

Abstract
Background/Aims
Dietary factors and intestinal bacteria play an important role in the rapidly increasing incidence of obesity and its associated conditions, such as steatosis and insulin resistance. In the current study, we evaluated the effect of probiotics, and their mechanisms on diet-induced obesity, steatosis and insulin resistance.
Methods
Wild-type male C57BL6 mice were fed either normal or high fat diets. Some mice received VSL#3 probiotics. Animal weight, hepatic steatosis, insulin resistance, and their relationship to hepatic NKT cell number and inflammatory signaling were evaluated.
Results
High fat diet induced a depletion of hepatic NKT cells thus leading to insulin resistance and steatosis. Oral probiotic treatment significantly improved the high fat diet-induced hepatic NKT cell depletion, insulin resistance and hepatic steatosis. This effect was NKT cell -dependant, resulted from the attenuation of the tumor necrosis factor-alpha and I Kappa B Kinase inflammatory signaling, and led to an improved sensitivity in insulin signaling.
Conclusions
Probiotics improve high fat diet-induced steatosis and insulin resistance. These effects of probiotic are likely due to increased hepatic NKT cell numbers and reduced inflammatory signaling.
Keywords: fatty liver, intestinal bacteria, inflammation, cytokines and obesity
Introduction
Obesity and related health problems, particularly those associated with fatty liver disease and insulin-resistance, are increasing rapidly (1–3). Of the various environmental factors that might contribute to the rising incidence of obesity-related diseases, changes in dietary habits merit particular consideration since diets that are enriched in certain macronutrients (e.g., saturated fats) induce both obesity and insulin resistance in experimental animals and humans (4, 5), while other studies showed improved hepatic steatosis with polyunsaturated fatty acid supplements(6, 7). This may explain why westernized diets are associated with an increased prevalence of obesity and type 2 diabetes. However, modification of dietary habits and lifestyles has not been well accepted and has not been successful in the general population in reducing the incidence of obesity and insulin resistance. Better and more easily accepted interventions are badly needed to change the current trend in obesity and insulin resistance. Therefore, it is crucial to understand the mechanisms of diet-induced obesity and to identify potential interventions. Intensive efforts aimed at clarifying mechanisms that underlie diet-induced obesity and insulin resistance have identified the inflammatory process and gut bacteria as important factors.
Growing evidence has pointed to a correlative and causative relationship between inflammation and insulin resistance. Human population studies have linked insulin resistance to systemic inflammation(8, 9). The pro-inflammatory TNF-α/IKK-β signaling pathway has been shown to mediate insulin resistance associated with obesity in many rodent models(10, 11). Constitutive activation of IKK-β in liver causes both hepatic and systematic insulin resistance (12). Conditional disruption of IKK-β in skeletal muscle fails to prevent obesity-induced insulin resistance(13). Taken together, these observations suggest that the liver may be the principal tissue responsible for inflammation-mediated insulin resistance. In the liver, the inflammatory process is, at least partially, regulated by a group of “unconventional” T cells that express both natural killer (NK) receptors and T cell receptors (NKT cell). These NKT cells balance the production of pro-inflammatory and anti-inflammatory cytokines(14). Therefore, alterations of hepatic NKT cell function might lead to the relative over-production of pro-inflammatory cytokines, such as TNF-α, hence, causing both tissue-specific and systemic inflammation and insulin resistance.
Intestinal bacteria are known to play a critical role in obesity and insulin resistance. Colonization of germ-free mice with conventional gut bacteria causes a significant increase in body fat, despite a decrease in food intake(15). Recently, Gordon and colleagues have demonstrated that germ-free mice colonized with gut bacteria from obese mice gained a greater amount of weight than mice colonized with gut bacteria from lean mice on the same diet (16). Germ-free mice are also resistant to high fat diet-induced obesity related metabolic changes (17). Although many physicochemical determinants, such as intra-luminal pH, may effect the composition of gut bacteria, it is the amount and type of substrate available for bacterial growth that has the most significant influence (18). There is an increased abundance of Bacteroidetes and decreased abundance of Firmicutes in obese humans subjected to a fat-restricted diet (19). Probiotics, that is, live microbial food supplements which help maintain the balance of intestinal micro-flora, have shown in a number of studies to be effective in the treatment of insulin resistance in both rodents (20), and humans (21). In our previous study, we showed that probiotic therapy improved steatohepatitis and insulin resistance in leptin deficient, ob/ob mice(20).
In the current study, we evaluated the effect of probiotics on high fat diet-induced obesity, fatty liver and insulin resistance. More importantly, we investigated mechanisms underlying the immuno-modulatory function of probiotics and their role in the inflammatory process and insulin resistance. Results from our current study may have profound therapeutic implications for the management of obesity-related fatty liver diseases and insulin-resistance.
