

Abstract
The role of nuclear factor kappa B (NF-κB) in oxidative stress, and most recently in pro- and anti-apoptotic related mechanistic pathways, has well been established. Because of the dual nature of NF-κB, the wide range of genes it regulates and the plethora of stimuli that activate it, various studies addressing the functional role of NF-κB proteins have resulted in a number of differing findings. The present study examined the effect of a stimulus-free environment on the frontal cortex of mice brain with the p50 subunit of NF-κB knocked out p50 (-/-). Homozygous p50 mice knockout (KO) and wild type (WT) were used, and at 7-9 weeks they were sacrificed and various brain regions dissected. We analyzed the levels of oxidation in the frontal cortex of both the p50 (-/-) and WT mice. There was a significant reduction in the levels of protein-bound 4-hydroxynonenal (HNE) [a lipid peroxidation product], 3-nitrotyrosine (3NT), and protein carbonyls in the p50 (-/-) mice when compared to the WT. A proteomic profile analysis identified ATP synthase gamma chain, ubiquinol-cyt-C reductase, heat shock protein 10 (Hsp10), fructose bisphosphate aldolase C, and NADH-ubiquinone oxidoreductase as proteins whose expressions were significantly increased in the p50 (-/-) mice compared to the WT. With the reduction in the levels of oxidative stress and the increase in expression of key proteins in the p50 (-/-) brain, this study suggests that the p50 subunit can potentially be targeted for the development of therapeutic interventions in disorders in which oxidative stress plays a key role.
Keywords: NF-Kappa B, Proteomics, Oxidative stress, p50 Knockout Mice
Introduction
Nuclear factor kappa B (NF-κB) is a key transcription factor that was earlier known to regulate immune and inflammatory processes and viral replication (Baeuerle and Henkel, 1994; Baldwin, 1996). The NF-κB family of transcription factors play key roles in the regulation of cell growth, activation, differentiation, and survival. In addition, NF-κB plays an important role in synaptic plasticity and long-term memory formation (Albensi and Mattson, 2000; Mattson et al., 2000; Santoro et al., 2003). The activation of NF-κB is responsible for transcription of various genes like: TNFα, interleukins, e.g., IL1, IL2, and IL6; chemokines, adhesion molecules; enzymes such as inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) and proliferation-related proteins such as cyclin D1 among many others (Karin et al., 2002; Ferrucci et al., 2004; Chung et al., 2005). In mammals, this family of NF-κB consists of several proteins, which include NF-κB1 (p50), NF-κB2 (p52), RelA (p65), c-Rel (Rel), and RelB (Hayden and Ghosh, 2004). A hallmark of this family of proteins is that they contain a highly conserved domain of ∼300 amino acids, termed the Rel homology domain, which contains sequences important for dimerization, DNA binding, and nuclear localization (Grilli et al., 1993; Hayden and Ghosh, 2004). The classical and well-studied NF-κB heterodimer, which is also a potent activator of gene expression, is composed of p50 and p65 subunits. Unlike p65, p50 and p52 subunits lack transactivation domains and are produced either by the proteolytic processing of the precursor molecules p105 and p100 or cotranslationally from incompletely synthesized molecules by the proteasome (Ghosh et al., 1998; Sun and Andersson, 2002; Moorthy et al., 2006).
NF-κB is expressed in many cell types in the nervous system and is constitutively active in subsets of cells in the cortex and hippocampus of the rodent brain at comparably low levels (Kaltschmidt et al., 1994; O'Neill and Kaltschmidt, 1997). In resting cells, the classical NF-κB heterodimer p65/p50 is complexed with the inhibitor protein IκB, of which there are 5 known isoforms; IκBα, IκBβ, IκBγ, IκBε, and Bcl-3. NF-κB is sequestered as an inactive complex in the cytoplasm by IκB, until activation or stimulation of NF-κB in the cells leads to the phosphorylation and immediate proteasomal-mediated degradation of IκB. This leads to the nuclear translocation of NF-κB which then binds to the regulatory region of DNA and leads to transcription of various responsive genes (May and Ghosh, 1998; Santoro et al., 2003; O'Donnell et al., 2005). NF-κB can be activated in both transcriptional activating and repressing forms (Beg and Baltimore, 1996; May and Ghosh, 1998). This dual role of NF-κB has indeed presented considerable complexities and difficulties in studies that try to examine the mechanisms of action of this transcription factor. Hence, the precise role of NF-κB and its related mechanistic pathways have not been completely established.
Other than the classical heterodimer composed of the p50 and p65 subunits, little is known about gene regulation by other hetero- and homodimeric forms of NF-κB (Gadjeva et al., 2004). NF-κB knock outs have provided the best avenue for the investigation of the various roles of these subunits in the activation of NF-κB. It is known that p50-deficient mice develop normally, though they display functional defects in immune response, (Beg et al., 1995). Neurons from p50 knock out mice survive well in culture and are no more sensitive or resistant to neuronal death than wild type neurons (Aleyasin et al., 2004),. On the other hand, p65 knockouts have been shown to have defects during development and to die prematurely of liver apoptosis (Beg et al., 1995; Hayden and Ghosh, 2004). As a result, p50 knockout mice are preferred in studies that try to elucidate various mechanisms of NF-κB-related activity.
