

Abstract
In oxygenic photosynthesis, photosystem II (PSII) is the multisubunit membrane protein responsible for the oxidation of water to O2 and the reduction of plastoquinone to plastoquinol. One electron charge separation in the PSII reaction center is coupled to sequential oxidation reactions at the oxygen-evolving complex (OEC), which is composed of four manganese ions and one calcium ion. The sequentially oxidized forms of the OEC are referred to as the Sn states. S1 is the dark-adapted state of the OEC. Flash-induced oxygen production oscillates with period four and occurs during the S3 to S0 transition. Chloride plays an important, but poorly understood role in photosynthetic water oxidation. Chloride removal is known to block manganese oxidation during the S2 to S3 transition. In this work, we have used azide as a probe of proton transfer reactions in PSII. PSII was sulfate-treated to deplete chloride and then treated with azide. Steady state oxygen evolution measurements demonstrate that azide inhibits oxygen evolution in a chloride-dependent manner and that azide is a mixed or noncompetitive inhibitor. This result is consistent with two azide binding sites, one at which azide competes with chloride and one at which azide and chloride do not compete. At pH 7.5, the Ki for the competing site was estimated as 1 mM, and the Ki′ for the uncompetitive site was estimated as 8 mM. Vibrational spectroscopy was then used to monitor perturbations in the frequency and amplitude of the azide antisymmetric stretching band. These changes were induced by laser-induced charge separation in the PSII reaction center. The results suggest that azide is involved in proton transfer reactions, which occur before manganese oxidation, on the donor side of chloride-depleted PSII.
INTRODUCTION
Photosystem II (PSII) is a chlorophyll-containing protein complex found in the thylakoid membrane of cyanobacteria, algae, and higher plants. PSII catalyzes the light-induced oxidation of water and reduction of plastoquinone (reviewed by Nelson and Yocum (1) and Yocum (2)). The water-splitting reactions provide molecular oxygen, which is necessary for the maintenance of aerobic life on earth. Chlorophyll (chl) is the primary donor during the light-induced electron transfer reactions, which lead to the production of a transmembrane charge-separated state. Two plastoquinone acceptors, QA and QB, are sequentially reduced on the stromal side of the PSII reaction center. On the PSII lumenal side, a chl cation radical, 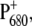 oxidizes tyrosine 161 (YZ) of the D1 polypeptide to produce a tyrosyl radical (YZ•). YZ• then oxidizes the oxygen-evolving complex (OEC), which is composed of four manganese ions and one calcium ion. X-ray diffraction has been used to determine the structure of PSII at 3.8-3.0 Å (3–7). However, x-ray induced damage to the OEC complicates interpretation of the manganese ligand environment in the current structures (5,8,9).
oxidizes tyrosine 161 (YZ) of the D1 polypeptide to produce a tyrosyl radical (YZ•). YZ• then oxidizes the oxygen-evolving complex (OEC), which is composed of four manganese ions and one calcium ion. X-ray diffraction has been used to determine the structure of PSII at 3.8-3.0 Å (3–7). However, x-ray induced damage to the OEC complicates interpretation of the manganese ligand environment in the current structures (5,8,9).
Four sequential light-induced charge separations are required to produce one oxygen molecule from two water molecules. These sequential reactions are stored as oxidation reactions at the OEC. Accordingly, the Mn4Ca+2 cluster cycles among five oxidation states in the production of molecular oxygen (10). The oxidation states are labeled S0–S4, where the subscript describes the number of oxidizing equivalents stored. The rate of OEC oxidation slows as charge is accumulated, and there is a period four pattern of oxygen release (11,12). Oxygen release occurs during the S3 to S0 transition, in which the transient S4 state is formed. Information about the S4 state has been obtained by x-ray absorption spectroscopy (12), electron paramagnetic resonance (EPR) spectroscopy (13), and transient infrared spectroscopy (14). UV spectroscopy has been used to probe the identity of S state intermediates accumulated at high oxygen pressure (15).
Chloride is required to achieve the maximum rate of PSII oxygen evolution activity (16–19). Although chloride is known to bind near the OEC (20,21), chloride has not yet been located in the PSII x-ray structures and is not an identified component in the Mn4Ca+2 cluster (3–7). Previously, chloride has been proposed to bind to amino acid side chains (22–24) or directly to metal ions (16).
Chloride depletion alters the functional properties of the OEC. Chloride depletion changes the S2 state EPR signals (25,26), and S-state transition-associated Fourier transform infrared (FT-IR) spectra (24,27). Chloride removal also inhibits manganese oxidation (28–34). Previously, chloride has been proposed to have a role in structural maintenance of the OEC (35), as a manganese ligand (16), as a facilitator of proton transfer (19), as an adjustor of the OEC midpoint potential (36), and/or as an activator of substrate (37).
In PSII, azide has been shown to be a reversible inhibitor (38). Evidence has been presented for azide interactions with both the PSII donor-side chloride site and the PSII acceptor-side. Evidence for a donor-side azide binding site near the OEC includes perturbation of the S2-state EPR (26) and electron spin-echo envelope modulation (ESEEM) (39) signals. In addition, azide-induced inhibition of semiquinone anion (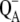 ) oxidation is partially reversed by bicarbonate (40). This result suggests a second, low affinity azide binding site on the PSII acceptor side.
) oxidation is partially reversed by bicarbonate (40). This result suggests a second, low affinity azide binding site on the PSII acceptor side.
It has been proposed that proton transfer precedes manganese oxidation on some of the S state transitions (12,14). To gain more insight into proton transfer reactions occurring in the OEC, we have used azide as a vibrational spectroscopic probe. The antisymmetric stretching vibration of azide is sensitive to changes in protonation and hydrogen bonding, because these interactions stabilize the triple-bonded, valence bond structures (41). In our experiments, PSII was chloride-depleted by treatment with sulfate (24,32) and was then treated with azide. Reaction-induced, rapid scan FT-IR spectroscopy and flash excitation were then used to step PSII through the accessible S state transitions, and the effect of light-induced electron transfer on the azide vibrational spectrum was assessed. Previous FT-IR studies (for previous examples, see (42–46)) have shown that long-lived conformational changes occurring during the S state cycle can be monitored, even on the slow (seconds) timescale of rapid scan FT-IR spectroscopy (47). Our work provides evidence that azide inhibition is due to changes in proton transfer reactions on the PSII donor side.