Methods
Animal experiments
Adult (age 6–8 week) male wild-type C57BL-6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). The mice were fed with commercial diets (BioServ, Inc., Frenchtown, NJ) containing either high amounts of fat (HF, #3282, 60% kcal from fat) or normal amounts of fat (ND, 11% kcal from fat) for 8–12 weeks. The mice were pair-fed between the ND and HF group to achieve isocaloric status. CD1d null mice of a C57BL/6 strain were kindly provided by Dr. Mark Soloski and maintained in our laboratory. All mice were maintained in a temperature- and light-controlled facility. Some HF- or ND-fed mice were given VSL#3 probiotics (a mixture of viable, lyophilized bifidobacteria, lactobacilli ands Streptococcus thermophilus, 1.5 × 109 colonies/mouse/day) by oral gavage for 4 weeks after the mice had been on HF or ND diets for 8 weeks. The probiotics were freshly resuspended in sterile water daily, just prior to use. Heat-killed probiotics were used as part of control. To study the insulin signaling pathway, mice were injected with either saline or 25mU/kg of insulin via the portal vein 5 min. before harvesting the liver. All animal experiments fulfilled NIH and JHU criteria for the humane treatment of laboratory animals.
Isolation and cell-surface labeling of hepatic mononuclear cells (HMNCs)
HMNCs were isolated as previously described (22). HMNCs were then labelled with a CD1d tetramer (NIH tetramer facility) loaded with a ligand (PBS-57, an analogue of α-GalCer) or anti-mouse fluorescent antibodies against CD8, CD4, NK1.1 (Pharmingen, San Diego, CA). After surface labeling, HMNCs were evaluated by flow cytometry (Becton Dickinson, Palo Alto, CA), and the data analyzed using Cell Quest software (Becton Dickinson).
Glucose tolerance test (GTT)
Following an overnight fast, mice were given a dose of glucose (1.5 gram/kg, i.p.). Blood glucose from the tail vein (2 µl of blood sample) was measured using an Accu-Check® glucometer with range of 20 – 600 mg/dl (Roche Diagnostics).
Liver Histology and Triglyceride content
Thin slices of liver tissue were stained with hematoxylin. Ten 200X light microscope fields were assessed on each section and scored for the severity of steatosis as previously described (20). The histology was evaluated by an experienced hepatologist (ZL) in a blind fashion. Total lipids were extracted from liver tissue according to published method(23). Triglyceride content was measured with a kit according to manufacturer’s instruction (Sigma-Aldrich #TR0100)
Cell purification and adoptive transfer
Donor NKT cells were isolated from normal livers of wild-type C57BL-6 mice. After isolation, HMNCs or splenic cells were labelled with surface marker antibodies (anti-TCRβ and anti-NK1.1). NKT cells (NK1.1+, TCRβ+) were isolated using a FACSVantage SE high speed sorter (Becton Dickenson), and injected into recipient mice (1×105 cells/mouse via tail vein injection) that had been on a HF diet for 12 weeks. Cells from the NKT cell negative fraction were injected into the control group. The effects of NKT adoptive transfer were evaluated after 3 days.
RNA Isolation and Evaluation of Hepatic Gene Expression
RNA was isolated as described previously(24, 25). cDNA was synthesized from 5 µg of total RNA using oligo(dT) as a template and the SuperScriptII kit (Invitrogen). Quantified PCR amplifications were performed using TagMan Gene Expression Assays PCR Master Mix and primer sets (Applied Biosystems, Foster City, CA). Negative controls were performed without cDNA in the reaction mixture. The results were normalized against glyceraldehyde-3-phosphate dehydrogenase gene expression.
Immunoprecipitation and Western Blot
Whole protein extracts of liver tissue were resolved either by 10% SDS-PAGE directly or immunoprecipitated with antibodies against the insulin receptor (IR), IRS-1 or IRS-2 (Cell Signaling, Beverly, MA) first and then resolved by 10% SDS-PAGE according to standard procedures. Samples were then transferred to 0.45-µm nitrocellulose membranes (Bio-Rad, Hercules, CA), and incubated overnight with various antibodies against total IκB-α, phosphorylated Iκ B-α (Cell Signaling), anti-phosphotyrosine (PY20), Akt(1/2) and activated Akt (pSer473) (Cell Signaling, Beverly, MA) at 4 °C. Chemiluminescent signals were quantitated by density measurement using ImageJ software (NIH).
ELISA-based assay for DNA binding activity of NF-κB
Nuclear extracts were obtained from mice liver tissues using the NE-PER nuclear and cytoplasmic extraction reagents kits (Pierce, Rockford, IL) according to the manufacturer’s instructions. The DNA binding activity of NF-κB was determined using the ELISA-based TransAM NF-κB Family kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol.