Considering the above, the current study involved further analysis of the p50 subunit of NF-κB. Since most studies on p50 knockout mice have so far been carried out using a wide range of stimuli (Weih et al., 1997; Iimuro et al., 1998; Pennypacker et al., 2001; Mabley et al., 2002; Gadjeva et al., 2004; Kassed and Herkenham, 2004), the present study provided a unique opportunity for investigating the role of p50 subunit in a stimulus-free experimental setting. We hypothesized that the homozygous knock out of the p50 (-/-) subunit could potentially be protective in a stimulus-free environment through an oxidative stress mechanism. To test our hypothesis, we measured the levels of oxidative stress biomarkers in the frontal cortex of WT and p50 (-/-) mice and observed a significant reduction in the levels of lipid peroxidation, as measured by the lipid peroxidation product HNE, and the levels of protein oxidation, as measured by the levels of 3-nitrotyrosine (3NT) and protein carbonyls, in the brains of p50 (-/-) mice compared to the WT. We further carried out a differential expression proteomic analysis on brain proteins of the p50 (-/-) mice, and we identified five proteins to be significantly increased in expression in the p50 (-/-) mice when compared to the WT. We determined that the lack of the p50 subunit makes the brain of mice less susceptible to oxidative stress and also activates the expression of key proteins involved in energy metabolism and antioxidant activity. Hence, this study has provided insights into the role of the p50 subunit of NF-κB and additional evidence on the mechanism of NF-κB activation.
Results
Levels of oxidative stress
The levels of protein carbonyls, 3NT and 4-hydroxnonenal (HNE) as indicators of oxidative stress were measured in the frontal cortex of p50 (-/-) mice and compared to WT. Fig.1 shows total protein oxidation measured by the accumulation of protein carbonyls and 3NT and also the levels of lipid peroxidation as measured by protein-bound HNE. There was a significant decrease in the levels of all oxidative stress parameters measured in brains of the p50 (-/-) mice compared to that of WT.
Figure 1.

Protein carbonyl, 3NT and HNE levels in the frontal cortex of p50 (-/-) compared to wild type. There was a significant reduction in the levels of protein carbonyls, 3NT and HNE in the p50 (-/-) mice compared to the wild type. Data are represented as % Wild type (WT); error bars indicate the SEM for each group measured (* p<0.05 n=5).
Protein expression levels
Two-dimensional electrophoresis offers an excellent tool for the screening of abundant protein changes in various disease states (Butterfield, 2004). To assess whether there were any changes in the proteomic profile in the brain of p50 (-/-) mice, we investigated the pattern of protein expression in the frontal cortex from the p50 (-/-) mice compared to the wild type. Comparing the densitometric intensities of individual spots on the gels, five proteins were expressed at significantly higher levels in the frontal cortex of p50 (-/-) mice compared to the wild-type. Fig 2a shows SYPRO ruby stained 2D gels of the p50 (-/-) mice (KO) vs. wild type (WT) groups, with identified protein boxed and labeled. Fig 2b provides an expanded view of select proteins showing their increased expression. The brain proteins identified with increased expression in the p50 (-/-) mice were: ATP synthase gamma chain, ubiquinol-cyt-C reductase, heat shock protein (Hsp10), fructose bisphosphate aldolase C and NADH-ubiquinone oxidoreductase. These proteins identified by mass spectrometry are shown in Table 1. Table 2 provides the changes in densitometric intensities of protein levels expressed as arbitrary units (A.U) ± S.E.M. Table 1 shows that the Mowse scores obtained are all highly significant and that the probability of a random identification using proteomics is exceedingly small (i.e., p < 8.0 × 10-7).
Figure 2.

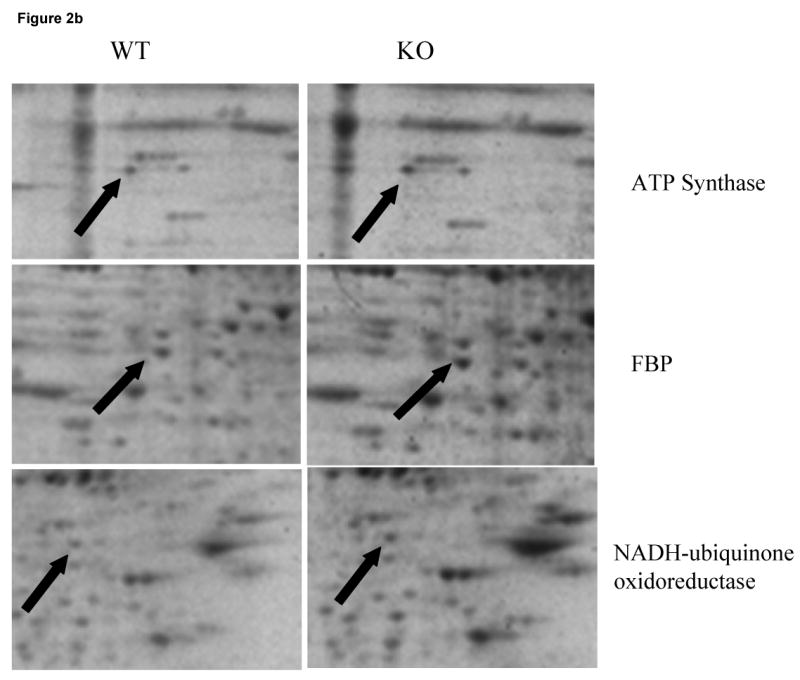
Figure 2a: SYPRO Ruby-stained 2D-gels maps of frontal cortex brain samples from Wild-type vs. p50 (-/-) mice. Proteins identified by mass spectrometry showing differential expression are presented as the boxed spots.
