

Abstract
Reduced birth weight and slow neonatal growth are risks correlated with the development of common diseases in adulthood. The Human Growth Hormone/Chorionic Somatomammotropin (hGH/CSH) gene cluster (48 kb) at 17q22-24, consisting of one pituitary-expressed postnatal (GH1) and four placental genes (GH2, CSH1, CSH2 and CSHL1) may contribute to common variation in intrauterine and infant growth, and also to the regulation of feto-maternal and adult glucose metabolism. In contrast to GH1, there are limited genetic data on the hGH/CSH genes expressed in utero. We report the first survey of sequence variation encompassing all five hGH/CSH genes. Resequencing identified 113 SNPs/indels (ss86217675-ss86217787 in dbSNP) including 66 novel variants, and revealed remarkable differences in diversity patterns among the homologous duplicated genes as well as between the study populations of European (Estonians), Asian (Han Chinese) and African (Mandenkalu) ancestries. A dominant feature of the hGH/CSH region is hyperactive gene conversion, with the rate exceeding tens to hundreds of times the rate of reciprocal crossing-over and resulting in near absence of linkage disequilibrium. The initiation of gene conversion seems to be uniformly distributed because the data do not predict any recombination hotspots. Signatures of different selective constraints acting on each gene indicate functional specification of the hGH/CSH genes. Most strikingly, the GH2 coding for placental growth hormone shows strong intercontinental diversification (Fst= 0.41-0.91; p<10−6) indicative of balancing selection, whereas the flanking CSH1 exhibits low population differentiation (Fst=0.03-0.09), low diversity (non-Africans π=8-9 × 10−5, Africans π= 8.2 × 10−4), and one dominant haplotype worldwide, consistent with purifying selection. The results imply that the success of an association study targeted to duplicated genes may be enhanced by prior resequencing of the study population in order to determine polymorphism distribution and relevant tag-SNPs.
Keywords: primate-specific duplicated genes, human Growth Hormone/Chorionic Somatomammotropin (hGH/CSH) gene cluster resequencing, population-specific and locus-specific variation, gene conversion, LD, selection
INTRODUCTION
Low birth weight and slow growth in infancy may lead to increased risk of adult diseases characterized by insulin resistance, such as metabolic syndrome, type 2 diabetes, cardiovascular disease, and obesity (reviewed by [Newsome et al., 2003] and [Stocker et al., 2005]). This observation has been explained by programming of metabolism due to undernutrition in utero [Barker et al., 1990; Hales et al., 1991] and alternatively by the contribution of hereditary factors. It has been proposed that common genetic variants which increase insulin resistance may predispose both to low insulin-mediated growth in utero and insulin resistance in adulthood [Hattersley and Tooke, 1999]. Among the known human diabetes-related loci, an association with birth weight has been shown for genes coding for insulin (INS) [Dunger et al., 1998], insulin-like growth factor 1 (IGF1) [Vaessen et al., 2002], glucokinase (GCK) [Weedon et al., 2005] and TCF7L2 [Freathy et al., 2007]. However, the contribution of INS and IGF1 variation to birth weight has not been consistently replicated and remains controversial [Bennett et al., 2004; Frayling et al., 2002]
The Human Growth Hormone/Chorionic Somatomammotropin (hGH/CSH) gene cluster is an excellent candidate for contributing to common variation in intrauterine and infant growth, and to the regulation of glucose metabolism. In most mammals including prosimians, a single gene (GH1) encodes the pituitary growth hormone (GH), whereas in anthropoids, five to eight GH-related genes arisen through successive events of duplication have been described [Li et al., 2005; Ye et al., 2005]. In great apes and Old World Monkeys the duplicated loci, apart from a couple of GH-related genes, have acquired a novel function and code for chorionic somatomammotropin (CSH genes), also known as placental lactogen, expressed in the placenta [Chen et al., 1989]. The human hGH/CSH gene cluster, located on chromosome 17q22-24 and spanning 48 kb consists of the GH1 (#MIM 139250), CSHL1 (chorionic somatomammotropin-like; #MIM 603515), CSH1 (#MIM 150200), GH2 (gene for placental growth hormone, PGH; #MIM 139250), and CSH2 (#MIM 118820) genes transcribed from the same chromosomal strand [Chen et al., 1989] (Fig. 1A). These duplicated genes are highly homologous at the DNA level (91-97 %; Supplementary Fig. S1), and each of them encodes for three to five protein isoforms generated by alternative splicing (Supplementary Fig. S2). Growth hormone (GH) deficiency (1:4000-1:10,000/births) is associated with short stature (reviewed by [Procter et al., 1998]) and there is evidence that substitutions in the GH1 gene may contribute to the determination of GH secretion as well as to variation in height [Esteban et al., 2007; Hasegawa et al., 2000; Millar et al., 2003]. In addition to a critical role in postnatal growth, GH contributes to the maintenance of glucose metabolism as a transcriptional regulator for IGF1 [Jorgensen et al., 2004; Rosenfeld, 2003]. The placenta-expressed CSH1, CSH2 and GH2 genes have key roles in the regulation of fetal glucose supply and growth as well as maternal metabolism, and are candidates for the developing of insulin resistance during pregnancy [Barbour et al., 2002; Fleenor et al., 2005; Handwerger and Freemark, 2000; Lacroix et al., 2002]. However, little is known about the impact of common genetic variation across the hGH/CSH region on growth and metabolism in utero and in early infancy, and susceptibility to metabolic and cardiovascular disease in adulthood. In contrast to the pituitary-expressed GH1, there are limited data on the normal or pathological variation of the human GH2, CSH1, CSH2 and CSHL1 expressed in placenta. Detailed research on the hGH/CSH cluster has been hindered by the complex genomic structure of the region rich in repetitive genes and intergenic sequence fragments evolved through multiple duplication events.
Figure 1.
