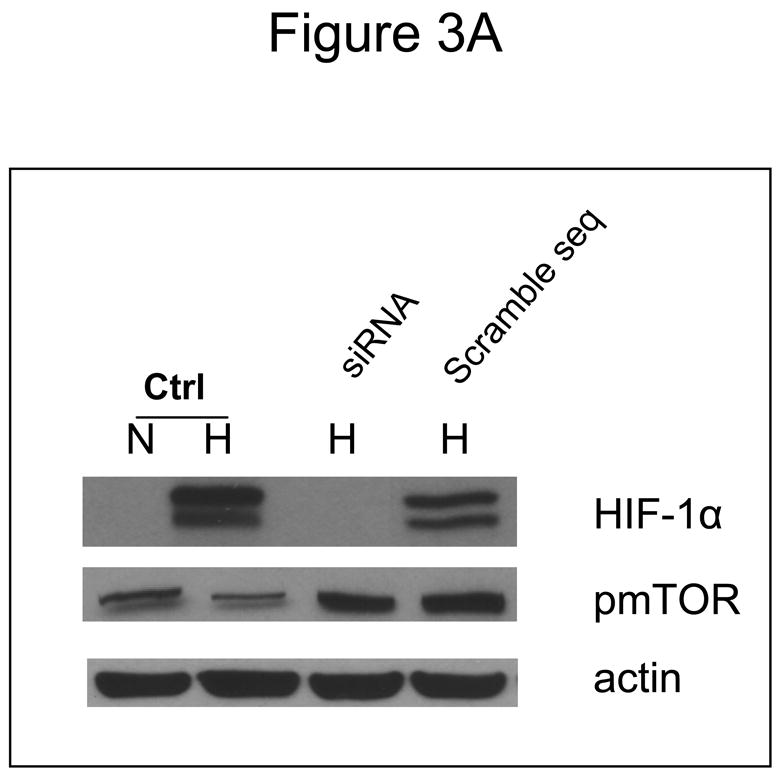

In cells where HIF-1α is down-regulated by siRNA, CCI-779 does not increase the toxicity of 2-DG in hypoxic cells. (A) Westernblot analysis of SCLC line B treated under normoxic (N) and hypoxic (H) conditions confirming the effectiveness of siRNA in down-regulating HIF-1α. (B, C) Under hypoxia, CCI-779 does not increase 2-DG toxicity in cells transfected with siRNA directed against HIF-1α. In contrast, CCI-779 increases 2-DG toxicity in cells treated with scrambled RNA. Each data point is the average of triplicate samples with S.D. *P=0.07, **p=0.016
Figure 4.


In mutant cells where HIF-1αis non- functional, CCI-779 does not increase 2-DG toxicity under hypoxic conditions. (A, B) Cytotoxicity assays in HEPA (HIF+) and C4 (HIF−) cells after exposure to various doses of 2-DG and 0.1μg/ml of CCI-779 under hypoxic conditions for 48h. Note the increase in cell death after treatment with 2-DG in combination with CCI-779 in the (A) HEPA cell line that expresses functional HIF but not in the (P<0.02) (B) C4 HIF-deficient mutant cell line (P<0.05). (C) Cytotoxicity assays in 4 SCLC vs. 2 NSCLC lines. Note that in NSCLC cells which do not express HIF under hypoxia, co-treatment with CCI-779 and 2-DG results in decreased toxicity. Each data point is the average of triplicate samples with S.D. *P=0.001,**P=0.002
In cells where HIF is absent, inhibiting mTOR results in reduced 2-DG toxicity
Surprisingly, in cells unable to mount a HIF response, co-treatment with CCI-779 and 2-DG resulted in decreased rather than increased toxicity (as shown above in Fig. 4C). In this figure, a 20%–25% decrease in cell death was observed in NSCLC cell lines growing under hypoxia (where HIF is not detectable) when CCI-779 was combined with 2-DG as compared to 2DG alone. This phenomenon was also observed in cells wherein HIF was inactive either through siRNA or a mutation in HIF-1β (C4 cell line). A possible explanation for this result could be that inhibition of mTOR with CCI-779 stimulates an energy-conservation effect that somewhat decreases 2-DG-induced ATP depletion under hypoxia. Indeed, it has previously been shown that blockage of mTOR by rapamycin in Lewis lung carcinoma cells growing under prolonged conditions of hypoxia (5days) results in conservation of energy as measured by increased ATP (21). In order to determine whether such activity of CCI-779 could account for the reduced cytotoxic effects of 2-DG in the conditions in which HIF is inactive, ATP levels were measured. As compared with 2-DG treatment alone, CCI-779 was found to increase ATP levels under hypoxia by 26 and 18 % in NSCLC cell line A treated with 2-DG at 4 and 6mM respectively (Fig 5A). Similarly CCI-779 increases ATP levels by 34 and 25 % in HIF- mutant hepatoma (C4) cell lines respectively as compared to these cells treated with 2-DG alone (Fig 5B). Overall, these data further support the idea that CCI-779 reduces the cytotoxic effect of 2-DG in cells in which HIF is inactive by increasing ATP levels.