MATERIALS AND METHODS
PSII membranes were isolated from market spinach as previously described (48). Activity was measured with a Clark-type electrode and either recrystallized 2,6-dichlorobenzoquinone (DCBQ) or recrystallized paraphenylbenzoquinone (PPBQ) as an electron acceptor (49). Chloride-depleted samples were prepared by sulfate treatment as previously described (24,32). Immediately before the experiment, PSII membranes were exchanged into pH 7.5 buffer (400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5) by centrifugation and resuspension. Exchanged samples were incubated in sulfate buffer (400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5, and 50 mM Na2SO4) for 15 min while shaking on ice and in darkness. After centrifugation, the chloride-depleted samples were suspended again in pH 7.5 buffer. Manganese-depleted samples were prepared as previously described using alkaline tris(hydroxymethyl)aminomethane (Tris) (50). Manganese-depleted samples were exchanged into pH 7.5 buffer, displayed no oxygen-evolving capability, and were stored at -70°C until use. The final Tris concentration for the manganese-depleted samples was estimated to be ≤6 mM. Azide-exchanged samples were prepared by suspension of chloride-depleted or manganese-depleted PSII into buffer containing 400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5, and indicated concentrations of sodium 14N-azide or terminally labeled (15N14N14N) sodium 15N-azide (98% 15N-enriched, Cambridge Isotope Laboratories, Andover, MA). Solvent isotope exchange was achieved by performing each step of the chloride depletion protocol in 2H2O-containing buffers (99.8% 2H enriched, Isotec, Miamisburg, OH). The p2H is reported as the uncorrected pH meter reading (51).
Reaction-induced FT-IR difference spectroscopy was performed at pH 7.5 as previously described (24,46,47). Chloride-depleted samples were concentrated with dry nitrogen gas to give an O-H stretching absorbance (3370 cm−1) to amide II absorbance ratio of ≥3. Manganese-depleted samples were concentrated with nitrogen for ten minutes. Data acquisition parameters were as follows: 8 cm−1 resolution; four levels of zero filling; Happ-Genzel apodization function; 60 KHz mirror speed; Mertz phase correction. Samples were given a single saturating 532 nm laser flash followed by 20 min of dark adaptation to set all reaction centers in the S1 state. Each subsequent flash was followed by 15 s of rapid scan data collection at 4°C. S state difference spectra were created by ratio of data taken before and after flash excitation, followed by conversion to absorbance. All data was normalized to an amide II intensity of 0.5 absorbance units (AU) to eliminate differences in sample pathlength (24,46,47). The amide II band intensity was determined from an infrared absorption spectrum, which was generated through the use of a blank background scan.
RESULTS
Table 1 presents representative steady state oxygen evolution rates for sulfate-treated PSII (see also (24)). Sulfate treatment is known to dissociate the 24 and 18 kDa extrinsic subunits and to decrease the chloride binding affinity, allowing for quantitative ion release (18,20,24,32,52). When assayed at pH 6.0 and pH 7.5 in the presence of DCBQ, untreated PSII had oxygen rates of >750 μmol O2 (mg chl-h)−1 and >500 μmol O2 (mg chl-h)−1, respectively. Sulfate treatment resulted in a complete suppression of oxygen evolution activity at pH 7.5 (≤30 μmol O2 (mg chl-h)−1). Table 1 demonstrates that addition of chloride resulted in the reconstitution of the majority of oxygen-evolving activity at pH 6.0 (460 μmol O2 (mg chl-h)−1) and pH 7.5 (240 μmol O2 (mg chl-h)−1). As expected (32), calcium addition had little effect on oxygen-evolving activity in sulfate-treated PSII (Table 1). Thus, sulfate treatment allows the effect of chloride depletion to be studied separately from the effects of calcium depletion (24,32). In addition, sulfate-treated samples reconstituted with chloride are known to produce a higher flash yield of the S state transitions, compared to other methods of chloride depletion (32).
TABLE 1.
Oxygen evolving activity of sulfate-treated PSII at pH 6.0 and 7.5
| 10 mM Ca+2 | 15 mM Cl− | pH 6.0* | pH 7.5† |
|---|---|---|---|
| — | — | 60 ± 30 | 30 ± 10 |
| + | + | 460 ± 60 | 240 ± 3 |
| — | + | 370 ± 60 | 200 ± 20 |
| + | — | 46 ± 7 | 9 ± 1 |
Oxygen evolving activity measured at 25°C and listed with units of μmol O2 (mg chl-h)−1 (24). Values are reported ±1 standard deviation (SD). All measurements are an average of at least three individual experiments. Each measurement performed with 1 mM ferricyanide and 0.5 mM DCBQ.
Conditions were 400 mM sucrose and 50 mM MES-NaOH (pH 6.0).
Conditions were 400 mM sucrose and 50 mM HEPES-NaOH (pH 7.5).
Fig. 1 and Fig. S1 (see the Supplementary Material, Data S1) demonstrate that azide inhibits PSII oxygen evolution in a chloride-dependent manner, both at pH 6.3 (Fig. 1 A and Fig. S1 A in Data S1) and 7.5 (Figs. 1 B and Fig. S1 B in Data S1). The direct plots (see Fig. S1 in Data S1) and the Lineweaver-Burk double reciprocal plots (1/vo versus 1/[Cl−]) give the dependence of the initial oxygen evolution velocity on the azide concentration. At pH 6.3, the Lineweaver-Burk results (Fig. 1 A) are consistent with a family of lines that intersect to the left of the 1/vo axis. This pattern provides evidence for reversible, noncompetitive (or mixed) inhibition between azide and chloride, in agreement with an earlier report (38). In a Michaelis-Menten interpretation, this type of inhibition occurs if azide simultaneously competes at the chloride site both in the enzyme (competitive site) and in the enzyme-substrate complex (uncompetitive site). Assuming that azide is a mixed inhibitor, fits to the direct plots (see Fig. S1 A in Data S1) were used to yield kinetics constants. At pH 6.3, the Km for chloride was derived as 0.7 mM, and the Vmax was derived as 360 μmol O2 (mg chl-h)−1. The Ki for azide binding to the competitive site was derived as 0.3 mM and the K′i for the uncompetitive site was derived as 2 mM. Note that the uncompetitive azide site may correspond to an acceptor side binding event (see Discussion section). Previously, Dixon and Cornish-Bowden plots were used to estimate the azide inhibition constants for the competitive and uncompetitive sites at pH 6.3 in sodium-chloride treated PSII samples (38), and indistinguishable kinetic constants were obtained.
FIGURE 1.