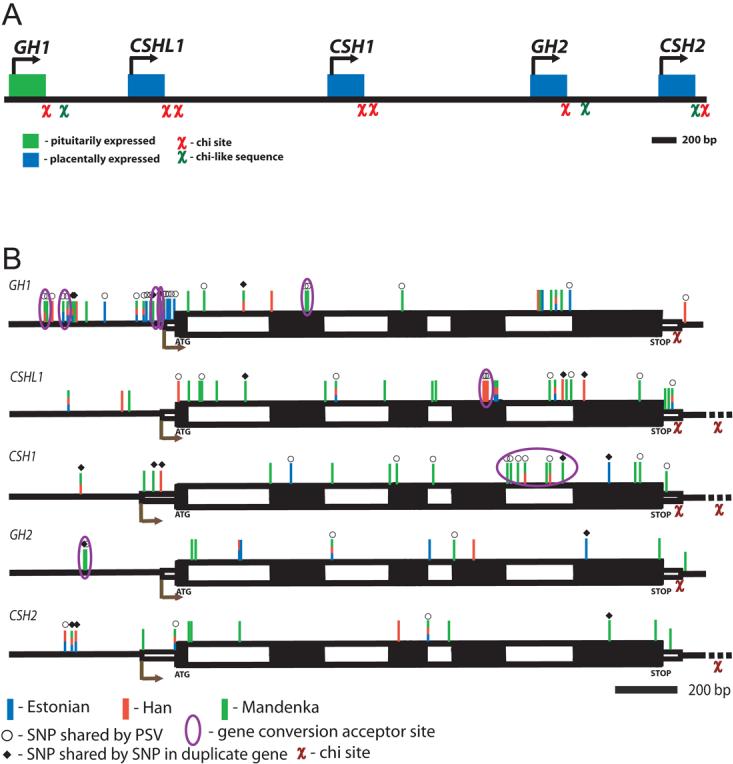
A: A schematic representation of the human GH/CSH gene cluster, located at 17q22-24. The χ-sites at the 3′ UTR and downstream regions of the genes may be associated with recombination and gene conversion initiation [Smith, 1988]. B. Polymorphisms identified in GH1, CSHL1, CSH1, GH2 and CSH2 genes. The main transcript of each gene is drawn to an approximate scale, including the 5′upstream and 3′ downstream regions (black lines), 5′ and 3′ UTRs (thin unfilled boxes), exons (thick black boxes) and introns (thick unfilled boxes). The blue (Estonians), red (Han Chinese), and green (Mandenkalu) bars represent the identified SNPs and indels in the three populations. SNPs located in the shared position with paralogus sequence variants (PSVs) are marked with open circles (○) and SNPs co-localizing among the genes with black squares (◆). The SNPs within identified gene conversion acceptor tracts are surrounded with a blue oval.
We have designed the first survey of the high resolution sequence variation of all five human GH/CSH genes using long-range PCR and the resequencing of 74 individuals of European (Estonians), Asian (Han Chinese) and African (Mandenkalu) ancestries. We address the data with the following questions: (1) What are the variation patterns of duplicated and highly homologous hGH/CSH genes across the loci and across the populations? (2) What are the roles of gene conversion and crossover in shaping the diversity and linkage disequilibrium (LD) of the hGH/CSH region? (3) Is there evidence for selective pressures on these recently evolved genes contributing to human reproduction and metabolism? (4) Does the distribution of population-specific gene variants have implications for the design of association studies?
In addition, the fine-scale analysis of the hGH/CSH region contributes to the general understanding of the forces shaping the genetic variation of segmentally duplicated regions, reported to cover 5%-10% of human genome [Bailey et al., 2002]. Currently, the lack of this knowledge may prevent the design of successful association studies involving duplicated genomic regions, densely packed with genes.
SUBJECTS AND METHODS
Population samples
The study has been approved by the Ethics Committee of Human Research of the University Clinic of Tartu, Estonia (permission no. 146/118, 27.02.2006). For the resequencing of the five genes within the hGH/CSH genome cluster, a total of 74 DNA samples from three populations were used: 25 Estonian (Europe), 25 Chinese Han (Asia) and 24 Mandenka (Africa) individuals. The Estonian samples originated from healthy blood donors around Estonia, collected upon informed consent. The Estonian population represents a North-European population [Mueller et al., 2005]; the Han Chinese are a major subpopulation of China; the Mandenkalu are a representative population of West-Africa [Rosa et al., 2007], used for decades in human population genetics studies. The Mandenka and Han samples were obtained from the HGDP-CEPH Human Genome Diversity Cell Line Panel (http://www.cephb.fr/HGDP-CEPH-Panel/) [Cann et al., 2002].
Gene-specific and nested PCR, and resequencing
Due to the complex and repetitive genomic structure of the hGH/CSH region, we carried out the amplification of the five individual genes using long-range PCR with primers pairs targeted to the most diverged genomic segments identified within ~ ±5 kb of a particular gene. The gene-specific product was obtained by an initial long-range PCR followed by nested PCR. The gene-specific and nested PCR primers for the GH1, CSHL1, CSH1, GH2, and CSH2 genes (Supplementary Table S1) were designed based on the Homo sapiens Growth Hormone locus sequence (NCBI GenBank database locus no NG_001334.1, http://www.ncbi.nlm.nih.gov), using the Web-based version of Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The PCR primer pairs were required to have at least on unique primer in the human genome, verified using NCBI-BLAST. Approximately 100 ng of genomic DNA were used in the PCR reactions. The amplified regions for GH1, CSHL1, CSH1, and CSH2 were 3590, 10,310, 6574, and 3559 bp, respectively. The amplification of the GH2 genomic region yielded a 3690 bp product for the Estonian and the Han samples, and 5628 bp for the Mandenka individuals. The binding site for the reverse primer for GH2 long-range PCR (G2EH-R, Supplementary Table S1) resides partially in an Alu sequence. This primer did not amplify the GH2 region in the Mandenkalu, potentially because the respective Alu sequence was either diverged or absent in these individuals. Therefore, an alternative primer (G2M-R, Supplementary Table S1) was designed resulting in successful PCR of the GH2 gene for 20 (out of 24) Mandenka samples (length 5628 bp). Primary long-range PCR fragments were further amplified by nested PCR. The amplified region included all of the coding sequence of the genes and a part of the flanking sequences: 2178 to 2247 bp for GH1, CSH1, GH2, and CSH2; and 4954 bp for CSHL1. Each gene was sequenced from both strands using ten sequencing primers (Supplementary Table S1), with the exception of GH2 (nine primers). Sequencing reactions (1.5 μL) were run on an ABI 377 Prism automated DNA sequencer (Applied Biosystems) using ReproGel 377 gels (Amersham Biosciences Inc.). PCR, purification of the PCR products and the resequencing reactions were carried out as previously described [Hallast et al., 2005].