Statistical analysis
All values are expressed as mean ±SE. Treatment related differences were evaluated by ANOVA. The paired-individual means were compared by t-test
Results
High fat diet- induced hepatic NKT cell depletion precedes the development of glucose intolerance and hepatic steatosis
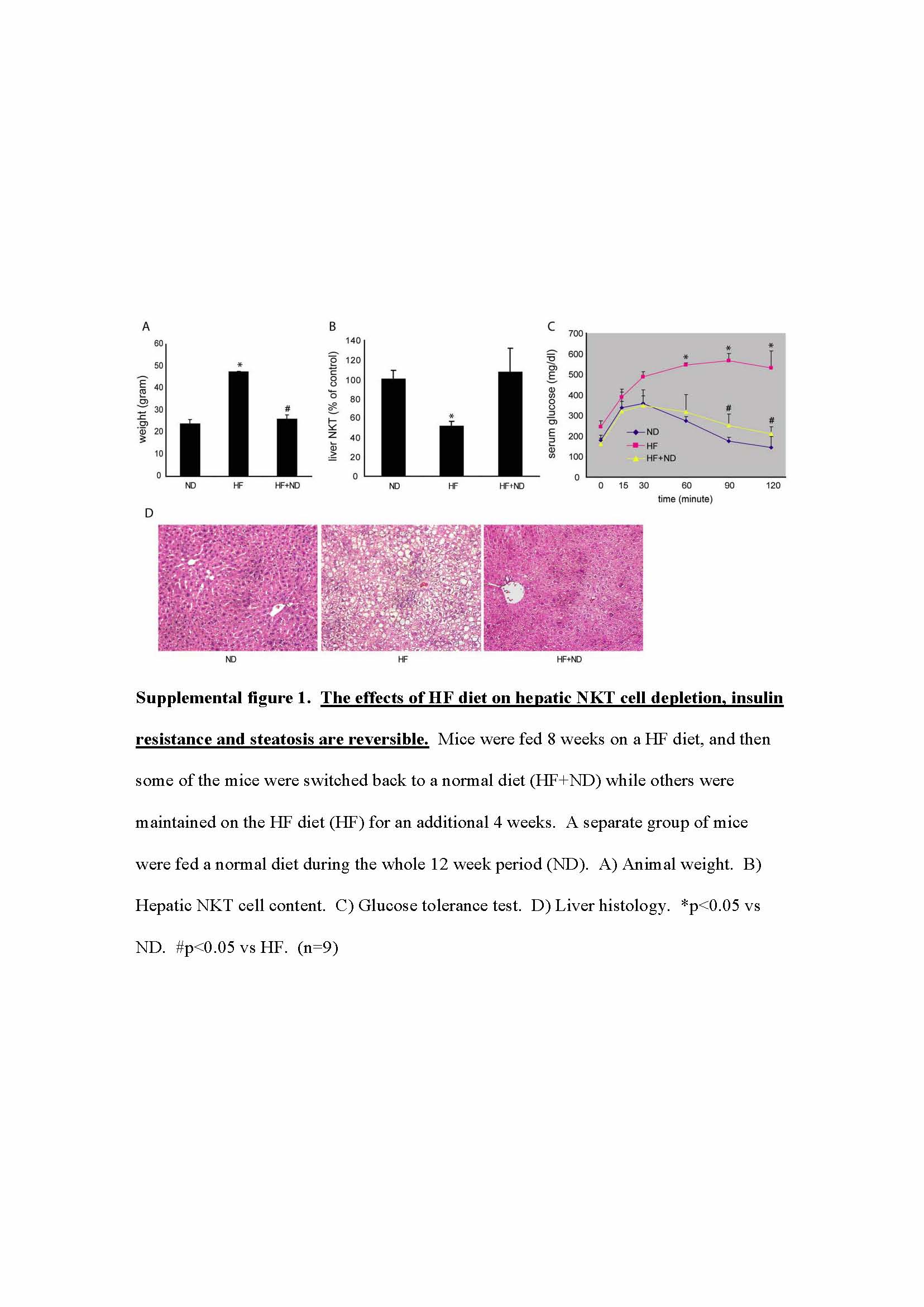
Our previous study showed that HF diet causes obesity, hepatic steatosis and NKT cell depletion (22). However, it was difficult to interpret whether hepatic NKT cell depletion was the cause or the consequence of steatosis. To better understand the causal relationship between hepatic NKT cell depletion and steatosis, a time course analysis of HF diet-induced hepatic NKT cell depletion and steatosis was performed. Our results showed an early onset of hepatic NKT cell (CD1d tetramer positive cells in Fig. 1A) depletion once the animals started the high fat diet. Because the number of total hepatic mononuclear cells (HMNC) remained unchanged, the decreased percentage of CD1d tetramer positive cells reflected the decreased amount of hepatic NKT cells. There was also a major overlap between hepatic CD1d tetramer positive and CD3+ NK1.1+ cells. The majority of CD1d tetramer positive NKT cells were CD3+ NK1.1+ and vice versa (Fig. 1B, C). The depletion of NKT cells appeared to be liver specific since the splenic NKT cells remained unchanged (Fig. 1D). In addition, CD4+ NKT cells were the predominant hepatic NKT cell fraction depleted (Fig. 1D). Mice receiving HF diet also developed insulin resistance and hepatic steatosis (Fig. 1E, F, Table 1). However, the insulin resistance and steatosis only became apparent after eight weeks of HF diet, therefore, hepatic NKT cell depletion preceded the development of insulin resistance and steatosis. The HF diet induced hepatic NKT cell depletion; insulin resistance and steatosis were reversible when the HF diet mice were switched back to a normal diet (Suppl. Fig. 1).