Lineweaver-Burk double-reciprocal plots, considering chloride as the substrate and showing the inhibitory effect of azide on PSII oxygen evolution activity. PSII was treated with sulfate to deplete chloride, and the PSII sample was then treated with 0 (red), 0.25 (orange), 0.38 (green), 0.50 (light blue), 0.75 (dark blue), or 1.0 (purple), 1.8 (black), and 2.5 (brown; pH 7.5 only) mM 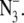 In A, the assay conditions were taken from reference (38) and employed 400 mM sucrose, 50 mM MES-NaOH, pH 6.3, and 2 mM recrystallized PPBQ. In B, the assay conditions were 400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5, and 2 mM recrystallized PPBQ. The concentrations of chloride and azide were adjusted by addition from 1 M NaCl and 100 mM NaN3 stock solutions, respectively, in the appropriate assay buffer. Error bars represent 1 SD. The color coded, superimposed lines were generated with Vmax, Km, Ki, and Ki′ values, derived from fits to the hyperbolic oxygen evolution plots (see Results section and the Supplementary Material, Data S1). The double reciprocal plots were fit with the equation
In A, the assay conditions were taken from reference (38) and employed 400 mM sucrose, 50 mM MES-NaOH, pH 6.3, and 2 mM recrystallized PPBQ. In B, the assay conditions were 400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5, and 2 mM recrystallized PPBQ. The concentrations of chloride and azide were adjusted by addition from 1 M NaCl and 100 mM NaN3 stock solutions, respectively, in the appropriate assay buffer. Error bars represent 1 SD. The color coded, superimposed lines were generated with Vmax, Km, Ki, and Ki′ values, derived from fits to the hyperbolic oxygen evolution plots (see Results section and the Supplementary Material, Data S1). The double reciprocal plots were fit with the equation 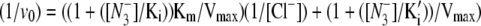
When pH 6.3 (Figs. 1 A and Fig. S1 A in Data S1) and pH 7.5 (Figs. 1 B and Fig. S1 B in Data S1) data were compared, a similar pattern of mixed inhibition was observed. Fits to the direct plots were again used to derive kinetic constants (Fig. S1 B in Data S1). At pH 7.5, the Km for chloride was derived as 3 mM, the Vmax was derived as 330 μmol O2 (mg chl-h)−1, the Ki for azide binding to the competitive site was derived as 1 mM, and the K′i for the uncompetitive site was derived as 8 mM. The Lineweaver-Burk plots exhibit some nonlinearity at high azide concentrations (Fig. 1 ≥2.5 mM azide). Azide inhibition was found to be reversible at pH 7.5 (data not shown), as previously reported at pH 6.3 (38). Taken together with previous work (38), Fig. 1 illustrates that the azide inhibition constant is similar in sulfate-treated PSII and other PSII preparations (38).
Fig. 1 suggests that azide can be used as a spectroscopic probe, which will bind on the donor side of sulfate-treated PSII. The azide antisymmetric stretching band has been used previously to monitor protonation reactions in other enzymes, including bacteriorhodopsin (53) and halorhodopsin (54). The frequency of the antisymmetric azide band falls outside the normal range of peptide and biological cofactor infrared absorption (1800–1200 cm−1), facilitating detection of azide even in complex biological systems (53,55).
To probe proton transfer reactions at the PSII azide site, we employed reaction-induced difference FT-IR spectroscopy. Fig. 2 A and B, show the results of three consecutive 532 nm laser pulses (red, 1 flash; green, 2 flashes; blue, 3 flashes) on the azide antisymmetric stretching vibration in oxygen-evolving PSII at pH 6.0 and 7.5, respectively. Chloride was not depleted in these samples before the addition of 15 mM azide. The difference spectra were constructed using data acquired after each flash and data acquired before the three laser flashes. In oxygen-evolving PSII, these spectra correspond to the S1 to S2 (one flash), S1 to S3 (two flashes), and S1 to S0 (three flashes) transitions. Our previously published work, analyzing the mid-infrared region of the spectrum, demonstrates that S state advancement is detectable under these conditions (24). Azide bands were not observed in PSII without flash excitation ( see Fig. 2 F) or in mixtures of azide, DCBQ acceptor and other buffer components, which did not contain PSII, but received flash excitation (data not shown).
FIGURE 2.

Difference FT-IR spectra showing flash-induced perturbation of the azide antisymmetric stretching band in untreated, manganese-depleted, or chloride-depleted PSII samples. Difference spectra were created by ratio of data taken before and after flash excitation, followed by conversion to absorbance. In A–E, the results of three consecutive 532 nm laser flashes to a preflashed, dark-adapted PSII sample are presented in red (flash 1), green (flash 2), and blue (flash 3). In A, untreated PSII contained 400 mM sucrose, 50 mM MES-NaOH, pH 6.0, and 15 mM NaCl (average of 12). In B, untreated PSII contained in 400 mM sucrose, 50 mM HEPES-NaOH, pH 7.5 (pH 7.5 buffer) (average of 15). In C, manganese-depleted PSII contained pH 7.5 buffer (average of 38). In D and E, chloride-depleted PSII contained pH 7.5 buffer (average of 39). In A-D, the sample also contained 15 mM 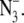 In E, chloride-depleted PSII in pH 7.5 buffer contained 15 mM [15N(14N)2]− (average of 21). In F, a dark-minus-dark control was generated from the data set in D. Delta absorbance on the y axis indicates that the spectra show changes in absorbance. All samples contained 1.5 mM recrystallized DCBQ. The tick marks on the y axis represent 5 × 10−4 AU.
In E, chloride-depleted PSII in pH 7.5 buffer contained 15 mM [15N(14N)2]− (average of 21). In F, a dark-minus-dark control was generated from the data set in D. Delta absorbance on the y axis indicates that the spectra show changes in absorbance. All samples contained 1.5 mM recrystallized DCBQ. The tick marks on the y axis represent 5 × 10−4 AU.
Fig. 2, A and B, show that PSII charge separation perturbs bands assignable to the azide antisymmetric stretching mode either at pH 6.0 (Fig. 2 A) or at pH 7.5 (Fig. 2 B). In both data sets, two band shifts at 2124 (−)/2111 (+) cm−1 and 2102 (−) /2088 (+) cm−1 are observed. These bands are significant compared to the noise in the measurement, as assessed by comparison to a dark-minus-dark spectrum (Fig. 2 F). The pH 7.5 spectrum (Fig. 2 B) displays additional negative bands at 2078 and 2062 cm−1, which are not present at pH 6.0 (Fig. 2 A). In the PSII samples used in Fig. 2, A and B, chloride has not been depleted. Therefore, we assign azide bands, observed under these conditions, to a PSII acceptor side binding site. In agreement with this interpretation, similar perturbations of azide bands were also observed in manganese-depleted PSII at pH 7.5 (Fig. 2 C). These manganese-depleted samples do not contain a chloride-binding site because the OEC has been removed by Tris treatment (20,52). The small increase in azide band amplitude on each flash in (Fig. 2, A–C) may be due to a light-induced conformational change, which allows increased access to the acceptor side binding site.