For each gene and for each population, the sequences were assembled into a contig as described [Hallast et al., 2005]. Polymorphisms were identified using the PolyPhred program (Version 4.2) (http://www.phrap.org/phredphrapconsed.html) [Nickerson et al., 1997] and confirmed by manual checking. A genetic variant was verified only if it was observed in both the forward and the reverse orientations. The consensus sequences of resequenced genomic regions of GH1, CSHL1, CSH1, GH2, and CSH2 genes (Supplementary Fig. S3) were used as references for the gDNA numbering of SNPs and indels (Supplementary Table S2). Allele frequencies were estimated and conformance to Hardy-Weinberg Equilibrium (HWE) was computed by an exact test (α = 0.01) using the Web version of the Genepop 3.3 program (http://genepop.curtin.edu.au/) [Rousset and Raymond, 1995]. A total of three SNPs in the Mandeka population were found to be deviating from HWE, apparently because of a small sample size. Before entering the statistical analysis, the specificity of the PCR products was controlled by the verification of the monomorphic status of gene-specific positions used as markers for each individual gene (Supplementary Fig. S1).
Statistical analyses
Haplotypes were inferred from unphased genotype data using the Bayesian statistical method in the program PHASE 2.1.1 (http://www.stat.washington.edu/stephens/) [Stephens et al., 2001b], using the model allowing recombination. The running parameters were: number of iterations = 1000, thinning interval = 1, burn-in = 100; the –X10 parameter was used for increasing the number of iterations of the final run of the algorithm.
Sequence diversity parameters and pairwise FST distances between populations (measures of genetic differentiation) were calculated with Arlequin version 3.1 (http://cmpg.unibe.ch/software/arlequin3/) [Excoffier et al., 2005] with the most probable phased haplotypes as an input sequence. The direct estimate of per-site heterozygosity (π) was derived from the average pairwise sequence differences, while Watterson's θ represents an estimate of the expected per-site heterozygosity based on the number of segregating sites (S). The Tajima's D statistic [Tajima, 1989] (DT) was calculated to determine if the observed patters of diversity in the three studied populations are consistent with the standard neutral model. The basis of the DT value is the difference between the π and θ estimates: under neutral conditions π = θ and DT = 0. The relationships between inferred haplotypes were investigated with NETWORK 4.201 software (http://www.fluxus-technology.com) [Bandelt et al., 1999] using the Median-Joining (MJ) network algorithm. For each of the genes, only SNPs located in the genomic region from the transcription initiation site until the end of the mRNA were included in network calculations. An additional network was drawn for the 5′ upstream region of the GH1 gene (for 407 bp upstream of the transcription start site). Singleton polymorphisms were excluded from network calculations (cannot be reliably phased), and the calculations were performed with default parameters.
The gene sequence variants derived from estimated haplotypes were used for gene conversion analysis. For the manual detection of gene conversion sites between a pair of hGH/CSH genes, the derived complete sequence variants were aligned using the Web-based ClustalW program (http://www.ebi.ac.uk/clustalw/). A minimum gene conversion site was defined as a region within an acceptor gene with ≥ 2 associated, motif-forming polymorphisms for which a potential donor gene could be defined. The direction of the gene conversion event was established based on the frequency of the conversion sequence motif in the two homologous loci. The converted tract in the acceptor gene represented a minor locus variant, whereas the donor gene was nearly monomorphic in the gene conversion region (Table 3). The maximum possible gene conversion tract covers the identical sequence between two compared genes on both sides of the minimum gene-conversion tract. Alternatively, the GENECONV algorithm (Version 1.81) (http://www.math.wustl.edu/~sawyer/geneconv/) [Sawyer, 1989] was used for the estimation of gene conversion tract length (as described in [Hallast et al., 2005]). Not relying on polymorphism data, this method searches for regions where pairs of sequences are unusually similar compared to overall similarity. The analysis predicts the fragments likely to have been converted between gene pairs, but it does not provide information about the directionality of the gene conversion events. Alignments were analyzed using the “g0” parameter, meaning that mismatches within fragments are not allowed and p < 0.05 from global fragments were considered as significant.
TABLE 3.
Detection of Gene Conversion Tracts from Aligned Sequences of hGH/CSH Genes Using SNPs as Markers
| Haplotype |
Gene Conversion tract length (bp) |
|||||
|---|---|---|---|---|---|---|
| Gene | Variant | Frequency | Sequencea | Min | Max | |
| GH2 | common | 98% | −322 | G…C −318 | 5 | 72 |
| acceptor | 0.7% | C…A | ||||
| CSHL1 | Donor | 100% | C…A | |||
| CSH1 | common | 96.6% | +1086 | C… …T… …T… …A… …T… …A +1228 | 142 | 302 |
| acceptor | 1.35% | A… …C… …G… …C… …C… …G | ||||
| CSH2 | Donor | 100% | A… …C… …G… …C… …C… …G | |||
| GH1 | common | 89.2% | −435 | G…………A −422 | 14 | 33 |
| acceptor | 1.35% | A…………G | ||||
| All others | Donor | 100% | A…………G | |||
| GH1 | common | 50% | −68 | A….A −63 | 6 | 95 |
| acceptor | 7.43% | G….T | ||||
| CSH2/CSH1 | Donor | 100%/99.3% | G….T | |||
| GH1 | common | 81.8% | −63 | A..G −60 | 4 | 63 |
| acceptor | 4.05% | C..C | ||||
| CSHL1 | Donor | 100% | C..C | |||
| GH1 | common | 83.1% | −370 | G……G −363 | 8 | 60 |
| acceptor | 15.5% | T……T | ||||
| GH2 | Donor | 100% | T……T | |||
| GH1 | common | 97.3% | +410 | C.T +412 | 3 | 23 |
| acceptor | 2.03% | A.A | ||||
| GH2 | Donor | 100% | A.A | |||
| CSHL1 | common | 99.3% | +944 | CC…A +949 | 6 | 79 |
| acceptor | 0.68% | TG…G | ||||
| CSH1/CSH2 | Donor | 100% | TG…G | |||
location of a gene conversion tract on genomic sequence relative to the position of ATG of the acceptor gene; A denotes +1.