Figure 1. High fat diet decreases hepatic NKT cells and leads to insulin resistance and fatty liver.

Wild type C57BL6 mice were fed either high fat diet (HF) or normal diet (ND). Hepatic mononuclear cells (HMNC) were isolated and NKT cells were identified with CD1d tetramer loaded with a ligand and co-labeled with CD4, CD8, CD3 and NK1.1. A) Histogram of CD1d tetramer labeling of HMNC from HF diet mice at different time points. Each panel represents a fixed number of HMNC counted at a certain time point. There is progressive NKT cell (CD1d tetramer positive at the right side peak) depletion after 2 weeks on HF diet. The total HMNC amount remained unchanged. The decreased percentage of CD1d tetramer positive cells reflected the decreased amount of hepatic NKT cells. B) Dot plot of hepatic CD3+ NK1.1+ cells that were gated on CD1d tetramer positive cells. C) Histogram of hepatic CD1d tetramer cells that were gated on CD3+ NK1.1+ cells. D) Flow cytometric analyses of hepatic and splenic NKT cells. Depletion of CD4+ NKT cells is more prominent than total NKT cells. There was no change in splenic NKT cells. E) Glucose tolerance tests. Significant glucose intolerance only appears after 8 weeks of HF diet. F) Liver histology. Hepatic steatosis is not apparent until 8 weeks of HF diet. (*p<0.05 vs ND) (n=3 at each time points with duplicated experiments).
Table 1.
Probiotics improve HF diet-induced insulin resistance and steatosis.
| Mean ± SD | Histological steatosis score |
Triglyceride mg/g of liver tissue |
HOMA-IR |
|---|---|---|---|
| ND | 0 ± 0** | 5.05 ± 0.92** | 0.78 ± 0.01* |
| HF | 2.8 ± 0.42 | 45.75 ± 6.81 | 1.58 ± 0.25 |
| HF+Probiotics | 1.0 ± 0.94** | 17.73 ± 10.19** | 0.80 ± 0.10* |
| HF+NKT | 1.9 ± 0.74** | 19.01 ± 9.06** | 0.76 ± 0.47 |
| CD1dko+ND | 0 ± 0 | 6.51 ± 2.08 | NA |
| CD1dko+HF | 2.25 ± 0.71§§ | 28.01 ± 6.58§§ | NA |
| CD1dko+HF+probiotics | 2.13 ± 0.64p1 | 22.60 ± 2.63p2 | NA |
Mice were treated the same as described in Figure Legend 2 and 3. Serum glucose and insulin levels were measured after overnight fasting. HOMA-IR, an established measure of insulin sensitivity(40), were calculated as glucose concentration X insulin concentration ÷ 22.5. Liver HE stained sections were evaluated under 200X magnification. On each section, findings from 10 high power fields (HPF) were averaged to derive the data point for that mouse. Hepatic triglyceride was extracted and measured. Mean values (±SD) for 3 mice of each group are shown.
p< 0.05
p< 0.01vs HF or ND
p<0.01 vs CD1dko+ND
0.71
0.17 vs CD1dko+HF
Probiotics improved HF diet-induced hepatic NKT cell depletion
Gut bacteria have been shown to be important regulators of diet-induced obesity and associated metabolic syndrome e(15, 16). Bacterial glycosphingolipids have also been shown to activate NKT cells in vivo(26, 27). Therefore, we evaluated whether probiotic treatment that modifies gut flora has any effect on hepatic NKT cells. We used VSL#3, a probiotic extensively tested in animal models of intestinal inflammation and demonstrated that viable VSL#3 bacteria strains can be cultured from the stool after the oral administration of VSL#3 probiotic to mice, rats, or human subjects(28, 29). Our results showed that probiotics significantly improved HF diet-induced hepatic NKT cell depletion (Fig. 2), but had little effect on hepatic NKT cells of ND-fed mice (Fig. 2). In addition, heatkilled probiotics had no effect on HF diet-induced hepatic NKT cell depletion (data not shown). These indicated that the effect of probiotics on hepatic NKT cells is likely by restoring alternations caused by HF diet, rather than by affecting the hepatic NKT cells directly.
Figure 2. Probiotics improve hepatic NKT cell depletion in HF diet mice.

Mice were fed HF or ND diets for 8 weeks, and then half the mice were treated with probiotics (HF+probiotics, ND+probiotics), and the other half were treated with vehicle (HF, ND) for an additional 4 weeks. All mice were maintained on either HF diet or ND diet. A) Representative flow cytometry dot plot of hepatic NKT cells. The total amounts of hepatic mononuclear cell (HMNC) remain similar between experimental groups. Therefore, the percentage of HMNC represents the amount of hepatic NKT cells in the liver. B) Mean (+SD) results from triplicate experiments are graphed (n=8/exp). *p<0.05 vs HF. C) Mice from different treatment groups consumed a similar amount of calories. Therefore the effects of probiotic were not due to decreased oral intake.