Figure 2b: An expanded view of SYPRO Ruby-stained 2D-gels maps for a select proteins identified whose expression was found to be significantly increasesd. Left panel shows the WT while right panel shows the KO gels. Proteins are marked by the solid arrows.
Table 1.
Differentially expressed protein levels from mice frontal cortex
| Identified Protein | GI accession number | Number of peptide matches identified | % Coverage of matched peptides | pI, MrW | Mowse score | Probability of a random identification |
|---|---|---|---|---|---|---|
| ATP Synthase gamma chain | gi|21263432 | 8 | 20 | 9.06,32900 | 61 | 8.0 ×10-7 |
| Ubiquinol-cyt-C reductase core protein 2 | gi|14548302 | 8 | 23 | 9.26,48200 | 77 | 2.0 ×10-8 |
| Heat shock protein, mitochondrial (Hsp10) | gi|2493662 | 14 | 43 | 8.18,10800 | 74 | 4.0 ×10-8 |
| Fructose Bisphosphate aldolase C | gi|32470593 | 9 | 22 | 6.79,39600 | 162 | 6.3 ×10-17 |
| NADH-ubiquinone oxidoreductase | gi|23396786| | 8 | 28 | 6.40,30300 | 102 | 6.3 ×10-11 |
Table 2.
Proteomic characterizations of differentially expressed NF-κB knockout mice brain proteins identified
| Identified Protein | WT(A.U. ± S.E.M) | KO (A.U. ± .E.M) | Fold Change | p value |
|---|---|---|---|---|
| ATP Synthase | 959 ± 160 | 1367 ± 88 | 1.4 | 0.05 |
| Ubiquinol cyt C reductase | 3514 ± 1482 | 8008 ± 1393 | 2.3 | 0.01 |
| HSP 10 | 1341 ± 542 | 2671 ± 155 | 1.9 | 0.04 |
| FBP | 1344 ± 84 | 1609 ± 35 | 1.2 | 0.01 |
| NADH-Ubiquinone oxidoreductase | 346 ± 137 | 781 ± 133 | 2.3 | 0.05 |
Discussion
In the present study we have observed that the complete knock out of the p50 subunit in a stimulus-free experimental setting leads to a reduction in the levels of oxidative damage in the p50 (-/-) mice brain when compared to that of the wild type mouse. We observed a significant reduction in the levels of oxidative stress biomarkers, i.e., protein carbonyls, 3NT and protein modification by the lipid peroxidation product HNE. In addition, following a proteomic profile analysis of the frontal cortex of the p50 (-/-) mice brain, there was a significant increase in the expression of proteins related to energy metabolism and antioxidant activity compared to wild type. These results are an intriguing finding and are discussed here with relevance to the mechanism of NF-κB transcription factor, and in particular the possible role of the p50 subunit in oxidative stress in a stimulus-free environment.
The disruption of the p50 subunit in various experimental settings has generated a number of interesting findings. These include increased oxidative stress, calcium dysregulation, and damage to striatal neurons following treatment with mitochondrial toxin 3-nitropropionic acid (3NP) (Yu et al., 1999); negative modulation of learning abilities and hippocampal response to brain injury (Kassed et al., 2002); higher constitutive expression of COX-2 following an acute inhalation of pulmonary irritant ozone (Mabley et al., 2002; Fakhrzadeh et al., 2004); and age-related neuronal degeneration (Lu et al., 2006) in p50 knock out mice. On the contrary, studies have shown that NF-κB p50-deficient mice show increased hippocampal neuronal survival and reduced infarct size after middle cerebral artery occlusion (Schneider et al., 1999; Pennypacker et al., 2001; Nurmi et al., 2004). Further, kainate induced excitotoxity by the AMPA-glutamate receptor subunit GluR1 in neuronal culture is inhibited by antisense knockdown of NF-κB p50 (Yu et al., 2002). The studies mentioned above provide two differing views resulting from the disruption of the p50 subunit of NF-κB. While one group shows a protective and beneficial effect, the other group shows a deleterious effect. To make matters more difficult to interpret, p50 knockout mice have elicited a wide range of immune responses to infections. The knockouts have shown to be more susceptible to some bacterial infections, such as Streptococcus pneumonie, respond normally to others, such as Escherichia coli, and are more resistant to viruses, such as murine encheplomyocarditis compared to wild type (Sha et al., 1995). These multiple findings present researchers with a difficult task of trying to conclusively elucidate the exact mechanisms of this key transcription factor (Lipton, 1997).