Fig. 2 D reveals the effect of PSII charge separation on azide bands in chloride-depleted samples. Reaction-induced FT-IR spectra were recorded at pH 7.5. While the S1 to S2 transition (first flash) proceeds in chloride-depleted PSII, the S2 to S3 transition (second flash) is blocked at the level of manganese oxidation (28–34). Thus, in chloride-depleted samples, two flashes generate the  state. This
state. This 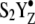 or S3′ state has a halftime of ∼0.5 s (32). Previous FT-IR studies of chloride-depleted samples have shown that structural changes occur in the OEC during the S2 to S3′ transition, even though manganese oxidation is blocked (24). These structural changes are long-lived and occur on the seconds timescale (24), as are other rapid-scan, FT-IR-detected structural changes during the normal S state cycle (46,47).
or S3′ state has a halftime of ∼0.5 s (32). Previous FT-IR studies of chloride-depleted samples have shown that structural changes occur in the OEC during the S2 to S3′ transition, even though manganese oxidation is blocked (24). These structural changes are long-lived and occur on the seconds timescale (24), as are other rapid-scan, FT-IR-detected structural changes during the normal S state cycle (46,47).
The reaction-induced spectra obtained with a single flash (S1 to S2 transition) (Fig. 2 D, red) are similar to data acquired in manganese-depleted PSII (Fig. 2 C, red). This can be ascertained in Fig. 3 A, in which these spectra subtract to give a flat baseline (compare to Fig. 3 C). This result suggests that there is no significant perturbation of the azide donor-side binding site on the first flash. Spectra acquired with a second (see Fig. 2 D, green) and third (see Fig. 2 D, blue) flash (S1 to (S2 YZ S3′ transition) are similar to each other and exhibit new positive bands at 2111 and 2040 cm−1 and increased negative intensity at ∼2062 cm−1, compared to manganese-depleted PSII. In these samples, both the second and third flashes correspond to the inhibited S1 to 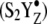 S3′ transition, because charge recombination occurs in the time between the flashes (56). The frequencies of all observed bands were downshifted when terminally 15N labeled azide (see Fig. 2 E) was employed, confirming the spectral assignment to the azide antisymmetric stretching band.
S3′ transition, because charge recombination occurs in the time between the flashes (56). The frequencies of all observed bands were downshifted when terminally 15N labeled azide (see Fig. 2 E) was employed, confirming the spectral assignment to the azide antisymmetric stretching band.
FIGURE 3.

Double difference FT-IR spectra showing perturbations of azide antisymmetric stretching bands. To identify any structural changes at the OEC azide site, manganese-depleted PSII data were subtracted from chloride-depleted PSII data. These double difference spectra were created by subtraction of data shown in Fig. 2, C and D. In A, subtracted spectra were acquired with one laser flash (Fig. 2, C and D, red). In B, subtracted spectra were acquired with three laser flashes (Fig. 2, C and D, blue). In C, a control double difference spectrum, in which no vibrational bands are expected, was generated by subtraction of one half of the data in (Fig. 2 D, red) from the other half of the data set and dividing by the square root of two. Delta absorbance on the y axis indicates that the spectra show changes in absorbance. All samples contained 1.5 mM recrystallized DCBQ. The tick marks on the y axis represent 5 × 10−4 AU.
A double difference spectrum (Fig. 3 B), chloride-depleted-minus-manganese-depleted, can be used to remove acceptor-side and any other non-OEC azide contributions. This method will identify azide bands, which are due to binding at the OEC azide site and are perturbed on the second and third flash. This double difference spectrum reveals three negative bands, at 2093, 2078, and 2066 cm−1, which shift both to higher and lower frequency with three flash excitation. The positive bands are observed at 2116, 2052, and 2040 cm−1. The intensities are significant compared to a control double difference spectrum, in which no vibrational bands are expected (Fig. 3 C). The observation of these unique azide spectral features demonstrates that light-induced charge separation perturbs the OEC azide binding site during the S1 to 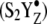 S3′ transition in chloride-depleted PSII. The observation of multiple bands may be consistent with multiple azide orientations in its PSII binding site. In the presence of buried charges, the orientation of azide can influence the vibrational frequency through a Stark effect. Alternatively, sulfate treatment or extrinsic subunit removal may introduce heterogeneity at the PSII donor side. Such heterogeneity may cause the structure of the azide binding site to vary slightly from one PSII complex to another.
S3′ transition in chloride-depleted PSII. The observation of multiple bands may be consistent with multiple azide orientations in its PSII binding site. In the presence of buried charges, the orientation of azide can influence the vibrational frequency through a Stark effect. Alternatively, sulfate treatment or extrinsic subunit removal may introduce heterogeneity at the PSII donor side. Such heterogeneity may cause the structure of the azide binding site to vary slightly from one PSII complex to another.
In the double difference spectrum (Fig. 3 B), negative bands correspond to bound azide in the dark-adapted S1 state. The antisymmetric stretching vibration of azide is expected at 2050 cm−1 in water (53). In hydrophobic solvents, such as dimethyl sulfoxide, the band shifts down to 2018 cm−1 (53). Protonation of azide upshifts these bands into the 2148-2129 cm−1 region (41,53,55). Hydrogen bonding is also expected to upshift the antisymmetric stretching vibration (41). Therefore, the characteristic frequencies of the observed negative bands in Fig. 3 B, at 2093, 2078, and 2066 cm−1, suggest that azide is bound in the S1 state in a hydrogen bonded, but deprotonated form, consistent with the expected azide pKA of ∼4.7 (57).
The data acquired with a single flash (see Fig. 2 D, red) show that no significant azide frequency perturbation occurs at the azide donor-side site during the S1 to S2 transition (see also double difference spectrum, Fig. 3 A). However, the second and third flashes, which generate the S1 to 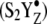 S3′ transition (Fig. 3 B), produce two new populations of azide, which are reflected as positive spectral bands. Based on the frequencies, both a protonated form (2116 cm−1) and nonhydrogen bonded anionic forms (2052 and 2040 cm−1) are generated.
S3′ transition (Fig. 3 B), produce two new populations of azide, which are reflected as positive spectral bands. Based on the frequencies, both a protonated form (2116 cm−1) and nonhydrogen bonded anionic forms (2052 and 2040 cm−1) are generated.