Probiotics improved HF diet- induced obesity, insulin resistance and steatosis
Our previous study showed that probiotic therapy improves steatohepatitis and insulin resistance in leptin deficient, ob/ob mice(20). Here, we evaluated the effect of probiotics in HF diet -induced obesity, insulin resistance and steatosis. Our results show that probiotics significantly reduced weight and improved insulin resistance (Fig. 3A, D, Table 1) and steatosis (Fig. 4A–C, Table 1) in animals fed HF diet, but had little effect on those fed ND diet (Fig. 3A, 3D and 4D). To investigate the role of hepatic NKT cells in the effect of probiotics, normal hepatic NKT cells were isolated and adoptive transferred to HF diet fed mice. There was no change of diet intake among mice that received NKT cell adoptive transfer (data not shown). Adoptive transfer NKT cells significantly improved HF diet induced obesity, insulin resistance (Fig. 3B, E, Table 1) and steatosis (Fig. 4E, F, Table 1), regardless of whether the donor NKT cells were isolated from the liver or spleen (data not shown). Previously, other investigators also showed that adoptive transfer of NKT cells ameliorates non-alcoholic steatohepatitis and glucose intolerance in leptin deficient ob/ob mice(30). To further confirm the pivotal role of NKT cells in the effect of probiotics, NKT cell deficient CD1d null mice(31) were fed HF diet and treated with probiotics as described above. The result showed that HF diet fed CD1d null mice had little improvement of their insulin resistance (Fig 3C, F) after probiotic treatment. There was a slight, but not statistically significant decrease of steatosis (Fig. 4G–I, table 1) indicating that, at least the majority of the effects of probiotics are NKT cell dependant, although we cannot rule out an additional action of probiotics over and above a NKT effect. There was also a statistical difference between the ND and HF + probiotic and the ND and HF + NKT groups indicating that, although probiotic and adoptive transfer treatments improve animal weight significantly, the treatment did not normalize animal weight. This could be either dose dependant or due to the involvement of additional unknown mechanisms.
Figure 3. Probiotics improve obesity and insulin resistance in HF diet mice via a NKT cell dependant manner.

Mice were treated the same as described in Figure 2. A) Animal weight. D) Glucose tolerance tests. (n=8/exp with triplicate experiments). In separate experiments, NKT cells were isolated from the livers of normal mice and then injected to recipient mice that had been on HF diet for 12 weeks (HF+NKT). Control HF mice received the same amount of splenic T cells (HF+T). The animals were then evaluated 4 days after adoptive transfer. B) Animal weight. E) Glucose tolerance tests. (n=6/exp with duplicated experiments). In addition, wild type (WT) and CD1d null (CD1dko) C57BL6 mice (not age and weight matched) were fed for 8 weeks on either ND or HF diets, and then some the HF diet mice were treated with probiotics (wt+HF+probiotics or CD1dko+HF+probiotics), and the other HF diet mice were treated with vehicle (wt+HF or CD1dko+HF) for an additional 4 weeks. ND diet mice all were fed ND diet continuously (CD1dko+ND). C) Animal weight change. F) Glucose tolerance tests. (n=6/exp with duplicated experiments). #p<0.05 vs ND, *p<0.05 vs HF. p=0.36 CD1dko+HF vs CD1dko+HF+probiotics.
Figure 4. Probiotics improve steatosis in HF diet mice via a NKT cell dependant manner.

Mice were treated the same as described in Figure 2 and 3. Representative HE stains of liver histology are shown.
Probiotics reduced inflammatory signaling in HF diet fed mice
Hepatic NKT cells regulate hepatic cytokine production and balance pro-inflammatory and anti-inflammatory response. To evaluate the effect of hepatic NKT cell depletion on hepatic cytokine expressions in HF diet mice, and more importantly, to evaluate the effect of probiotics treatment and NKT cell adoptive transfer, the expression of TNF-α and IL-4 expressions were evaluated in the liver of ND and HF diet mice as well as HF diet mice treated with probiotics and NKT cell adoptive transfer. TNF-α has been identified as a key pro-inflammatory cytokine that mediates insulin resistance(10, 11). IL-4, on the other hand, is the key anti-inflammatory cytokine that mediates NKT cell dependant insulin resistance in NOD mice(32). HF diet significantly increased the expression of TNF-α and reduced the expression of IL-4 (Fig. 5A). Probiotics and adoptive transfer NKT cell treatment improved the elevated TNF-α expression and the suppressed IL-4 expression in HF diet mice (Fig. 5A). TNF-α activates IKK-β, a prerequisite for wild-type mice with diet-induced obesity to develop insulin resistance(33). Following this lead, the hepatic IKK-β activities in mice fed HF and ND diet were evaluated, as well as the effects of probiotic treatment and NKT cell adoptive transfer. IKK-β phosphorylates Iκ Bα. Therefore, the ratio of phospho-Iκ Bα/total-Iκ Bα represents IKK-β activity. The expression of total-IκBα in whole liver extract was measured by Western blot. The phospho-IκBα expression was then measured by stripping and re-probing the same membrane. HF diet fed mice have significantly increased IKK-β activity, as indicated by an increased ratio of phospho-IκBα/total-Iκ Bα (Fig. 5B). Probiotic treatment and adoptive transfer of NKT cells reduced IKK-β activity (Fig. 5B). We further evaluated NF-κB binding activities, the down stream signal for IκBα, with similar findings. HF diet fed mice have significantly increased NF-κB binding activity. Probiotic treatment and adoptive transfer of NKT cells reduced NF-κB binding activity (Fig. 5C).