For the estimation of the rate of recombination, we calculated the population crossing-over parameter ρ= 4Nerbp, where Ne equals effective population size and rbp the crossing-over rate per base pair per generation, using three alternative algorithms. The Li and Stephens' [Li and Stephens, 2003] method is based on the “Product of Approximate Conditionals” (PAC) model, which considers all loci simultaneously, allowing variation of recombination rate across the region of interest and thus estimation of putative recombination hotspots. The average background recombination rate (ρPAC) and the factor (λ) by which the ρPAC between loci exceeds the average background rate were estimated from unphased genotype data using PHASE 2.1.1 software [Li and Stephens, 2003; Stephens et al., 2001b]. Within this model, a λ value of 1 corresponds to an absence of recombination rate variation, while values of λ >1 indicate an increase in crossover activity. The running parameters for the PHASE program are described above. Alternatively, we used two methods that simultaneously estimate the population recombination parameter ρbp and the population gene-conversion rate γbp = 4Necbp, where cbp denotes the probability of a gene-conversion event per base pair per generation. These two approaches assume that gene-conversion and crossing-over are alternative resolutions of the Holliday junction and that the conversion tract length is geometrically distributed with the mean length L. Padhukasahasram et al's “pattern matching” (PM) method (http://people.cornell.edu/pages/bp85/) [Padhukasahasram et al., 2006] utilizes summary statistics and matches the frequencies of multilocus patterns of nucleotide sites with coalescent simulations with gene-conversion. This method was implemented by simulating datasets conditional on the observed number of segregating sites at exactly the same positions and roughly similar minor allele frequencies (MAF) for each position as in real data. In Hudson's “composite likelihood” (CL) method (http://home.uchicago.edu/~rhudson1/source/maxhap.html) [Hudson, 2001], estimates are based on the products of full-likelihoods for smaller subsets (i.e. all possible SNP pairs) in the data. We obtained estimates for ρ and γ from phase-estimated data using the Maxhap program as well as with the method of Padhukasahasram et al. [Padhukasahasram et al., 2006] for gene-conversion tract lengths of L = 100 bp and 500 bp. The choices of L values were based on reports from human single-sperm analysis [Jeffreys and May, 2004] and the lengths of gene-conversion tracts identified for the hGH/CSH genes. We also obtained estimates using both these methods for models with crossing-over alone (L=0). All calculations of ρ and γ considered SNPs with MAF ≥10%. The descriptive statistic of linkage disequilibrium (LD), r2 was calculated for pairs of common SNPs (MAF ≥10%) and summarized using the Haploview 3.32 program (http://www.broad.mit.edu/mpg/haploview/index.php) [Barrett et al., 2005].
RESULTS
Dissimilar diversity patterns among highly homologous hGH/CSH genes
We resequenced all five of the hGH/CSH genes and part of the flanking sequences (approx. 400 bp upstream and 200 bp downstream of each gene, a total of 10,786 bp per individual) in three population samples: the Estonians (E; European origin, n=25), the Chinese Han (H; Asian origin, n=25), and the Mandenkalu (M; African origin, n=24). 113 segregating sites were identified in total (Fig. 1B, Table 1, Supplementary Table S2; ss86217675-ss86217787 in dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/), including three 1 bp indels and 108 SNPs; 66 sequence variants (58.4%) were novel, previously not described in the dbSNP database. Only two sequence variants identified in the GH/CSH genes overlapped with polymorphic HAPMAP SNPs (http://www.hapmap.org/; release April 2007): rs20770680:T>C in CSHL1 (information available only for the CEPH individuals) and an African-specific (Yorubans in HAPMAP, Mandenkalu in this study) rs9909776:T>C in CSH1. Three GH1 and five GH2 positions genotyped in HAPMAP were monomorphic; CSH2 was not covered by HAPMAP. In addition to the SNPs and indels we also identified a polymorphic microsatellite in intron 1 of GH2, formed by the combination of a variation in an (AG)n dinucleotide (n=7–8; g.647G>A in Supplementary Table S2) and a flanking (A)n mononucleotide repeat (n=10-11; g.648del). This polymorphism was excluded from the current analysis due to the unreliability of genotype calling of microsatellite alleles from sequencing data.
TABLE 1.
Sequence Parameters of GH1, CSHL1, CSH1, GH2 and CSH2 Genomic Regions
| Analyzed regiona |
Segregating sites |
SNPs shared with |
Population genetics parameters |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) |
Pb | All | Commonc | Singleton | Indels (1bp) |
dbSNP | PSVd | SNP in duplicate gene |
πe | θf | DT | DT p-value |
|
|
GH1 promoter |
407 | E | 10 | 6 | 0 | 1 | 9 | 8 | 2 | 0.00689 | 0.00550 | 1.09 | 0.124 |
| H | 8 | 3 | 4 | 1 | 7 | 5 | 2 | 0.00371 | 0.00385 | −0.09 | 0.498 | ||
| M | 13 | 9 | 2 | 1 | 11 | 8 | 4 | 0.00861 | 0.00777 | 0.87 | 0.164 | ||
| All | 17 | 1 | 2 | 13 | 11 | 4 | 0.00691 | 0.00755 | |||||
| GH1 gene | 1636 | E | 7 | 1 | 4 | 0 | 7 | 5 | 0 | 0.00050 | 0.00096 | −1.26 | 0.095 |
| H | 4 | 1 | 2 | 0 | 2 | 0 | 1 | 0.00034 | 0.00055 | −0.88 | 0.179 | ||
| M | 11 | 5 | 3 | 0 | 3 | 5 | 1 | 0.00119 | 0.00152 | −0.63 | 0.326 | ||
| All | 18 | 6 | 0 | 9 | 9 | 1 | 0.00079 | 0.00197 | |||||
| CSHL1 | 2160 | E | 6 | 6 | 0 | 0 | 6 | 2 | 0 | 0.00135 | 0.00062 | 2.98 | 0.005 |
| H | 13 | 5 | 6 | 0 | 6 | 6 | 2 | 0.00119 | 0.00134 | −0.34 | 0.438 | ||
| M | 22 | 7 | 5 | 0 | 8 | 6 | 1 | 0.00179 | 0.00230 | −0.71 | 0.257 | ||
| All | 28 | 10 | 0 | 8 | 10 | 3 | 0.00152 | 0.00233 | |||||
| CSH1 | 2067 | E | 2 | 0 | 1 | 0 | 1 | 1 | 2 | 0.00009 | 0.00022 | −1.02 | 0.17 |
| H | 4 | 0 | 4 | 0 | 0 | 2 | 2 | 0.00008 | 0.00043 | −1.86 | 0.003 | ||
| M | 17 | 3 | 2 | 1 | 3 | 9 | 2 | 0.00082 | 0.00185 | −1.75 | 0.018 | ||
| All | 20 | 4 | 1 | 4 | 10 | 5 | 0.00033 | 0.00174 | |||||
| GH2 | 2040 | E | 3 | 1 | 0 | 1 | 1 | 1 | 0 | 0.00032 | 0.00033 | −0.06 | 0.496 |
| H | 2 | 0 | 1 | 0 | 1 | 1 | 0 | 0.00009 | 0.00022 | −1.02 | 0.161 | ||
| M | 7 | 0 | 4 | 0 | 1 | 3 | 1 | 0.00026 | 0.00081 | −1.87 | 0.012 | ||
| All | 10 + 1 MS |
5 | 1 | 1 | 3 | 2 | 0.00057 | 0.00098 | |||||
| CSH2 | 2062 | E | 5 | 5 | 0 | 0 | 3 | 3 | 2 | 0.00113 | 0.00054 | 2.62 | 0.004 |
| H | 6 | 6 | 0 | 0 | 3 | 3 | 2 | 0.00136 | 0.00065 | 2.77 | 0.003 | ||
| M | 10 | 7 | 1 | 0 | 3 | 2 | 2 | 0.00108 | 0.00109 | −0.22 | 0.475 | ||
| All | 13 | 1 | 0 | 5 | 3 | 3 | 0.00142 | 0.00122 | |||||
Genomic sequences used for calculations included the entire gene + the resequenced upstream region. For GH1, the calculations were performed separately for the upstream and gene region.