To test this interpretation, reaction-induced FT-IR spectra were acquired in azide-containing, chloride-depleted PSII, either in 1H2O (Fig. 4 A) or 2H2O buffers (Fig. 4 B). Solvent isotope exchange is expected to downshift the frequencies of protonated and hydrogen bonded forms of azide (58–60). The effect of solvent isotope exchange is shown in a double difference, 1H2O-minus-2H2O, spectrum (Fig. 4 C). A positive band at ∼2106 cm−1 is observed to shift to 2079 cm−1 and a negative band at 2065 cm−1 is observed to shift to 2048 cm−1. These shifts are consistent with assignment of the positive 2116 cm−1 band (see Fig. 3 B) to protonated azide and the negative 2066 cm−1 band (see Fig. 3 B) to hydrogen bonded azide, as described below. The isotope shifts for the other positive and negative bands are not discernable from the isotope-edited spectrum. These results suggest that, during the S1 to  S3′ transition, azide acts both as a proton acceptor and a proton donor at the OEC azide site.
S3′ transition, azide acts both as a proton acceptor and a proton donor at the OEC azide site.
FIGURE 4.

Difference FT-IR spectra showing the effect of solvent isotope exchange on the azide antisymmetric stretching band in chloride-depleted PSII samples. Difference spectra were created by ratio of data taken before and after flash excitation, followed by conversion to absorbance. In A and B, the results of three consecutive 532 nm laser flashes to a preflashed, dark adapted PSII sample are presented in red (flash 1), green (flash 2), and blue (flash 3). In A, chloride-depleted PSII in 1H2O p1H 7.5 buffer contained 15 mM 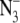 (average of 39). In B, chloride-depleted PSII in 2H2O p2H 7.5 buffer contained 15 mM
(average of 39). In B, chloride-depleted PSII in 2H2O p2H 7.5 buffer contained 15 mM 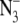 (average of 26). In C, an isotope-edited, 1H2O-minus-2H2O, double difference spectrum was generated by subtracting (B, flash 3, blue) from (A, flash 3, blue). In D, a dark-minus-dark control was generated from the data set in A. In E, a control double difference spectrum, in which no vibrational bands are expected, was generated by subtraction of one half of the data in A from the other half of the data set and dividing by the square root of two. Delta absorbance on the y axis indicates that the spectra show changes in absorbance. All samples contained 1.5 mM recrystallized DCBQ. The tick marks on the y axis represent 5 × 10−4 AU.
(average of 26). In C, an isotope-edited, 1H2O-minus-2H2O, double difference spectrum was generated by subtracting (B, flash 3, blue) from (A, flash 3, blue). In D, a dark-minus-dark control was generated from the data set in A. In E, a control double difference spectrum, in which no vibrational bands are expected, was generated by subtraction of one half of the data in A from the other half of the data set and dividing by the square root of two. Delta absorbance on the y axis indicates that the spectra show changes in absorbance. All samples contained 1.5 mM recrystallized DCBQ. The tick marks on the y axis represent 5 × 10−4 AU.
DISCUSSION
Azide has been used previously to study proton transfer reactions in enzymes. In PSII, azide acts as an inhibitor (38). In other proteins, azide addition may be stimulatory, due to effects on hydrogen bonding networks in proton transfer pathways (61). For example, azide has been observed to stimulate proton transfer in site-directed mutants of bacteriorhodopsin, halorhodopsin, and hydroxysteroid dehydrogenase (53,62–67). Previously, azide has also been shown to promote electron and proton transfer to the quinone acceptors in photosynthetic reaction center mutants (57).
We have used the azide antisymmetric stretching vibration as a vibrational probe (53,55). The antisymmetric stretching frequency is perturbed by changes in hydrogen bonding, polarity, and metal coordination (41,53,55). Azide vibrational band frequencies are also sensitive to changes in electric fields (68). Previously, azide has been used as a vibrational probe in bacteriorhodopsin. An increase in 2132 cm−1 amplitude was attributed to the transient protonation of azide during proton transfer (53). Also, in metmyoglobin, shifts in azide bands at 2046 cm−1 and 2023/2018 cm−1 led to an estimation of the thermal spin equilibrium for high- and low-spin heme, respectively (69,70).
In our work, reaction-induced difference FT-IR spectra show that the antisymmetric stretching mode of azide is perturbed by PSII charge separation. These data provide evidence for two azide binding sites in PSII. At pH 7.5, perturbation of azide gave rise to derivative-shaped bands, with both positive and negative components, at 2124 (−) /2111 (+), 2102 (−) /2088 (+) cm−1 and negative bands at 2078 and 2062 cm−1. We attribute these bands to an acceptor side or other non-OEC azide binding site. These bands were observed both in manganese-depleted PSII, in which the OEC has been removed by Tris treatment, and in oxygen-evolving preparations, which had not been depleted of chloride.
Previous EPR and ESEEM measurements demonstrate that azide also binds to the chloride site near the OEC (26,39). In chloride-depleted PSII, we have detected unique azide vibrational bands when a second or third flash induces the S1 to 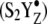 S3′ transition. We assign these bands to donor-side bound azide, because the vibrational frequencies are not observed in manganese-depleted PSII or in PSII, which has not been depleted of chloride. The azide species, which are present in the S1 state, absorb at 2093 (−), 2078 (−), and 2066 (−) cm−1. These are frequencies characteristic of hydrogen bonded, anionic azide. When the S3′ state is formed, three positive bands are observed. The frequency of the 2116 cm−1 band suggests assignment to protonated azide, and the frequencies of the 2052 and 2040 cm−1 bands suggest assignment to nonhydrogen bonded or weakly hydrogen bonded anionic azide. Solvent isotope exchange identified a 27 cm−1, 17 cm−1, and nondetectable isotope shift for the positive 2116, negative 2066, and positive 2040 cm−1 bands. Previous studies of azide have revealed a deuterium isotope shift of ∼30 cm−1 for the protonated species (59,60) and an ∼8 cm−1 shift for the hydrogen bonded, anionic species (58). Therefore, the results of solvent isotope exchange are consistent with the attribution of the 2116, 2066, and 2040 cm−1 bands to protonated azide, hydrogen bonded anionic azide, and nonhydrogen bonded (or weakly hydrogen bonded) anionic azide, respectively.
S3′ transition. We assign these bands to donor-side bound azide, because the vibrational frequencies are not observed in manganese-depleted PSII or in PSII, which has not been depleted of chloride. The azide species, which are present in the S1 state, absorb at 2093 (−), 2078 (−), and 2066 (−) cm−1. These are frequencies characteristic of hydrogen bonded, anionic azide. When the S3′ state is formed, three positive bands are observed. The frequency of the 2116 cm−1 band suggests assignment to protonated azide, and the frequencies of the 2052 and 2040 cm−1 bands suggest assignment to nonhydrogen bonded or weakly hydrogen bonded anionic azide. Solvent isotope exchange identified a 27 cm−1, 17 cm−1, and nondetectable isotope shift for the positive 2116, negative 2066, and positive 2040 cm−1 bands. Previous studies of azide have revealed a deuterium isotope shift of ∼30 cm−1 for the protonated species (59,60) and an ∼8 cm−1 shift for the hydrogen bonded, anionic species (58). Therefore, the results of solvent isotope exchange are consistent with the attribution of the 2116, 2066, and 2040 cm−1 bands to protonated azide, hydrogen bonded anionic azide, and nonhydrogen bonded (or weakly hydrogen bonded) anionic azide, respectively.