Figure 5.

In cells unable to mount a HIF response, CCI-779 increases resistance to 2-DG which correlates with increased ATP levels. (A, B) ATP assays in NSCLCA and C4 cells after exposure to various doses of 2-DG and 0.1μg/nl of CCI-779 under hypoxic conditions for 24h. Note the increase in ATP levels in both (A) NSCLC line A and (B) hepatoma (HIF−) cell line, C4, after treatment with CCI-779. Each data point is the average of triplicate samples with S.D. P<0.05.
DISCUSSION
Previously it was shown that treatment with siRNA specific for HIF-1α increased sensitivity to 2-DG which could be explained by correspondingly decreasing amounts of hexokinase, the enzyme which 2-DG interacts with to block glycolysis (5). Since the techniques for effectively applying siRNA in patients remain to be elucidated, it was not clear how this information could be used clinically. However, CCI-779 which inhibits the translation of HIF-1α by blocking mTOR, has recently been approved for patient use as an anti-cancer agent. Therefore, this compound could have activity as a HIF inhibitor, and when applied clinically could thereby increase tumor cell sensitivity to 2-DG under hypoxic conditions. Indeed in Fig. 1D and E, we show that under hypoxia, CCI-779 blocks mTOR activity which in turn leads to down-regulation of HIF-1α and HKII. It should be noted that mTOR activity is measured by the phosphorylated levels of its downstream effector molecule P70S6K. Thus, loss of the phosphorylated form of this protein when cells are treated with CCI-779 indicates the blockage of mTOR activity.
Moreover, the levels of HIF-1α were further reduced when 2-DG was combined with CCI-779 in SCLC line B (Fig. 1D). Thus, although the mechanism remains unclear, it appears that by lowering ATP levels with 2-DG in hypoxic cells as previously shown (5), impacts favorably on the effectiveness of CCI-779 in further down-regulating HIF-1α (and perhaps other mTOR targets). This idea is supported by previous results which showed that inadequate levels of ATP trigger AMPK phosphorylation which in turn suppresses mTOR activity in cells under hypoxia (22–24). As can be seen in supplemental dataS1, 2-DG increases the phosphorylation levels of AMPK which correlates with attenuation of mTOR activity as reflected by reduced levels of p-P70S6K. Overall, the lowering of HIF-1α as a consequence of co-treatment with 2-DG and CCI-779 corresponds to increased toxicity by 2-DG when CCI-779 (at a non-toxic dose) is combined with this glycolytic inhibitor in hypoxic tumor cells (Fig. 2A, 3C, and 4A). Thus, CCI-779 may prove to be useful in the clinical setting as an anti-HIF agent for increasing the effectiveness of 2-DG in killing the hypoxic cell population found in most solid tumors.