Population samples: (E) Estonians (chromosomes 2N=50); (H) Han (2N=50); (M) Mandenkalu (2N=48 for GH1, CSHL1, CSH1, and CSH2; n=40 for GH2).
MAF>10%
paralogous sequence variant
Estimate of nucleotide diversity per site from average pairwise difference among individuals
Estimate of nucleotide diversity per site from number of segregating sites (S); DT – Tajima's D statistic, significant (p<0.05) estimates are highlighted in bold; MS - a complex microsatellite polymorphism detected in GH2 intron 1
A majority of the segregating sites (n=84; 75.7%) was specific for one population sample. All three population samples shared only 14.4% of the identified SNPs (6 in GH1, 6 in CSHL1, 3 in CSH2 and 1 in GH2) and an additional 11 SNPs were represented in subjects of two sample sets. The Mandenka chromosomes were found to have the highest number of polymorphisms (n=83; Han n=38; Estonians n=33) and the highest fraction of population sample specific SNPs (M, n=58; H, n=15 and E, n=11).
Sequence diversity, characterized by the mean nucleotide diversity parameter π (calculated per bp) differed severely between the individual genes. The diversity of CSHL1 (0.00152) and CSH2 (0.00142) was up to 5 times higher compared to the two other placental genes CSH1 (0.00033) and GH2 (0.00057) as well as compared to the reported average for 292 autosomal genes (π=0.00058) [Stephens et al., 2001a] (Table 1). It is noteworthy that there was a 9-fold difference in the diversity for the GH1 promoter (π=0.00691) compared to the GH1 gene region (π=0.00079), transcribed in the pituitary. This polarity between the genomic sequence of the gene and its promoter was not observed for the placenta-expressed genes (data not shown).
Allelic variants leading to non-synonymous substitutions in GH1, CSH1, GH2 and CSH2 protein isoforms
Indicative of functional constraint, most of the detected protein-altering sequence variants were rare (<10%) and population sample specific: 13 found only in Mandenkalu, 2 in Han, and 1 in Estonians (Table 2). The exception was p.Phe70Leu variant in CSH2 isoforms 1 and 2 with a frequency of 28% among the Han. The ratio between non-synonymous (ns) to synonymous (s) substitutions may imply selective forces acting upon a locus. In cases where ns>s, non-synonymous sites could be evolving faster, and the evidence for positive selection is supported. Notably, for GH2 (ns=4/s=0) and CSH1 (ns=4/s=3 for isoform 1; ns=11/s=3 including the isoform 2 coding region) the ns/s ratio was >>1. For GH1 (ns=2/s=2) and CSH2 (ns=1/s=1) the number of identified allelic variants leading to non-synonymous and synonymous substitutions was equal (Table 2; Supplementary Table S2).
TABLE 2.
Identified non-synonymous changes in GH1-CSHL1-CSH1-GH2-CSH2 genes
| Gene | SNPa | Exon | Amino acid change | SP/ MPb | Samplec | MAFd (%) |
|---|---|---|---|---|---|---|
| GH1 | g.863T>A | 2 | p.Phe51Tyr | MP | M | 8.3 |
| g.1166G>C | 3 | p.Glu82Aspe p.Glu67Aspf |
MP | M | 2.1 | |
| CSHL1h | g.457G>A | 1 | p.Ala3Thr | na | H | 2.0 |
| g.1394C>T/g.1395C>G | 4 | p.Thr127Met | na | H | 2.0 | |
| g.1399A>G | 4 | p.Thr129Gly | na | H | 2.0 | |
| g.1437C>A | 4 | p.Asp141Glu | na | E M H |
46.0 4.2 30.0 |
|
| CSH1 | g.733G>A | 2 | p.Arg6Trp | SP | M | 6.3 |
| g.1114A>G | 3 | p.Glu58Gly | MP | M | 4.2 | |
| g.1158G>A | 3 | p.Asp73Asn | MP | M | 4.2 | |
| g.1537C>A | 4f | p.Ser168Xf | MP | M | 4.2 | |
| g.1557T>C | 4f | p.Tyr175Hisf | MP | M | 4.2 | |
| g.1573T>G | 4f | p.Phe180Cysf | MP | M | 4.2 | |
| g.1581A>C | 4f | p.Lys183Glnf | MP | M H |
4.2 2.0 |
|
| g.1632C>A | 4f | p.Pro200Thrf | MP | M | 4.2 | |
| g.1642T>C | 4f | p.Leu203Prof | MP | M H |
4.2 2.0 |
|
| g.1679A>G | 4f | p.Ile215Metf | MP | M | 4.2 | |
| g.1912C>A | 5 | p.Cys208X | MP | M | 2.1 | |
| GH2 | g.1331G>A | 4 | p.Arg103His | MP | M | 2.5 |
| g.1369G>T | 4 | p.Val116Leu | MP | H | 2.0 | |
| g.1744T>C | 4f/5g | p.Trp241Argf p.Met155Thrg |
MP | E | 6.0 | |
| g.2002C>G | 5 | p.Pro241Alag | MP | M | 7.5 | |
| CSH2 | g.1150C>A | 3 | p.Phe70Leue,f | MP | H | 28.0 |
A full description of each SNP in available in Supplementary Table S2. Reference sequences for each resequenced gene region are in Supplementary Fig. 3
SP – signal peptide/MP – mature peptide; na – not applicable
E-Estonians, M-Mandenkalu, H-Chinese Han
MAF – minor allele frequency
Amino acid change specific to either isoform 1 precursor (see Supplementary Fig. S2)
Amino acid change specific to either isoform 2 precursor (see Supplementary Fig. S2)
Amino acid change specific to either isoform 3 precursor (see Supplementary Fig. S2)
Changes in CSHL1 are shown based on the predicted hypothetical protein
We detected apparent gene function-disrupting mutations. An African-specific substitution in the CSH1 gene creates a premature stop codon in exon 5 of the major transcript (p.Cys208X; Table 2). Another example is a consequence of a gene conversion (see below) event introducing seven changes (from g.1537C>A to g.1679A>G, Supplementary Table S2) including p.Ser168X in CSH1 isoform 2 precursor (Table 2; Supplementary Fig. S2). The latter is an alternatively spliced mRNA variant for the expression of a non-secreted protein isoform that presumably localizes in the cell membrane [Untergasser et al., 2000]. An alternative conversion event (from g.861C>A to g.863T>A, Supplementary Table S2) on Mandenka chromosomes has resulted in the p.Phe51Tyr variant in GH1, replacing a non-polar hydrophobic with a polar hydrophilic amino acid. Several other rare CSH1 and GH2 amino acid replacement mutations may alter either the polarity or the charge of the proteins, which in turn may have consequences on the hormone structure and functional activity.