These results suggest that a heterogeneous proton-coupled electron transfer reaction occurs at the OEC azide site during the S1 to 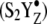 S3′ transition. Bound, hydrogen bonded azide molecules both protonate and deprotonate. Although manganese oxidation does not occur on the second flash or third flash in chloride-depleted samples (28–34), the proton transfer reactions may be driven by conformational changes, linked to YZ oxidation. Therefore, our results are consistent with the conclusion that proton transfer reactions occur before the manganese oxidation reaction, as previously suggested (12,14). During the S1 to S2 transition, bands attributable to protonation of azide are not observed in our chloride-depleted samples. This result is consistent with previous suggestions that the S1 to S2 transition involves uncompensated electron transfer and no net proton transfer (37,71,72).
S3′ transition. Bound, hydrogen bonded azide molecules both protonate and deprotonate. Although manganese oxidation does not occur on the second flash or third flash in chloride-depleted samples (28–34), the proton transfer reactions may be driven by conformational changes, linked to YZ oxidation. Therefore, our results are consistent with the conclusion that proton transfer reactions occur before the manganese oxidation reaction, as previously suggested (12,14). During the S1 to S2 transition, bands attributable to protonation of azide are not observed in our chloride-depleted samples. This result is consistent with previous suggestions that the S1 to S2 transition involves uncompensated electron transfer and no net proton transfer (37,71,72).
Taken together, our results suggest that azide may establish a perturbed hydrogen bond network near the OEC. We propose that azide inhibits oxygen evolution by disruption or rearrangement of the normal hydrogen bond network, which is necessary for proton-coupled electron transfer reactions in the OEC. Because azide exhibits mixed inhibition and competes at the chloride site, this interpretation may also imply a role for chloride in PSII proton transfer reactions. Finally, our data support the interpretation that proton transfer reactions at the OEC azide site precede manganese oxidation reactions (12,14,24), at least in chloride-depleted PSII.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work supported by the National Science Foundation (MCB 03-55421).
Editor: Janos K. Lanyi.
References
- 1.Nelson, N., and C. F. Yocum. 2006. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 57:521–565. [DOI] [PubMed] [Google Scholar]
- 2.Yocum, C. F. 2008. The calcium and chloride requirements of the O2 evolving complex. Coord. Chem. Rev. 252:296–305. [Google Scholar]
- 3.Zouni, A., H.-T. Witt, J. Kern, P. Fromme, N. Krauß, W. Saenger, and P. Orth. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]
- 4.Kamiya, N., and J.-R. Shen. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc. Natl. Acad. Sci. USA. 100:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biesiadka, J., B. Loll, J. Kern, K.-D. Irrgang, and A. Zouni. 2004. Crystal structure of cyanobacterial photosystem II at 3.2 Å resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 6:4733–4736. [Google Scholar]
- 6.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1837. [DOI] [PubMed] [Google Scholar]
- 7.Loll, B., J. Kern, W. Saenger, A. Zouni, and J. Biesiadka. 2005. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 438:1040–1044. [DOI] [PubMed] [Google Scholar]
- 8.Yano, J., J. Kern, K.-D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni, and V. K. Yachandra. 2005. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA. 102:12047–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grabolle, M., M. Haumann, C. Muller, P. Liebisch, and H. Dau. 2006. Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10–300 K. J. Biol. Chem. 281:4580–4588. [DOI] [PubMed] [Google Scholar]
- 10.Kok, B., B. Forbush, and M. McGloin. 1970. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem. Photobiol. 11:457–475. [DOI] [PubMed] [Google Scholar]
- 11.Dekker, J. P., H. J. van Gorkom, J. Wensink, and L. Ouwehand. 1984. Absorbance difference spectra of the successive redox states of the oxygen-evolving apparatus of photosynthesis. Biochim. Biophys. Acta. 767:1–9. [Google Scholar]
- 12.Haumann, M., P. Liebisch, C. Muller, M. Barra, M. Grabolle, and H. Dau. 2005. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 310:1019–1021. [DOI] [PubMed] [Google Scholar]
- 13.Razeghifard, M. R., and R. J. Pace. 1999. EPR kinetic studies of oxygen release in thylakoids and PSII membranes: a kinetic intermediate in S3 to S0 transition. Biochemistry. 38:1252–1257. [DOI] [PubMed] [Google Scholar]
- 14.Barry, B. A., I. B. Cooper, A. De Riso, S. H. Brewer, D. M. Vu, and R. B. Dyer. 2006. Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc. Natl. Acad. Sci. USA. 103:7288–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clausen, J., and W. Junge. 2004. Detection of an intermediate of photosynthetic water oxidation. Nature. 430:480–483. [DOI] [PubMed] [Google Scholar]
- 16.Sandusky, P. O., and C. F. Yocum. 1984. The chloride requirement for photosynthetic oxygen evolution: Analysis of the effects of chloride and other anions on amine inhibition of the oxygen evolving complex. Biochim. Biophys. Acta. 766:603–611. [Google Scholar]
- 17.Sandusky, P. O., and C. F. Yocum. 1986. The chloride requirement for photosynthetic oxygen evolution: Factors affecting nucleophilic displacement of chloride from the oxygen-evolving complex. Biochim. Biophys. Acta. 849:85–93. [Google Scholar]