Hexokinase, the enzyme which 2DG interacts with to block glycolysis, has been shown to be reduced when HIF is inhibited by siRNA (5). This result was found to correlate with increased toxicity of 2-DG in cells under hypoxia (5). Here, we find similarly that when HIF is inhibited via blockage of mTOR by CCI-779, hexokinase (HKII) is reduced by 30% (Fig. 1E). Interestingly, when 2DG is combined with CCI-779 a further decrease in HKII is observed. A possible explanation for this result is that the binding of 2-DG to HKII leads to dissociation of this enzyme from its normal mitochondrial localization hence resulting in cytosolic degradation (25). Additionally, we find that HKII is reduced in hypoxic cells treated with 2DG alone (data not shown), which further supports the possibility that binding of HKII by 2-DG lowers its levels. Recently, it has been reported that HIF-1α increases mitochondrial respiration in cells under low oxygen tension (26). This latter result presents the possibility that HIF-1α may render hypoxic cells resistant to glycolytic inhibitors by increasing mitochondrial ATP production. The contributions of HIF-induced increases in hexokinase and efficiency to utilize oxygen in hypoxic cells for conferring resistance to glycolytic inhibitors are currently being investigated in our laboratory to determine the importance of each.
It has been shown that blocking mTOR with rapamycin or its analogs leads to suppression of the translation of numerous proteins (27–31). In order to address whether the increased sensitivity to 2-DG by treatment with CCI-779 is indeed due to down-regulation of HIF-1α, cells transfected with siRNA directed against HIF-1α as well as a HIF- mutant cell line, were co-treated with these two agents. The results which demonstrate that CCI-779 did not increase the sensitivities of these cell lines to 2-DG when grown under hypoxia clearly implicate the down-regulation of HIF as the primary mechanism by which CCI-779 augments 2-DG toxicity in wild type cells.
Interestingly, mTOR activity was found to be highly expressed intrinsically in 4 lung cancer cell lines which corresponded to high HIF-1α levels and relative resistance to 2-DG when cells were grown under hypoxia (Fig. 1C). In contrast, mTOR activity was significantly lower in 2 lung cancer cell lines correlating with barely or non- detectable levels of HIF-1α (Fig. 1B) and increased sensitivity to 2-DG under hypoxic conditions (Fig. 1C). Furthermore, the level of resistance to 2-DG in the cell lines over expressing pAKT, pmTOR and HIF-1α could be overcome by co-treating them with CCI-779 (Fig. 4C). Similar to the experiments described above for the HIF− mutant and HIF knockdown cells, when the lung cancer cell lines expressing extremely low or non-detectable levels of HIF protein were co-treated with CCI-779, increased sensitivity to 2-DG was not observed (Fig. 2B, 3B, 4B ). Thus, the data in lung cancer cell lines support our findings with hepatoma which indicate that down-regulation of HIF is the mechanism by which CCI-779 sensitizes hypoxic cells to glycolytic inhibitors. Additionally, the amount of 2-DG induced cell death by CCI-779 in HIF+ cells compares favorably to the amount of cell death in HIF− mutant, HIF-1α siRNA knockdown cells and lung cancer cell lines intrinsically deficient in HIF-1α, when treated with 2-DG alone. Thus, these results highlight the importance of HIF in attenuating tumor cell sensitivity to glycolytic inhibitors, which is in agreement with our previous findings (5).