Active gene conversion and near absence of linkage disequilibrium
We searched for traces of past gene conversion (GC) events between homologous hGH/CSH genes, both by visual inspection of clustered SNP motifs shared by gene-pairs and by using the GENECONV [Sawyer, 1989] algorithm. The alignment of complete nucleotide sequence variants of all possible hGH/CSH gene pairs resulted in the identification of 8 possible GC sites encompassing at least 2 SNPs (Table 3; Fig. 1B), and the GENECONV analysis predicted 16 GC tracts (Supplementary Table S3). The GC tract lengths from the visual inspection (minimum ranging 3-142 bp, the mean 24 bp; and maximum up to 302 bp, mean 91 bp) were consistent with the prediction by the computer algorithm (62 – 861 bp, mean 130 bp). The GH1 locus, and especially its promoter region, was identified as the most active gene conversion partner in both analyses. The dominant direction of the GC events appears to be from the placental genes (mostly donors) towards the ancestral GH1 locus. The major role of GC in generating the extremely high diversity of the GH1 promoter was also reflected by the complex and intricate haplotype network, rich in rare population sample specific variants (Fig. 2, Supplementary Table S4F). Similar results for GH1 upstream region have been previously reported [Giordano et al., 1997; Horan et al., 2003]. A high number of SNPs shared by the gene copies and the co-existence of paralogous gene variants provided additional support for active gene conversion. Still, independently co-occurring mutations cannot be ruled out.
Figure 2.
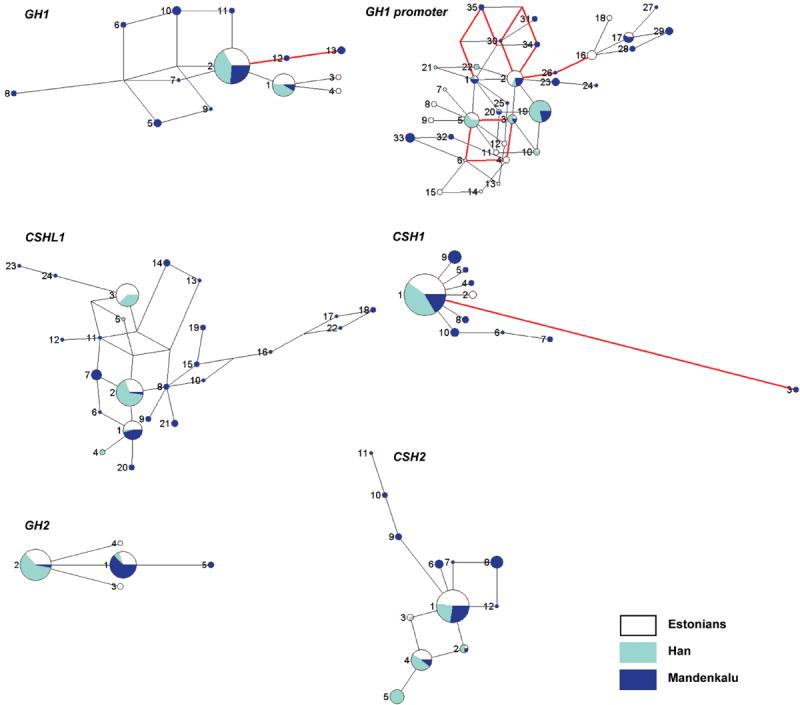
Median-Joining (MJ) networks for predicted haplotypes of the hGH/CSH genes and the 5′ upstream region of GH1. Singleton polymorphisms were excluded from the analysis, because they could not be reliably phased. The size of each node is proportional to the haplotype frequency in the total dataset. The relative distribution of each haplotype among the individuals of Estonian, Chinese Han and Mandenka origin is indicated by white, light blue and dark blue, respectively. Haplotype nomenclature is shown in Supplementary Table S4A-S4F. Red lines designate identified gene conversion tracts (Table 3) – note that several haplotypes may have been formed because of gene conversion.