It has been shown that NF-κB is redox regulated and that oxidative stress is an activator of NF-κB. NF-κB is a known regulator of gene transcription and most agents that activate NF-κB trigger the formation of reactive oxygen species (ROS), or are oxidants themselves (Laskin and Pendino, 1995; Flohe et al., 1997; Kim et al., 2000; Chung et al., 2005). In the present study, we observed a significant decrease (20-30%) in the levels of oxidative stress as indexed by oxidative stress biomarkers, i.e., protein carbonyls, 3NT and lipid peroxidation product HNE in the brains of p50 (-/-) mice compared to those of the WT. Previously, 4-hydroxyhexenal (HHE), a reactive aldehyde similar to HNE, was reported to activate NF-κB, which further activates p38, MAPK, and extracellular signal regulated kinase (ERK) leading to various signaling cascades (Je et al., 2004). Reduction in the levels of oxidative damage in various neurodegenerative models, for example through the use of antioxidants or related compounds, has strong implications in various physiological functions (Farr et al., 2003; Poon et al., 2004b). As a result, the reduced levels of HNE and other oxidative stress biomarkers in the p50 (-/-) mice observed in the present study could thus inhibit the activation of NF-κB leading to a possible protective mechanism.
Since the p65 but not the p50 subunit of NF-κB has a transactivation domain, there is a possibility that the observed protective effects of knocking out the p50 subunit could be a result of a compensatory mechanism elicited by p65. This compensatory phenomenon has previously been observed in a number of p50 knockout studies (Beg et al., 1995; Iimuro et al., 1998; Kato et al., 2002; Gadjeva et al., 2004). We propose that p65's compensatory role would lead to a more functional NF-κB transcription factor and enhance its activity leading to the increased transcription of protective genes that eventual result in the reduction of the levels of oxidative stress observed in the current study.
It is also probable that other pathways in the activation of NF-κB could be playing a role in the mechanisms leading to the reduction in oxidative stress levels observed in the p50 (-/-) mice. One potential pathway could be the involvement of nuclear factor-erythroid 2–related factor 2 (Nrf2). Nrf2 regulates the basal and inducible expression of various antioxidant and other cytoprotective genes (Motohashi and Yamamoto, 2004). A possible cross-talk between these redox-sensitive transcription factors has been proposed (Yang et al., 2005). Nrf2 regulates NF-κB activation largely by modulating its upstream signaling components since just like NF-κB, Nrf2 is also redox regulated (Thimmulappa et al., 2006). Thus, the deletion of the p50 subunit would possibly play a role in the interaction of these two transcription factors leading to the transcription of a broad range of antioxidants resulting in the decreased levels of oxidative stress observed. We should, however, say that this is still a speculation.
In addition to a reduction in the levels of oxidation observed in the p50 (-/-) mice, we also observed a significant increase in the expression of key proteins related to energy metabolism and cytoprotective activities. This is not surprising since it has previously been shown that oxidative stress induces activation of NF-κB leading to the up-regulation of antioxidant enzymes such as glutathione peroxidase and catalase among others (Messina et al., 2006). There was a significant increase in the expression of key energy metabolism-related proteins in the p50 (-/-) mice compared to the wild type. Among these was NADH:ubiquinone oxidoreductase, also known as Complex I. It couples the oxidation of NADH and the reduction of ubiquinone, leading to the generation of a proton gradient which is then used for generation of ATP in the mitochondria (Weiss et al., 1991). Another elevated protein in p50 (-/-) mice was ubiquinol-cytochrome-C reductase, also known as the bc1 complex or complex III. This protein is the central redox enzyme in the ETC and catalyzes the oxidation of diverse quinols by high potential redox-carriers. In the process, a proton gradient is generated that is utilized for the generation of ATP (Mulkidjanian, 2005). ATP synthase, also known as Complex V, which is a mitochondrial enzyme that utilizes the electrochemical proton gradient established across the inner mitochondrial membrane by the ETC for synthesis of ATP (Leyva et al., 2003), is another elevated protein in p50 (-/-) mice. Fructose bisphosphate aldolase C (FBP), which is a glycolytic enzyme that catalyses the reversible aldol cleavage or condensation of fructose-1, 6-bisphosphate into dihydroxyacetone-phosphate and glyceraldehyde 3-phosphate (Perham, 1990), is also elevated in brains of p50 (-/-) mice. The increase in expression of these proteins would result in efficient mechanisms for the generation of a proton gradient, improved glycolytic function, and an overall increase in energy metabolism, thereby enhancing the crucial cellular process of ATP generation and energy dependent process in the p50 (-/-) mice.
We also observed a significant increase in the expression of heat shock protein 10 (Hsp10), also known as chaperonin 10 in the p50 (-/-) mice compared to the wild type. Heat shock proteins (HSP) are a family of molecules that are highly conserved during evolution and involved in many cellular functions. HSP 10 is a mitochondrial protein particularly involved in protein folding. Increased expression of HSPs has recently been reported in neurodegenerative disorders (Poon et al., 2004a). As a result, the increase in the expression of HSP10 in the p50 (-/-) mice compared to the wild type observed here could mean enhanced activity leading to proper folding of proteins and more protection against oxidative stress, thereby contributing to the significant reduction of oxidative stress observed in these p50 (-/-) mice. Thus, the increase in expression of three key proteins of the ETC, a glycolytic enzyme, and a chaperone protein discussed above ensures not only sufficient generation of adequate ATP that would lead to improved cellular activities and hence survival in the p50 (-/-) mice, but also the proper folding of proteins.