- 18.Lindberg, K., and L.-E. Andreasson. 1996. A one-site, two-state model for the binding of anions in photosystem II. Biochemistry. 35:14259–14267. [DOI] [PubMed] [Google Scholar]
- 19.Olesen, K., and L.-E. Andreasson. 2003. The function of the chloride ion in photosynthetic oxygen evolution. Biochemistry. 42:2025–2035. [DOI] [PubMed] [Google Scholar]
- 20.Lindberg, K., T. Vanngard, and L.-E. Andreasson. 1993. Studies of the slowly exchanging chloride in photosystem II of higher plants. Photosynth. Res. 38:401–408. [DOI] [PubMed] [Google Scholar]
- 21.Clemens, K. L., D. A. Force, and R. D. Britt. 2002. Acetate binding at the photosystem II oxygen evolving complex: an S2-state multiline signal ESEEM study. J. Am. Chem. Soc. 124:10921–10933. [DOI] [PubMed] [Google Scholar]
-
22.Hasegawa, K., Y. Kimura, and T.-a. Ono. 2004. Oxidation of the Mn cluster induces structural changes of
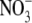 functionally bound to the Cl− site in the oxygen-evolving complex of photosystem II. Biophys. J. 86:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
functionally bound to the Cl− site in the oxygen-evolving complex of photosystem II. Biophys. J. 86:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar] - 23.Haumann, M., M. Barra, P. Loja, S. Loscher, R. Krivanek, A. Grundmeier, L.-E. Andreasson, and H. Dau. 2006. Bromide does not bind to the Mn4Ca complex in its S1 state in Cl−-depleted and Br−-reconstituted oxygen-evolving photosystem II: Evidence from X-ray absorption spectroscopy at the Br K-edge. Biochemistry. 45:13101–13107. [DOI] [PubMed] [Google Scholar]
- 24.Cooper, I. B., and B. A. Barry. 2007. Perturbations at the chloride site during the photosynthetic oxygen-evolving cycle. Photosynth. Res. 92:345–354. [DOI] [PubMed] [Google Scholar]
- 25.van Vliet, P., and A. W. Rutherford. 1996. Properties of the chloride-depleted oxygen-evolving complex of photosystem II studied by electron paramagnetic resonance. Biochemistry. 35:1829–1839. [DOI] [PubMed] [Google Scholar]
- 26.Haddy, A., R. A. Kimel, and R. Thomas. 2000. Effects of azide on the S2 state EPR signals from photosystem II. Photosynth. Res. 63:35–45. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa, K., Y. Kimura, and T.-a. Ono. 2002. Chloride cofactor in the photosynthetic oxygen-evolving complex studied by Fourier transform infrared spectroscopy. Biochemistry. 41:13839–13850. [DOI] [PubMed] [Google Scholar]
- 28.Itoh, S., C. T. Yerkes, H. Koike, H. H. Robinson, and A. R. Crofts. 1984. Effects of chloride depletion on electron donation from the water-oxidizing complex to the photosystem II reaction center as measured by the microsecond rise of chlorophyll fluorescence in isolated pea chloroplasts. Biochim. Biophys. Acta. 766:612–622. [Google Scholar]
- 29.Ono, T.-a., J. L. Zimmermann, Y. Inoue, and A. W. Rutherford. 1986. EPR evidence for a modified S state transition in chloride-depleted photosystem II. Biochim. Biophys. Acta. 851:193–201. [Google Scholar]
- 30.Deak, Z., I. Vass, and S. Styring. 1994. Redox interaction of tyrosine-D with the S-states of the water-oxidizing complex in intact and chloride-depleted photosystem II. Biochim. Biophys. Acta. 1185:65–74. [Google Scholar]
- 31.Ono, T.-a., T. Noguchi, Y. Inoue, M. Kusunoki, H. Yamaguchi, and H. Oyanagi. 1995. XANES spectroscopy for monitoring intermediate reaction states of Cl−-depleted Mn cluster in photosynthetic water oxidation enzyme. J. Am. Chem. Soc. 117:6386–6387. [Google Scholar]
- 32.Wincencjusz, H., H. J. van Gorkom, and C. F. Yocum. 1997. The photosynthetic oxygen evolving complex requires chloride for its redox state S2⇒S3 and S3⇒S0 transitions but not for S0⇒S1 or S1⇒S2 transitions. Biochemistry. 36:3663–3670. [DOI] [PubMed] [Google Scholar]
- 33.Wincencjusz, H., C. F. Yocum, and H. J. van Gorkom. 1998. S-state dependence of chloride binding affinities and exchange dynamics in the intact and polypeptide-depleted O2 evolving complex of photosystem II. Biochemistry. 37:8595–8604. [DOI] [PubMed] [Google Scholar]
- 34.Wincencjusz, H., C. F. Yocum, and H. J. van Gorkom. 1999. Activating anions that replace Cl− in the O2-evolving complex of photosystem II slow the kinetics of the terminal step in water oxidation and destabilize the S2 and S3 states. Biochemistry. 38:3719–3725. [DOI] [PubMed] [Google Scholar]
- 35.Homann, P. H. 1987. The relations between chloride, calcium, and polypeptide requirements of photosynthetic water oxidation. J. Bioenerg. Biomembr. 19:105–123. [DOI] [PubMed] [Google Scholar]
- 36.Boussac, A., and A. W. Rutherford. 1994. Electron transfer events in chloride-depleted photosystem II. J. Biol. Chem. 269:12462–12467. [PubMed] [Google Scholar]
- 37.McEvoy, J. P., and G. W. Brudvig. 2004. Structure-based mechanism of photosynthetic water oxidation. Phys. Chem. Chem. Phys. 6:4754–4763. [Google Scholar]
- 38.Haddy, A., J. A. Hatchell, R. A. Kimel, and R. Thomas. 1999. Azide as a competitor of chloride in oxygen evolution by photosystem II. Biochemistry. 38:6104–6110. [DOI] [PubMed] [Google Scholar]
- 39.Yu, H., C. P. Aznar, X. Xu, and R. D. Britt. 2005. Evidence that azide occupies the chloride binding site near the manganese cluster in photosystem II. Biochemistry. 44:12022–12029. [DOI] [PubMed] [Google Scholar]
- 40.Cao, J., and Govindjee. 1990. Anion effects on the electron acceptor side of Photosystem II in a transformable cyanobacterium Synechocystis 6803. In Current Research in Photosynthesis. M. Baltscheffsky, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. 515–518.