During recombination, gene conversion events are alternative solutions to crossovers, resulting in the non-reciprocal exchange of chromosomal fragments. We addressed the recombination landscape across the hGH/CSH cluster by calculating the population crossing-over parameter ρbp= 4Nerbp and gene conversion parameter γbp=4Necbp where Ne is the effective population size; rbp and cbp are the crossing-over rate and gene conversion rate per generation between adjacent nucleotide positions, respectively. We used three algorithms: (1) the Li and Stephens' [Li and Stephens, 2003] “Product of Approximate Conditionals” (PAC) likelihood method which calculates ρ, and in addition, allows the estimation of putative recombination hotspots; (2) Padhukasahasram's [Padhukasahasram et al., 2006] “pattern matching” (PM) and (3) Hudson's [Hudson, 2001] “composite likelihood” (CL) method, both of which allow the simultaneous estimation of crossing-over and gene conversion rate. The average recombination rate per base pair across the entire studied region, with estimates varying slightly between the three methods (Table 4; Estonians, ρ(L=0)= 2.59 × 10−4 – 4.43 × 10−4; Chinese Han, ρ(L=0)= 2.48 × 10−4 − 4.32 × 10−4; Mandenkalu, ρ(L=0) = 31.31 × 10−4 – 33.96 × 10−4) fell in the range published for a set of 74 genes [Crawford et al., 2004]. The higher ρ values for the Mandenkalu are consistent with the idea that African populations maintained a larger long-term effective population size than did non-Africans. When gene conversion (tract lengths L=100 or 500 bp) was incorporated into the estimations using CL and PM methods, the ρ values dropped (Table 4; Estonians, ρbp = 5.4 × 10−6 – 1.19 × 10−4; Chinese Han, ρbp = 2.2 × 10−5 – 2.16 × 10−4; Mandenkalu, ρbp = 2.12 × 10−4 – 10.61 × 10−4). For models with gene conversion, estimates of ρ were almost independent of the assumed tract length using Hudson's method, whereas for Padhukasahasram's method, ρ estimates changed 2-3 fold, depending on tract length. The estimated γ values decrease with longer conversion tracts because shorter tracts are likely to contain fewer SNPs than longer ones; therefore, such conversion events have to occur at a higher rate to create the same effects on linkage disequilibrium as longer tracts. Strikingly, the gene conversion rate (γ) estimates were tens to hundreds of times higher than the rate of crossing over (ρ; Table 4), which may indicate that in the duplicated hGH/CSH gene cluster Holliday junctions formed during recombination process are favorably resolved by non-reciprocal gene conversions compared to reciprocal crossovers.
TABLE 4.
Estimation of Population Crossing-over and Gene Conversion Parameters (× 104) Using SNPs with MAF ≥10 %
| Gene conversion tract lengtha |
Recombination parameters ρbp, γbp b |
Estonians | Han | Mandenkalu |
|---|---|---|---|---|
| Maximum PAC likelihood estimates [Li and Stephens, 2003] | ||||
| NA | ρbp | 4.43 | 2.58 | 32.2 |
| Pattern Matching method estimates [Padhukasahasram et al., 2006] | ||||
| L=0b | ρbp | 5.0 | 4.0 | 40 |
| L=100 | ρbp | 1.5 | 2.0 | 12 |
| γbp | 120 | 70 | 500 | |
| L=500 | ρbp | 0.6 | 0.8 | 4.0 |
| γbp | 40 | 40 | 220 | |
| Composite likelihood (CL) estimates [Hudson, 2001] | ||||
| L=0 | ρbp | 2.59 | 2.48 | 31.31 |
| L=100 | ρbp | 1.19 | 1.4 | 10.61 |
| γbp | 107 | 65.9 | 372 | |
| L=500 | ρbp | 0.86 | 1.18 | 9.02 |
| γbp | 27.6 | 21.4 | 117 | |
L is the gene conversion tract length
The population crossing-over rate per base pair, ρbp= 4Nerbp; the population gene-conversion rate per base pair, γbp=4Necbp.
The analysis of recombination rate variation across the entire hGH/CSH region predicted no recombination hotpot (Fig. 3), usually considered to be the initiator of gene conversion activity20. The whole region was characterized by relatively uniform recombination activity and minimal extent of LD measured by the correlation coefficient between alleles, r2 (Fig. 3). The ability to initiate recombination throughout the region may be promoted by the abundance of the hotspot motif CCTCCCT [Myers et al., 2005] (n=13 for sense and for n=22 antisense strand) compared to the motif with a “suppressor” mutation CCCCCCT (n=2/ n=4). In the Mandenkalu, LD was practically non-existent (median r2 0.033; range 0-1); it was weak for the Estonians and Han (median r2 0.2325; range 0.001-1; median r2 0.33; range 0.014-1, respectively), with strongest allelic associations in CSHL1 and in the upstream region of CSH2 (Fig. 3). This landscape of LD in the gene cluster supports the idea that gene conversion is a major force in breaking down allelic associations within duplicated regions.
Figure 3.

The r2-blot LD structure across the hGH/CSH gene cluster for SNPs with MAF > 10% in Estonians (left), Chinese Han (middle), and Mandenkalu (right), with estimation of crossing-over activity (measured by ρbp = 4Nerbp) and potential recombination hotspots (measured by λ = [ρ between a locus pair]/[average ρ for the analyzed region]).
Neutrality tests
In her seminal work, Tomoko Ohta raised the hypothesis that the duplicated primate GH/CSH genes may have evolved rapidly due to positive, directional Darwinian selection rather than relaxation of selective constraint [Ohta, 1993]. The current dataset allowed, for the first time, the exploration of possible selective constraints on hGH/CSH genes, through use of human polymorphism data. We are aware that when using population variation data one has to keep in mind that demographic history may confound inferences of natural selection because both history and selection shape the distribution of genetic variation. However, demographic history is a genome-wide force that is expected to affect patterns of variation at all loci in a genome similarly, whereas natural selection acts upon specific loci.
Neutrality of the hGH/CSH genes was addressed by Tajima's D (DT) [Tajima, 1989] statistics for each gene (Table 1) and FST values between population pairs (Table 5). Positive DT values indicate an excess of intermediate-frequency alleles in a population, being consistent with either balancing selection or a genetic drift in a small population, whereas negative DT values result from an excess of rare SNPs indicative of either recent directional selection or an increase in population size. In hGH/CSH cluster DT values were skewed towards the negative direction for the GH1 gene (DT= −1.26; −0.88 and −0.63, for Estonians (E), Han (H) and Mandenkalu (M), respectively), the CSH1 gene (DT= E −1.02; H −1.86, p=0.003; M −1.75, p=0.018), and the GH2 gene (DT= E −0.06; H −1.02; M −1.87, p=0.018). In contrast, in the CSH2 gene, exhibiting 97% sequence identity with CSH1 (Supplementary Fig. S1), all identified SNPs in Estonian and Han samples had MAF >10% (E, n=5; H, n=6; Table 1), resulting in the significantly positive DT estimates (E 2.62, p=0.004; H 2.77, p=0.003). In Estonians, a population-specific high and significant DT (2.98, p=0.005; other populations DT<0) was also detected for the CSHL1 locus. The non-uniform landscape of DT estimates was consistent with variable heterozygosity patterns described above.