Figure 5. Probiotics reduces pro-inflammatory signaling in HF diet fed mice.

Mice were treated the same as described in Figure 2 and 3. A) TNF-α and IL-4 expressions were determined by quantitative RT-PCR and normalized with GAPDH expression. B) Western blot of IκBα performed on whole liver extract. Representative autoradiographs are shown. Mean (±SD) results from triplicate experiments are graphed. C) ELISA based NF-κB binding activity assay. *p<0.05 vs HF. **p<0.01 vs HF. (n=8/exp with duplicated experiments)
Probiotics improve hepatic insulin signaling sensitivity in HF diet fed mice
We further evaluated the effect of HF diet-induced NKT cell depletion on the hepatic insulin signaling pathway. Insulin activates the insulin receptor tyrosine kinase (IR), which phosphorylates and recruits substrate adaptors, such as IRS-1 and IRS-2. Tyrosine phosphorylated IRS then displays binding sites for numerous signaling partners. Among them, PI3K has a major role in insulin function, mainly via the activation of the Akt/PKB cascades(34, 35). Protein extracts were prepared from whole liver tissues of ND and HF diet fed mice as well as HF mice treated with probiotics and adoptive transfer NKT cells. The protein extracts were immunoprecipitated with antibodies against IRS-1 or IRS2 and immunoblotted with the antibody specific for phosphotyrosine (PY). Akt activation was determined by immunoblotting of the original lysates with antibodies specific for total Akt and phospho-Akt (pSer473) and then comparing the ratio of phosphor-Akt/total-Akt. Insulin stimulation failed to induce IRS phosphorylation and Akt activation in HF diet fed mice compared to their ND controls, indicating hepatic insulin resistance (Fig. 6). Adoptive transfer NKT cells and probiotics treatment restored the insulin signaling capacity in HF diet fed mice (Fig. 6).
Figure 6. Probiotics improve insulin signaling sensitivity in HF diet mice.

Mice were treated the same as described in figure 2 and 3. In addition, mice received either saline or insulin injection before harvesting liver tissue. A) Western blot of phosphotyrosine (PY), IRS-1 and IRS-2 performed on immunoprecipitates of IRS-1 and IRS2. B) Western blot of p-Akt and Akt1/2 performed on whole liver extract. Representative autoradiographs are shown. Mean (±SD) results are graphed. *p<0.05, **p<0.01 vs HF. (n=8/exp with duplicated experiments)
Discussion
Despite mounting evidence associating dietary factors with obesity-related diseases, the mechanisms underlying their pathogenesis are not well understood. Recently, we reported that a HF diet that induces obesity, insulin resistance and hepatic steatosis also leads to hepatic NKT cell depletion. However, it was not clear whether the depletion of hepatic NKT cells was the cause or the consequence of overall metabolic dysfunction. In the current study, we obtain several lines of evidence to support the theory that the hepatic NKT cell is the key mediator of HF diet-induced metabolic abnormalities: (a) NKT cell depletion is observed long before steatosis and insulin resistance can be detected (Fig. 1), (b) adoptive transfer of NKT cells from a normal donor reverses HF diet-induced steatosis and insulin resistance (Fig. 3, 4 and Table 1), (c) the effects of probiotic treatment on insulin resistance and steatosis are NKT cell dependant (Fig. 3, 4 and Table 1) and (d) methionine-choline deficient (MCD) diet fed mice, which also develop significant steatosis but no insulin resistance(36), do not have hepatic NKT cell depletion despite steatosis (Suppl. Fig. 2). Based on this evidence, we believe that HF diet-induced NKT cell depletion is a contributing factor for steatosis and insulin resistance, rather than a consequence of these pathologies. Recently, Cani and colleagues reported that a high-fat diet increases plasma lipopolysaccharide (LPS) level, which also contributes to the pathogenesis of insulin resistance and increased liver triglyceride content(37). It is possible that the endotoxinemia caused by high fat diet reduces intrahepatic NKT cells and leads to worsened or amplified insulin resistance.