- 41.Garcia-Viloca, M., K. Nam, C. Alhambra, and J. L. Gao. 2004. Solvent and protein effects on the vibrational frequency shift and energy relaxation of the azide ligand in carbonic anhydrase. J. Phys. Chem. B. 108:13501–13512. [Google Scholar]
- 42.Noguchi, T., and M. Sugiura. 2001. Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry. 40:1497–1502. [DOI] [PubMed] [Google Scholar]
- 43.Hillier, W., and G. Babcock. 2001. S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry. 40:1503–1509. [DOI] [PubMed] [Google Scholar]
- 44.Yamanari, T., Y. Kimura, N. Mizusawa, A. Ishii, and T.-a. Ono. 2004. Mid- to low-frequency Fourier transform infrared spectra of S-state cycle for photosynthetic water oxidation in Synechocystis sp. PCC 6803. Biochemistry. 43:7479–7490. [DOI] [PubMed] [Google Scholar]
- 45.Debus, R. J., M. A. Strickler, L. M. Walker, and W. Hillier. 2005. No evidence from FTIR difference spectroscopy that aspartate-170 of the D1 polypeptide ligates a manganese ion that undergoes oxidation during the S0 to S1, S1 to S2, or S2 to S3 transitions in photosystem II. Biochemistry. 44:1367–1374. [DOI] [PubMed] [Google Scholar]
- 46.Barry, B. A., C. Hicks, A. DeRiso, and D. L. Jenson. 2005. Calcium ligation in photosystem II under inhibiting conditions. Biophys. J. 89:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeRiso, A., D. L. Jenson, and B. A. Barry. 2006. Calcium exchange and structural changes during the photosynthetic oxygen evolving cycle. Biophys. J. 91:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berthold, D. A., G. T. Babcock, and C. F. Yocum. 1981. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134:231–234. [Google Scholar]
- 49.Barry, B. A. 1995. Tyrosyl radicals in photosystem II. Methods Enzymol. 258:303–319. [DOI] [PubMed] [Google Scholar]
- 50.Yamomoto, Y., M. Doi, N. Tamura, and N. Nishimura. 1981. Release of polypeptides from highly active O2-evolving photosystem-2 preparation by Tris treatment. FEBS Lett. 133:265–268. [Google Scholar]
- 51.Jenson, D. L., A. Evans, and B. A. Barry. 2007. Proton-coupled electron transfer and tyrosine D of photosystem II. J. Phys. Chem. B. 111:12599–12604. [DOI] [PubMed] [Google Scholar]
- 52.Lindberg, K., T. Wydrzynski, T. Vanngard, and L.-E. Andreasson. 1990. Slow release of chloride from 36Cl-labeled photosystem II membranes. FEBS Lett. 264:153–155. [Google Scholar]
- 53.Le Coutre, J., J. Tittor, D. Oesterhelt, and K. Gerwert. 1995. Experimental evidence for hydrogen-bonded network proton transfer in bacteriorhodopsin shown by Fourier-transform infrared spectroscopy using azide as catalyst. Proc. Natl. Acad. Sci. USA. 92:4962–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guijarro, J., M. Engelhard, and F. Siebert. 2003. Binding of anions to halorhodopsin from Natronobacterium pharaonis studied by static and time-resolved FTIR spectroscopy. Biophys. J. 84:270A. (Abstr.) [Google Scholar]
- 55.Agrell, I. 1971. The infra-red spectra of some inorganic azide compounds. Acta Chem. Scand. A. 25:2965–2974. [Google Scholar]
- 56.Ono, T.-a., H. Conjeaud, H. Gleiter, Y. Inoue, and P. Mathis. 1986. Effect of preillumination on the P-680+ reduction kinetics in chloride-free photosystem II membranes. FEBS Lett. 203:215–219. [Google Scholar]
- 57.Takahashi, E., and C. A. Wraight. 1991. Small weak acids stimulate proton transfer events in site-directed mutants of the two ionizable residues, GluL212 and AspL213, in the QB-binding site of Rhodobacter sphaeroides reaction center. FEBS Lett. 283:140–144. [DOI] [PubMed] [Google Scholar]
- 58.Li, M., J. Owrutsky, M. Sarisky, J. P. Culver, A. Yodh, and R. M. Hochstrasser. 1993. Vibrational and rotational relaxation times of solvated molecular ions. J. Chem. Phys. 98:5499–5507. [Google Scholar]
- 59.Bendtsen, J., and G. Guelachvili. 1994. High-resolution infrared absorption spectrum of the ν2 band of hydrazoic acid (HN3). J. Mol. Spectrosc. 165:159–167. [Google Scholar]
- 60.Hansen, C. S., J. Bendtsen, and F. M. Nicolaisen. 1996. Analyses of the high-resolution infrared absorption spectra of the ν2 and ν3 bands of deuterated hydrazoic acid (DN3). J. Mol. Spectrosc. 175:239–245. [Google Scholar]
- 61.Steinhoff, H.-J., M. Pfeiffer, T. Rink, O. Burlon, M. Kurz, J. Riesle, E. Heuberger, K. Gerwert, and D. Oesterhelt. 1999. Azide reduces the hydrophobic barrier of the bacteriorhodopsin proton channel. Biophys. J. 76:2702–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tittor, J., C. Soell, D. Oesterhelt, H.-J. Butt, and E. Bamberg. 1989. A defective proton pump, point-mutated bacteriorhodopsin asp96 ⇒ asn is fully reactivated by azide. EMBO J. 8:3477–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Váró, G., L. S. Brown, R. Needleman, and J. K. Lanyi. 1996. Proton transport by halorhodopsin. Biochemistry. 35:6604–6611. [DOI] [PubMed] [Google Scholar]
- 64.Misra, S., R. Govindjee, T. G. Ebrey, N. Chen, J. X. Ma, and R. K. Crouch. 1997. Proton uptake and release are rate-limiting steps in the photocycle of the bacteriorhodopsin mutant E204Q. Biochemistry. 36:4875–4883. [DOI] [PubMed] [Google Scholar]
- 65.Schmies, G., B. Lüttenberg, I. Chizhov, M. Engelhard, A. Becker, and E. Bamberg. 2000. Sensory rhodopsin II from the haloalkaliphilic Natronobacterium pharaonis: light-activated proton transfer reactions. Biophys. J. 78:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hein, M., A. A. Wegener, M. Engelhard, and F. Siebert. 2003. Time-resolved FTIR studies of sensory rhodopsin II (NpSRII) from Natronobacterium pharaonis: Implications for proton transport and receptor activation. Biophys. J. 84:1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang, Y. H., L. Y. Chuang, and C. C. Hwang. 2007. Mechanism of proton transfer in the 3 alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. J. Biol. Chem. 282:34306–34314. [DOI] [PubMed] [Google Scholar]
- 68.Suydam, I. T., and S. G. Boxer. 2003. Vibrational Stark effects calibrate the sensitivity of vibrational probes for electric fields in proteins. Biochemistry. 42:12050–12055. [DOI] [PubMed] [Google Scholar]
- 69.Bogumil, R., C. L. Hunter, R. Maurus, H.-L. Tang, H. Lee, E. Lloyd, G. D. Brayer, M. Smith, and A. G. Mauk. 1994. FTIR analysis of the interaction of azide with horse heart myoglobin variants. Biochemistry. 33:7600–7608. [DOI] [PubMed] [Google Scholar]
- 70.Maurus, R., R. Bogumil, N. T. Nguyen, A. G. Mauk, and G. Brayer. 1998. Structural and spectroscopic studies of azide complexes of horse heart myoglobin and the His-64⇒Thr variant. Biochem. J. 332:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Messinger, J. 2004. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys. Chem. Chem. Phys. 6:4764–4771. [Google Scholar]
- 72.McEvoy, J. P., and G. W. Brudvig. 2006. Water-splitting chemistry of photosystem II. Chem. Rev. 106:4455–4478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.