Another possible explanation for the increase in the expression of the aforementioned proteins is a retrograde signaling pathway from the mitochondria in a vain attempt to recruit the knocked out p50 subunit to the nucleus for transcription in a compensatory mechanism. It has been shown that calcineurin (Cn), a calcium dependent phosphatase, can dephosphorylate the NF-κB inhibitor IκBβ when the mitochondrion is stressed (Biswas et al., 2003). The current study did not involve using a stimulus, but the up-regulation of mitochondrial ETC proteins, a glycolytic enzyme to help provide eventual substrates for ETC, and a mitochondrial specific heat shock protein conceivably suggests that the mitochondria is trying to signal for transcription of p50 target genes. Due to the diverse nature of the NF-κB signaling pathway this hypothesis is not outside the realm of possibility, but is presently speculative.
The present study and those mentioned in this manuscript underscore the complexities surrounding the mechanisms of NF-κB. The ultimate biological effect of NF-κB is highly dependent on the stimulus responsible for its activation or repression, the subunit composition of the gene targets, and cell type among many other factors (Culmsee et al., 2003). With these many variables to consider, a wide scope in the interpretation of results would be expected. Not much is known about the association between the p50 subunit of NF-κB and oxidative stress, particularly in a stimulus-free environment. It should be noted that whereas all these studies mentioned in this manuscript had various stimuli, in the present study no stimulus was given. We have shown that there is a reduction in the levels of oxidative stress and that there is also a significant increase in the expression of brain proteins related to energy metabolism and cytoprotective activities in the frontal cortex of the p50 (-/-) mice compared to wild type. The proposed mechanisms leading to the observed effects are currently speculative and require additional studies. Recognizing that additional studies are required to elucidate the functional consequence of the observed phenomenon, this study still provides insightful findings on role of NF-κB transcription factor, and in particular its p50 subunit, in a stimulus-free environment.
Experimental Procedures
Animals
Male wild-type (n=5) and NF-κB p50 -/- mice [B6, 129P-Nfkb1] (n=5) were received from Jackson Laboratory at 4-6 weeks and were sacrificed at 7-9 weeks. They were housed in a temperature and humidity controlled room with a 12:12 h light: dark cycle, with food and water available ad libitum. All procedures and protocols were approved by the Institutional Animal Care and Use Committee. Neural degeneration has been described for p50-/- mice at 6 and 10 months of age (Lu et al., 2006).
Measurement of protein carbonyls
Protein carbonyls are an index of protein oxidation and were determined as described previously (Butterfield and Stadtman, 1997). Briefly, samples (5 μg of protein) were derivatized with 10 mM 2, 4-dinitrophenylhydrazine (DNPH) in the presence of 5 μL of 12% sodium dodecyl sulfate for 20 min at room temperature (23°C). The samples were then neutralized with 7.5 μL of the neutralization solution (2 M Tris in 30% glycerol). Derivatized protein samples were then blotted onto a nitrocellulose membrane with a slot-blot apparatus (250 ng per lane). The membrane was then washed with wash buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) and blocked by incubation in the presence of 5% bovine serum albumin, followed by incubation with rabbit polyclonal anti-DNPH antibody (1: 100 dilution) as the primary antibody for 1 h. The membranes were washed with wash buffer and further incubated with alkaline phosphatase-conjugated goat anti-rabbit antibody as the secondary antibody for 1 h. Blots were developed using fast tablet (BCIP/NBT; Sigma-Aldrich) and quantified using Scion Image (PC version of Macintosh-compatible NIH Image) software. No non-specific background binding of the primary or secondary antibodies was found.
Measurement of 3-nitrotyrosine (3-NT)
Nitration of proteins is another form of protein oxidation (Castegna et al., 2003). The nitrotyrosine content was determined immunochemically as previously described (Drake et al., 2003). Briefly, samples were incubated with Laemmli sample buffer in a 1:2 ratio (0.125 M Trizma base, pH 6.8, 4% sodium dodecyl sulfate, 20% glycerol) for 20 min. Protein (250 ng) was then blotted onto the nitrocellulose paper using the slot-blot apparatus and immunochemical methods as described above for protein carbonyls. The mouse anti-nitrotyrosine antibody sigma (5: 1000 dilutions) was used as the primary antibody and alkaline phosphatase-conjugated anti-mouse secondary antibody was used for detection. Blots were then scanned using scion imaging and densitometric analysis of bands in images of the blots was used to calculate levels of 3-NT. No non-specific binding of the primary or secondary antibodies was found.
Measurement of 4-hydoxynonenal (HNE)
HNE is a marker of lipid oxidation and the assay was performed as previously described (Lauderback et al., 2001). Briefly, 10 μl of sample were incubated with 10 μl of Laemmli buffer containing 0.125 M Tris base pH 6.8, 4 % (v/v) SDS, and 20% (v/v) Glycerol. The resulting sample (250 ng) was loaded per well in the slot blot apparatus containing a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 3% (w/v) bovine serum albumin (BSA) in phosphate buffered saline containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST) for 1 h and incubated with a 1:5000 dilution of anti-4-hydroxynonenal (HNE) polyclonal antibody (sigma) in PBST for 90 min. Following completion of the primary antibody incubation, the membranes were washed three times in PBST. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8000 in PBST and added to the membrane. The membrane was washed in PBST three times and developed using Sigmafast Tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop, and quantified by Scion Image. A small background of the primary antibody binding to the membrane was found, but this was the same in both control and subject blots.