From the data presented in this manuscript, it appears that NSCLC are fundamentally different than SCLC in that they have lower mTOR, AKT activity and reduced HIF expression under hypoxia. However, in another study in which 110 NSCLC tissue samples were evaluated from patients, it was noted that pAKT levels were expressed in 51% of them (32). Thus, our findings of differences in pAKT between NSCLC and SCLC appear to be due to small sampling. It is clear however, that intrinsic sensitivity to 2-DG under hypoxia correlates with lower levels of this upstream pathway of mTOR. Moreover, in the SCLC cell lines in which we find increased levels of pAKT, inhibiting mTOR with CCI-779 is more effective in increasing sensitivity to 2-DG than in cells that express very low levels of this oncogenic protein. Increases in pAKT have been implicated in many different tumors and it has been suggested that cancer cells over-expressing this signaling molecule are more sensitive to mTOR inhibitors compared with those expressing lower levels (19, 33). Taken together, intrinsic differences in pAKT, pmTOR, and HIF found in lung cancer tumors could dictate future decisions on whether to use CCI-779 in combination with 2-DG when treating patients with either of this type of cancer.
A possible disadvantage of using mTOR inhibitors as anti-cancer agents directed at killing aerobically proliferating cells is that rapamycin has been shown to protect Lewis lung carcinoma cells from hypoxic-induced death (21). The explanation for this effect is that under hypoxia mTOR is not completely shut down and therefore addition of rapamycin will further block the function of this protein resulting in lowered energy consumption and thereby increased survival. If this is a general phenomenon of rapamycin then this agent would be expected to decrease 2-DG toxicity in hypoxic cells by a similar mechanism Since, however we find that CCI-779 increases 2-DG toxicity in hypoxic cells by decreasing HIF, it appears that the role of this agent in attenuating HIF supercedes its effects on the conservation of energy resulting in greater inhibition of glycolysis and overall cell death. This idea may also explain our observation that when CCI-779 is applied to cells deficient in HIF their survival to 2-DG under hypoxia is slightly increased (Fig. 2B and 4B) via its activity as an energy conserver which is reflected in increased ATP levels (Fig 5A,B). In support of this data is the result where the level of 2-DG toxicity in SCLC treated with siRNA against HIF-1α went down approximately 15–20% when CCI-779 was added (Fig 3B).
Our findings that the combinations of CCI-779 with 2-DG increases killing of hypoxic cells suggest that 2-DG may be an important additive when rapamycin and or its analogs are to be used to treat cancer patients. Moreover, our data indicate that the addition of mTOR inhibitors to clinical protocols that include 2-DG should increase the killing of the hypoxic tumor cell population by decreasing HIF.
Supplementary Material
Supplemental Data S1. Effects of 2-DG and CCI-779 on pAMPK and p-P70S6K. Under hypoxia (48h), 4mM of 2-DG significantly increases pAMPK in SCLC line B which correlates with decreased levels of p-P70S6K a downstream target of pmTOR.
Acknowledgments
This work was supported in part by the following: NIH/NCI RO1CA037109 grant and Threshold Pharmaceuticals Award to TJL and VA Research Merit Award to NS.
References
- 1.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 2.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;(9 Suppl 5):10–17. doi: 10.1634/theoncologist.9-90005-10. [DOI] [PubMed] [Google Scholar]
- 4.Maxwell PH, Pugh CW, Ratcliffe PJ. The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Adv Exp Med Biol. 2001;502:365–376. [PubMed] [Google Scholar]
- 5.Maher JC, Wangpaichitr M, Savaraj N, et al. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol Cancer Ther. 2007;6:732–741. doi: 10.1158/1535-7163.MCT-06-0407. [DOI] [PubMed] [Google Scholar]
- 6.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 7.Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 8.Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 9.Mamane Y, Petroulakis E, LeBacquer O, et al. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 10.Abraham RT. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr Top Microbiol Immunol. 2004;279:299–319. doi: 10.1007/978-3-642-18930-2_18. [DOI] [PubMed] [Google Scholar]