The ability of probiotics to restore hepatic NKT cells and improve HF diet-induced insulin resistance and fatty liver are novel findings and intriguing. A previous study showed that one probiotic, butyrivibrio fibrisolvens, increased splenic NKT cells(38). NKT cells regulation of inflammatory processes and chronic inflammation is known to be associated with insulin resistance and fatty liver disease(33). Our observation proves the hypothesis that probiotics, through modulation of hepatic NKT cells, reduce inflammatory signaling and ultimately lead to improved insulin resistance and to improved fatty liver disease. Our data show that probiotic treatment has no effect on fatty liver and insulin resistance in CD1d null mice, indicating that the response to probiotics involves NKT cell activity. Interestingly, the CD1d null mice do not exhibit insulin resistance, fatty liver or inflammation on the normal diet, suggesting that absence of NKT cells alone is not sufficient to cause obesity-related disorders. It indicates that there is a critical interaction between losing NKT cells, rather than just their absence, and the development of HF diet-induced insulin resistance and fatty liver. We hypothesize that CD1d null mice have developed compensatory mechanisms to regulate inflammatory processes other than by NKT cells. Since the anti-inflammatory activities of probiotics are mediated by NKT cells, which are lost in CD1d null mice, we are currently investigating the mechanisms of probiotic modulation of NKT cells and inflammation.
As discussed earlier, intestinal bacteria are the key mediators that regulate food energy harvest and utilization. In the current study, we demonstrate that intestinal bacteria also play an important role in regulating hepatic inflammatory processes. Modification of intestinal bacteria by probiotics significantly suppressed HF diet-induced activation of the TNF-α/IKK-β signaling pathway, which is the critical signaling for diet-induced insulin resistance. These data suggest that strategies designed to down regulate inflammatory mediators with probiotics have promising potential in patients with NAFLD. We also demonstrate that the effects of probiotics are mediated by hepatic NKT cells, a key component of the innate immune system. In the liver, NKT cells regulate cytokine production and balance pro-inflammatory and anti-inflammatory responses. NKT cells recognize a group of glycolipid antigens bound to the MHC class I-like molecule, CD1d, expressed with β2 micro globulin on various antigen presenting cells(39). Recently, bacterial glycosphingolipids from Gram-negative bacteria have also been shown to activate NKT cells in vivo(26, 27). Therefore, it is possible that intestinal bacteria or probiotics carry antigens that can modulate hepatic NKT cells directly, or indirectly by affecting non-parenchymal cells, such as Kupffer cells, that express CD1 and present antigen to NKT cells. Our current study does not distinguish the direct stimulating effect of probiotics from the indirect effect of probiotics through intestinal bacterial modulation, nor address the possibility of involvement of non-parenchymal cells. However, the lack of effect of probiotics on hepatic NKT cells in ND diet-fed mice implies that the effect of probiotics is through modulation of gut flora. Further studies are needed to separate these two mechanisms.
In conclusion, based on our observations in the current study, we hypothesize that dietary factors, through interaction with intestinal bacteria, modulate hepatic NKT cells, thus causing an inflammatory process which contributes to insulin resistance and steatosis. Further study of the mechanisms of our hypothesis will help to elucidate the pathogenesis of diet induced obesity, insulin resistance and NAFLD and may have profound implications in identifying targets for therapy in obesity-related diseases.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Mr. James Potter for his insightful suggestions and editorial assistance, the Hopkins Digestive Disease Basic Research Development Center (R24 DK064388-04) for providing technical support, and the NIH Tetramer Core Facility for providing CD1d tetramer. This work was supported by the AGA Roche Research Scholar Award in Liver Diseases (ZL) and a research grant from NIH/NIDDK R01DK075990-01 (ZL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003;289:76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. Jama. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 3.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–967. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 4.Harte RA, Kirk EA, Rosenfeld ME, LeBoeuf RC. Initiation of hyperinsulinemia and hyperleptinemia is diet dependant in C57BL/6 mice. Horm Metab Res. 1999;31:570–575. doi: 10.1055/s-2007-978797. [DOI] [PubMed] [Google Scholar]
- 5.Vessby B, Unsitupa M, Hermansen K, Riccardi G, Rivellese AA, Tapsell LC, et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia. 2001;44:312–319. doi: 10.1007/s001250051620. [DOI] [PubMed] [Google Scholar]
- 6.Capanni M, Calella F, Biagini MR, Genise S, Raimondi L, Bedogni G, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther. 2006;23:1143–1151. doi: 10.1111/j.1365-2036.2006.02885.x. [DOI] [PubMed] [Google Scholar]
- 7.Alwayn IP, Andersson C, Zauscher B, Gura K, Nose V, Puder M. Omega-3 fatty acids improve hepatic steatosis in a murine model: potential implications for the marginal steatotic liver donor. Transplantation. 2005;79:606–608. doi: 10.1097/01.tp.0000150023.86487.44. [DOI] [PubMed] [Google Scholar]