Two-dimensional electrophoresis
Sample preparation
Brain samples (200 μg) were incubated with 4 volumes of 2N HCl at room temperature for electrophoresis. Proteins were then precipitated by the addition of ice-cold 100% trichloroacetic acid (TCA) to obtain a final concentration of 15% TCA. Samples were then placed on ice for 10 min and precipitates centrifuged at 16,000 g for 3min. The resulting pellet was then washed three times with a 1:1(v/v) ethanol/ethyl acetate solution. The samples were then suspended in 200 μl of rehydration buffer composed of a 1:1 ratio (v/v) of the Zwittergent solubilization buffer (7M urea, 2M thiourea, 2% Chaps, 65 mM DTT, 1% Zwittergent 0.8% 3-10 ampholytes and bromophenol blue) and ASB-14 solubilization buffer (7M urea, 2M thiourea 5Mn TCEP, 1% (w/v) ASB-14, 1% (v/v) Triton X-100, 0.5% Chaps, 0.5% 3-10 ampholytes) for 1 h.
First dimension electrophoresis
For the first-dimension electrophoresis, 200 μL of sample solution was applied to a 110-mm pH 3–10 ReadyStrip™ IPG strips (Bio-Rad, Hercules CA). The strips were then actively rehydrated in the protean Isoelectric focusing (IEF) cell (Bio-Rad) at 50 V for 18 h. The isoelectric focusing was performed in increasing voltages as follows; 300 V for 1 h, then linear gradient to 8000 V for 5 h and finally 20 000 V/h. Strips were then stored at −80 °C until the 2nd dimension electrophoresis was to be performed.
Second dimension electrophoresis
For the second dimension, the IPG® Strips, pH 3–10, were equilibrated for 10 min in 50 mM Tris–HCl (pH 6.8) containing 6 M urea, 1% (w/v) sodium dodecyl sulfate (SDS), 30% (v/v) glycerol, and 0.5% dithiothreitol, and then re-equilibrated for 15 min in the same buffer containing 4.5% iodacetamide instead of dithiothreitol. Linear gradient precast criterion Tris–HCl gels (8–16%) (Bio-Rad) were used to perform second dimension electrophoresis. Precision Protein™ Standards (Bio-Rad, CA) were run along with the sample at 200 V for 65 min.
SYPRO ruby staining
After the second dimension electrophoresis, the gels were incubated in fixing solution (7% acetic acid, 10% methanol) for 20 min and stained overnight at room temperature with 50ml SYPRO Ruby gel stain (Bio-Rad). The SYPRO ruby gel stain was then removed and gels stored in DI water. It has been previously reported that 2D gel protein spots as low as 1 ng can be detected with Sypro Ruby stain (Nishihara and Champion, 2002) over a dynamic range of 3-orders of magnitude (Nishihara and Champion, 2002).
Image analysis
SYPRO ruby-stained gel images were obtained using a STORM phosphoimager (Ex. 470 nm, Em. 618 nm, Molecular Dynamics, Sunnyvale, CA, USA) and also saved in TIFF format. Gel imaging was software-aided using PD-Quest (Bio-Rad) imaging software. Briefly, a master gel was selected followed by normalization of all gels (WT and KO) according to the total spot density. Gel to gel analysis was then initiated in two parts. First, manual matching of common spots that could be visualized among the differential 2D gels was done. After obtaining a significant number of spots the automated matching all spots was then initiated. This generated a large pool of data, and only proteins showing significant differential expression between the two groups being analyzed were considered
In-gel trypsin digestion
Protein spots statistically different than controls were digested in-gel by trypsin using protocols previously described and modified by (Thongboonkerd et al., 2002). Briefly, spots of interest were excised using a clean blade and placed in Eppendorf tubes, which were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at room temperature for 15 min. Acetonitrile was then added to the gel pieces and incubated at room temperature for 15 min. This solvent mixture was then removed and gel pieces dried. The protein spots were then incubated with 20 μL of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C for 45 min. The DTT solution was removed and replaced with 20 μL of 55 mM iodoacetamide in 0.1 M NH4HCO3. The solution was then incubated at room temperature for 30 min. The iodoacetamide was removed and replaced with 0.2 mL of 50 mM NH4HCO3 and incubated at room temperature for 15 min. Acetonitrile (200 μL) was added. After 15 min incubation, the solvent was removed, and the gel spots were dried in a flow hood for 30 min. The gel pieces were rehydrated with 20 ng/μL-modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 with the minimal volume enough to cover the gel pieces. The gel pieces were incubated overnight at 37 °C in a shaking incubator.