- 11.Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wouters BG, van den Beucken T, Magagnin MG, et al. Control of the hypoxic response through regulation of mRNA translation. Semin Cell Dev Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Tang H, Hornstein E, Stolovich M, et al. Amino acid-induced translation of TOP mRNAs is fully dependent on phosphatidylinositol 3-kinase-mediated signaling, is partially inhibited by rapamycin, and is independent of S6K1 and rpS6 phosphorylation. Mol Cell Biol. 2001;21:8671–8683. doi: 10.1128/MCB.21.24.8671-8683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jefferies HB, Fumagalli S, Dennis PB, et al. Rapamycin suppresses 5′ TOP mRNA translation through inhibition of p70s6k. Embo J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 16.Pause A, Belsham GJ, Gingras AC, et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 17.Savaraj N, Wu C, Wangpaichitr M, et al. Overexpression of mutated MRP4 in cisplatin resistant small cell lung cancer cell line: collateral sensitivity to azidothymidine. Int J Oncol. 2003;23:173–179. [PubMed] [Google Scholar]
- 18.Xu J, Tyan T, Cedrone E, et al. Detection of 11q13 amplification as the origin of a homogeneously staining region in small cell lung cancer by chromosome microdissection. Genes Chromosomes Cancer. 1996;17:172–178. doi: 10.1002/(SICI)1098-2264(199611)17:3<172::AID-GCC5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 19.Wu C, Wangpaichitr M, Feun L, et al. Overcoming cisplatin resistance by mTOR inhibitor in lung cancer. Mol Cancer. 2005;4:25. doi: 10.1186/1476-4598-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood SM, Gleadle JM, Pugh CW, et al. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J Biol Chem. 1996;271:15117–15123. doi: 10.1074/jbc.271.25.15117. [DOI] [PubMed] [Google Scholar]
- 21.Hamanaka Y, Mukai M, Shimamura M, et al. Suppression of PI3K/mTOR pathway rescues LLC cells from cell death induced by hypoxia. Biochem Biophys Res Commun. 2005;330:318–326. doi: 10.1016/j.bbrc.2005.02.163. [DOI] [PubMed] [Google Scholar]
- 22.Kimura N, Tokunaga C, Dalal S, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 23.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Kimball SR. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Med Sci Sports Exerc. 2006;38:1958–1964. doi: 10.1249/01.mss.0000233796.16411.13. [DOI] [PubMed] [Google Scholar]
- 25.Robey RB, Hay N. Akt, hexokinase, mTOR: Targeting cellular energy metabolism for cancer therapy. Drug Discover Today:Disease Mechanisms. 2005;2:239–246. [Google Scholar]
- 26.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 27.Crespo JL, Hall MN. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2002;66:579–591. doi: 10.1128/MMBR.66.4.579-591.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dennis PB, Fumagalli S, Thomas G. Target of rapamycin (TOR): balancing the opposing forces of protein synthesis and degradation. Curr Opin Genet Dev. 1999;9:49–54. doi: 10.1016/s0959-437x(99)80007-0. [DOI] [PubMed] [Google Scholar]
- 29.Easton JB, Kurmasheva RT, Houghton PJ. IRS-1: auditing the effectiveness of mTOR inhibitors. Cancer Cell. 2006;9:153–155. doi: 10.1016/j.ccr.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 30.Rubio-Viqueira B, Hidalgo M. Targeting mTOR for cancer treatment. Adv Exp Med Biol. 2006;587:309–327. doi: 10.1007/978-1-4020-5133-3_24. [DOI] [PubMed] [Google Scholar]
- 31.Janus A, Robak T, Smolewski P. The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cell Mol Biol Lett. 2005;10:479–498. [PubMed] [Google Scholar]
- 32.Balsara BR, Pei J, Mitsuuchi Y, et al. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004;25:2053–2059. doi: 10.1093/carcin/bgh226. [DOI] [PubMed] [Google Scholar]
- 33.Dancey JE. Therapeutic targets: MTOR and related pathways. Cancer Biol Ther. 2006;5:1065–1073. doi: 10.4161/cbt.5.9.3175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data S1. Effects of 2-DG and CCI-779 on pAMPK and p-P70S6K. Under hypoxia (48h), 4mM of 2-DG significantly increases pAMPK in SCLC line B which correlates with decreased levels of p-P70S6K a downstream target of pmTOR.