- 8.Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41:1241–1248. doi: 10.1007/s001250051058. [DOI] [PubMed] [Google Scholar]
- 9.Grimble RF. Inflammatory status and insulin resistance. Curr Opin Clin Nutr Metab Care. 2002;5:551–559. doi: 10.1097/00075197-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 10.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 11.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 12.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohl M, Pasparakis M, Baudler S, Baumgartl J, Gautam D, Huth M, et al. Conditional disruption of I{kappa}B kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest. 2004;113:474–481. doi: 10.1172/JCI18712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki S, Habu Y, Kawamura T, Takeda K, Dobashi H, Ohkawa T. The liver as a crucial orgain in the first line of host defense: the role of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells and T helper 1 immune responses. Immunol Rev. 2000;174:35–46. doi: 10.1034/j.1600-0528.2002.017404.x. [DOI] [PubMed] [Google Scholar]
- 15.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. PNAS. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 17.Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. PNAS. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freter R. In: Probiotics: The Scientific Basis. Fuller R, editor. London: Chapman and Hall; 1992. pp. 111–144. [Google Scholar]
- 19.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: Human gut microbes associated with obesity. Nature. 2006;444:1022. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 21.Loguercio C, DeSimone T, Federico A, Terracciano F, Tuccillo C, DiChicco M, et al. Gut-liver axis: a new point of attack to treat chronic liver damage? Am J Gastroenterol. 2002;97:2144–2146. doi: 10.1111/j.1572-0241.2002.05942.x. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42:880–885. doi: 10.1002/hep.20826. [DOI] [PubMed] [Google Scholar]
- 23.Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:457–509. [PubMed] [Google Scholar]
- 24.Yang SQ, Lin HZ, Diehl AM. Fatty liver vulnerability to lipopolysaccaride despite NF-kB induction and caspase 3 inhibition. Am J Physiol. 2001;281:G382–G392. doi: 10.1152/ajpgi.2001.281.2.G382. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidine thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–161. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 27.Mattner J, DeBord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 28.Brigidi P, Vitali B, Swennen E, Altomare L, Rossi M, Matteuzzi D. Specific detection of bifidobacterium strains in a pharmaceutical probiotic product and in human feces by polymerase chain reaction. Syst Appl Microbiol. 2000;23:391–399. doi: 10.1016/S0723-2020(00)80070-3. [DOI] [PubMed] [Google Scholar]
- 29.Venturi A, Gionchetti P, Rizzello F, Johansson R, Zucconi E, Brigidi P, et al. Impact on the composition of the faecal flora by a new probiotic preparation: preliminary data on maintenance treatment of patients with ulcerative colitis. Aliment Pharmacol Ther. 1999;13:1103–1108. doi: 10.1046/j.1365-2036.1999.00560.x. [DOI] [PubMed] [Google Scholar]
- 30.Elinav E, Pappo O, Sklair-Levy M, Margalit M, Shibolet O, Gomori M, et al. Amelioration of non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice by oral immune regulation towards liver-extracted proteins is associated with elevated intrahepatic NKT lymphocytes and serum IL-10 levels. J Pathol. 2006;208:74–81. doi: 10.1002/path.1869. [DOI] [PubMed] [Google Scholar]
- 31.Mendiratta SK, Martin WD, Hong S, Boesteanu A, Joyce S, Van Kaer L. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 1997;6:469–477. doi: 10.1016/s1074-7613(00)80290-3. [DOI] [PubMed] [Google Scholar]
- 32.Laloux V, Beaudoin L, Jeske D, Carnaud C, Lehuen A. NK T cell-induced protection against diabetes in V alpha 14-J alpha 281 transgenic nonobese diabetic mice is associated with a Th2 shift circumscribed regionally to the islets and functionally to islet autoantigen. J Immunol. 2001;166:3749–3756. doi: 10.4049/jimmunol.166.6.3749. [DOI] [PubMed] [Google Scholar]
- 33.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li Z-W, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IKKbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 34.Johnston AM, Pirola L, Van Obberghen E. Molecular mechanisms of insulin receptor substrate protein-mediated modulation of insulin signalling. FEBS Lett. 2003;546:32–36. doi: 10.1016/s0014-5793(03)00438-1. [DOI] [PubMed] [Google Scholar]
- 35.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 36.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 37.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 38.Ohkawara S, Furuya H, Nagashima K, Asanuma N, Hino T. Oral administration of butyrivibrio fibrisolvens, a butyrate-producing bacterium, decreases the formation of aberrant crypt foci in the colon and rectum of mice. J Nutr. 2005;135:2878–2883. doi: 10.1093/jn/135.12.2878. [DOI] [PubMed] [Google Scholar]
- 39.Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med. 1994;180:1097–1106. doi: 10.1084/jem.180.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radziuk J. Insulin sensitivity and its measurement: structural commonalities among the methods. J Clin Endocrinol Metab. 2000;85:4426–4433. doi: 10.1210/jcem.85.12.7025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.