Mass spectrometry
A MALDI-TOF mass spectrometer in the reflectron mode was used to generate peptide mass fingerprints. Peptides resulting from in-gel digestion with trypsin were analyzed on a 384 position, 600 μm AnchorChip™ Target (Bruker Daltonics, Bremen, Germany) and prepared according to AnchorChip recommendations (AnchorChip Technology, Rev. 2, Bruker Daltonics, Bremen, Germany). Briefly, 1 μL of digestate was mixed with 1 μL of alpha-cyano-4-hydroxycinnamic acid (0.3 mg/mL in ethanol: acetone, 2:1 ratio) directly on the target and allowed to dry at room temperature. The sample spot was washed with 1 μL of a 1% TFA solution for approximately 60 seconds. The TFA droplet was gently blown off the sample spot with compressed air. The resulting diffuse sample spot was recrystallized (refocused) using 1 μL of a solution of ethanol: acetone: 0. 1 % TFA (6:3:1 ratio). Reported spectra are a summation of 100 laser shots. External calibration of the mass axis was used for acquisition and internal calibration using either trypsin autolysis ions or matrix clusters and was applied post acquisition for accurate mass determination.
Analysis of peptide sequences
Peptide mass fingerprinting was used to identify proteins from tryptic peptide fragments by utilizing the MASCOT search engine based on the entire NCBI and SwissProt protein databases. Database searches were conducted allowing for up to one missed trypsin cleavage and using the assumption that the peptides were monoisotopic, oxidized at methionine residues, and carbamidomethylated at cysteine residues. Mass tolerance of 150 ppm, 0.1 Da peptide tolerance and 0.2 Da fragmentation tolerance was the window of error allowed for matching the peptide mass values (Butterfield and Castegna, 2003). Probability-based MOWSE scores were estimated by comparison of search results against estimated random match population and were reported as -10*log10 (p), where p is the probability that the identification of the protein is a random event. MOWSE scores greater than 63 were considered to be significant (p < 0.05). All protein identifications were in the expected size and isoelectric point (pI) range based on the position in the gel.
Statistical analysis
Statistical analysis of differentially expressed protein levels matched with spots on 2D-gels from frontal cortex brain samples of the p50 (-/-) and wild type (WT) were used and carried out using Student's t-tests. Here we report a comparative proteomics analysis of p50 (-/-) vs. WT. A value of p < 0.05 was considered statistically significant. Only proteins that are considered significantly different by Student's t-test were subjected to in-gel trypsin digestion and subsequent proteomic analysis.
Acknowledgments
This work was supported in part by grants from NIH to D.A.B. [AG-10836; AG-05119].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Aleyasin H, Cregan SP, Iyirhiaro G, O'Hare MJ, Callaghan SM, Slack RS, Park DS. Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage. J Neurosci. 2004;24:2963–2973. doi: 10.1523/JNEUROSCI.0155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Biswas G, Anandatheerthavarada H, Zaidi M, Avadhini N. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFκB/Rel activation through calcineurin-mediated inactivation of IκBβ. J Cell Biol. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA. Proteomics: a new approach to investigate oxidative stress in Alzheimer's disease brain. Brain Res. 2004;1000:1–7. doi: 10.1016/j.brainres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–191. [Google Scholar]
- Butterfield DA, Castegna A. Proteomics for the identification of specifically oxidized proteins in brain: technology and application to the study of neurodegenerative disorders. Amino Acids. 2003;25:419–425. doi: 10.1007/s00726-003-0027-7. [DOI] [PubMed] [Google Scholar]
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- Chung HY, Jung KJ, Yu BP. Oxidative stress, Inflammation and Health. Marcel Dekker; NY: 2005. Molecular inflammation as an underlying mechanism of aging: The anti-inflammatory action of calorie restriction; pp. 387–419. [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donninger H, Bonome T, Radonovich M, Pise-Masison CA, Brady J, Shih JH, Barrett JC, Birrer MJ. Whole genome expression profiling of advance stage papillary serous ovarian cancer reveals activated pathways. Oncogene. 2004;23:8065–8077. doi: 10.1038/sj.onc.1207959. [DOI] [PubMed] [Google Scholar]
- Drake J, Sultana R, Aksenova M, Calabrese V, Butterfield DA. Elevation of mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects mitochondria against peroxynitrite-induced oxidative stress. J Neurosci Res. 2003;74:917–927. doi: 10.1002/jnr.10810. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Laskin DL. Ozone-induced production of nitric oxide and TNF-alpha and tissue injury are dependent on NF-kappaB p50. Am J Physiol Lung Cell Mol Physiol. 2004;287:L279–285. doi: 10.1152/ajplung.00348.2003. [DOI] [PubMed] [Google Scholar]
- Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Ble A, Bandinelli S, Lauretani F, Suthers K, Guralnik JM. A flame burning within. Aging Clin Exp Res. 2004;16:240–243. doi: 10.1007/BF03327390. [DOI] [PubMed] [Google Scholar]
- Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- Gadjeva M, Tomczak MF, Zhang M, Wang YY, Dull K, Rogers AB, Erdman SE, Fox JG, Carroll M, Horwitz BH. A role for NF-kappa B subunits p50 and p65 in the inhibition of lipopolysaccharide-induced shock. J Immunol. 2004;173:5786–5793. doi: 10.4049/jimmunol.173.9.5786. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Grilli M, Chiu JJ, Lenardo MJ. NF-kappa B and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest. 1998;101:802–811. doi: 10.1172/JCI483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Je JH, Lee JY, Jung KJ, Sung B, Go EK, Yu BP, Chung HY. NF-kappaB activation mechanism of 4-hydroxyhexenal via NIK/IKK and p38 MAPK pathway. FEBS Lett. 2004;566:183–189. doi: 10.1016/j.febslet.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-kappa B activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Herkenham M. NF-kappaB p50-deficient mice show reduced anxiety-like behaviors in tests of exploratory drive and anxiety. Behav Brain Res. 2004;154:577–584. doi: 10.1016/j.bbr.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Willing AE, Garbuzova-Davis S, Sanberg PR, Pennypacker KR. Lack of NF-kappaB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol. 2002;176:277–288. doi: 10.1006/exnr.2002.7967. [DOI] [PubMed] [Google Scholar]
- Kato A, Edwards MJ, Lentsch AB. Gene deletion of NF-kappa B p50 does not alter the hepatic inflammatory response to ischemia/reperfusion. J Hepatol. 2002;37:48–55. doi: 10.1016/s0168-8278(02)00068-5. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim KW, Yu BP, Chung HY. The effect of age on cyclooxygenase-2 gene expression: NF-kappaB activation and IkappaBalpha degradation. Free Radic Biol Med. 2000;28:683–692. doi: 10.1016/s0891-5849(99)00274-9. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Pendino KJ. Macrophages and inflammatory mediators in tissue injury. Annu Rev Pharmacol Toxicol. 1995;35:655–677. doi: 10.1146/annurev.pa.35.040195.003255. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Leyva JA, Bianchet MA, Amzel LM. Understanding ATP synthesis: structure and mechanism of the F1-ATPase (Review) Mol Membr Biol. 2003;20:27–33. doi: 10.1080/0968768031000066532. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Janus faces of NF-kappa B: neurodestruction versus neuroprotection. Nat Med. 1997;3:20–22. doi: 10.1038/nm0197-20. [DOI] [PubMed] [Google Scholar]
- Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience. 2006;139:965–978. doi: 10.1016/j.neuroscience.2005.12.062. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Hasko G, Liaudet L, Soriano F, Southan GJ, Salzman AL, Szabo C. NFkappaB1 (p50)-deficient mice are not susceptible to multiple low-dose streptozotocin-induced diabetes. J Endocrinol. 2002;173:457–464. doi: 10.1677/joe.0.1730457. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol. 2006;168:918–926. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy AK, Savinova OV, Ho JQ, Wang VY, Vu D, Ghosh G. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. Embo J. 2006;25:1945–1956. doi: 10.1038/sj.emboj.7601081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Mulkidjanian AY. Ubiquinol oxidation in the cytochrome bc1 complex: reaction mechanism and prevention of short-circuiting. Biochim Biophys Acta. 2005;1709:5–34. doi: 10.1016/j.bbabio.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Nishihara JC, Champion KM. Quantitative evaluation of proteins in one- and two-dimensional polyacrylamide gels using a fluorescent stain. Electrophoresis. 2002;23:2203–15. doi: 10.1002/1522-2683(200207)23:14<2203::AID-ELPS2203>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- O'Donnell SM, Hansberger MW, Connolly JL, Chappell JD, Watson MJ, Pierce JM, Wetzel JD, Han W, Barton ES, Forrest JC, Valyi-Nagy T, Yull FE, Blackwell TS, Rottman JN, Sherry B, Dermody TS. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Pennypacker KR, Kassed CA, Eidizadeh S, Saporta S, Sanberg PR, Willing AE. NF-kappaB p50 is increased in neurons surviving hippocampal injury. Exp Neurol. 2001;172:307–319. doi: 10.1006/exnr.2001.7817. [DOI] [PubMed] [Google Scholar]
- Perham RN. The fructose-1,6-bisphosphate aldolases: same reaction, different enzymes. Biochem Soc Trans. 1990;18:185–187. doi: 10.1042/bst0180185. [DOI] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004a;59:478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- Poon HF, Joshi G, Sultana R, Farr SA, Banks WA, Morley JE, Calabrese V, Butterfield DA. Antisense directed at the Abeta region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Res. 2004b;1018:86–96. doi: 10.1016/j.brainres.2004.05.048. [DOI] [PubMed] [Google Scholar]
- Santoro MG, Rossi A, Amici C. NF-kappaB and virus infection: who controls whom. Embo J. 2003;22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune response. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Sun Z, Andersson R. NF-kappaB activation and inhibition: a review. Shock. 2002;18:99–106. doi: 10.1097/00024382-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongboonkerd V, McLeish KR, Arthur JM, Klein JB. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int. 2002;62:1461–1469. doi: 10.1111/j.1523-1755.2002.kid565.x. [DOI] [PubMed] [Google Scholar]
- Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. p50-NF-kappaB complexes partially compensate for the absence of RelB: severely increased pathology in p50(-/-)relB(-/-) double-knockout mice. J Exp Med. 1997;185:1359–1370. doi: 10.1084/jem.185.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss H, Friedrich T, Hofhaus G, Preis D. The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur J Biochem. 1991;197:563–576. doi: 10.1111/j.1432-1033.1991.tb15945.x. [DOI] [PubMed] [Google Scholar]
- Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Cheng G, Wen X, Wu GD, Lee WT, Pleasure D. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism. Neurobiol Dis. 2002;11:199–213. doi: 10.1006/nbdi.2002.0530. [DOI] [PubMed] [Google Scholar]