TABLE 5.
Estimates of FST Parameters Between Studied Population Samples for Each Individual hGH/CSH Cluster Gene
| Genea | Estonians / Chinese Han |
Estonians / Mandenkalu |
Chinese Han / Mandenkalu |
|---|---|---|---|
| GH1 | 0.017 (NS) | 0.180 | 0.151 |
| CSHL1 | 0.327 | 0.078 | 0.295 |
| CSH1 | 0.031 (NS) | 0.092 | 0.089 |
| GH2 | 0.829 | 0.410 | 0.908 |
| CSH2 | 0.201 | 0.140 | 0.316 |
| GH1 5′ upstreamb | 0.101 | 0.062 | 0.155 |
Note – P< 10−6 for all FST estimates except denoted with NS.
the region covering genomic sequence from mRNA start to end
the resequenced genomic region 5′ of the GH1 mRNA start site (407 bp)
Despite the joint evolutionary, genomic and functional relatedness of the hGH/CSH genes we detected notable differences among the individual genes in the intercontinental genetic diversification quantified by the FST statistic (Table 5). The most striking result was for GH2: FST estimates range from 0.410 (E/M) to 0.829 (E/H) and 0.908 (H/M). The high FST estimates resulted from the distinct GH2 haplotype distributions for each study sample (Fig. 2; Supplementary Table S4D). More than >90% of the Han individuals carried haplotype (HAP) no. 2 (Fig. 2; Supplementary Table S4D). The Mandenkalu were enriched for the alternative variant (87.5%, GH2-HAP 1) and the Estonians possessed both GH2 variants (GH2-HAP 1, 34%; HAP2 56%). The two major worldwide GH2 variants differ by 1 bp in intron 2 (g.943C>A; Supplementary Table S4D). This high FST among human populations may indicate population-specific directional selection for GH2 variants or alternatively, an accelerated genetic drift. The CSH2 gene with one worldwide (CSH2-HAP 1), one Eurasian (CSH2-HAP4) and three population sample specific common haplotypes (Han, CSH2-HAP5; Mandenkalu CSH2-HAP6 and CSH2-HAP8) (Fig. 2; Supplementary Table S4E) also showed significant genetic structuring among humans (FST E/H 0.201; E/M 0.140; H/M 0.316; p<10−6; Table 5). In contrast to distinct sample-specific allelic distributions for GH2 and CSH2, there was no genetic differentiation for CSH1 (FST <0.1) among the studied populations or for GH1 among Eurasians (E/H; FST <0.02; Table 5). For GH1, the African Mandenkalu exhibited moderate differentiation from the Eurasians (H/M FST=0.151; E/M FST=0.180; p<10−6).
All analyses conducted for the GH1 promoter region have reflected a situation of intensive gene conversion [Giordano et al., 1997; Horan et al., 2003] and relaxed selective constraint, unique to human GH1 compared to other mammals [Krawczak et al., 1999]. The diversity patterns for the CSHL1 pseudogene (Tables 1 and 5; Fig. 2; Supplementary Table S4B) most probably reflect a mixture of random effects from evolutionary (drift), meiotic (gene conversion) and demographic events, both species-wide and population-specific.
DISCUSSION
This study showed that the duplicated hGH/CSH genes involved in growth and glucose metabolism exhibit substantial heterogeneity in their diversity patterns. The resequencing of the individual genes resulted in a high level (>75%) of population sample specific SNPs. There may be three (mutually inclusive) reasons for this. First, it may reflect population histories leading to genetic drift, e.g. by a deep bottleneck followed by demographic expansion. Second, it may indicate relaxed selection pressures on duplicated sequences (discussed below). The low level of shared variation among populations may be caused by the young age of most SNPs and the rapid turnover of the majority of the polymorphisms predating the out-of-Africa expansion of modern humans. Third, it may be an outcome of a continuous extensive gene conversion shuffling the SNP patterns. The last two mechanisms are expected to lead to high diversity. Indeed, the active gene conversion acceptor region in the GH1 promoter has >10 times higher heterozygosity (π=0.00691) compared to the human genome average (π=0.00058 [Stephens et al., 2001a]). In contrast, the GH1 gene is represented by only one worldwide (56% in Estonians, 68% in Chinese Han and 45.8% Mandenkalu) variant and two regional haplotypes enriched in either non-Africans or Africans. Whether this polarized polymorphism pattern within the ~2 kb sequence results from sharp borders for the gene conversion acceptor region or from selective constraints on the GH1 gene region remains to be targeted by further analyses. Previously we have detected significant positive correlation between the gene conversion acceptor activity and heterozygosity level within another primate-specific gene cluster, the hLHB/CGB region [Hallast et al., 2005].
A remarkable feature of the hGH/CSH region is near absence of linkage disequilibrium and no estimated crossing over hotspots. This could be attributed to hyperactive gene conversion, with the rate (γ) exceeding tens to hundreds of times the rate of reciprocal crossing over (ρ; Table 4). The excess of gene conversion compared to crossover events support the idea that the former may also arise through an independent mechanism [Allers and Lichten, 2001]. In a recent study exploring a number of D. melanogaster, D. simulans and Zea mays loci, gene conversion was estimated to contribute roughly twice as much as crossing over to total recombination, but the ratio of the two parameters (ƒ) was as high as 59.6 for individual loci [Morrell et al., 2006]. Similar to the non-uniform rate of crossovers across the genome, there seems to be a large variation in gene conversion activity among different duplicated regions.
In primates, the temporal proximity of the gene duplication events within the GH gene cluster and accelerated rate of sequence changes has led to the hypotheses of either relaxation of functional constraints or positive selection [Ohta, 1993; Wallis, 1994]. Interestingly, the burst of rapid evolution in the primate lineage has promoted independent duplication events of the ancestral primate GH gene in New World Monkeys (only GH-like genes) and Old World Monkeys/hominoids (GH-like genes and emergence of CSH) [Li et al., 2005]. For both of these parallel-emerged gene clusters, there is evidence for variability in evolutionary rates among the gene copies and for different selective constraints acting on different members of the gene family [Li et al., 2005; Ye et al., 2005].
There is evidence from this study that the hGH/CSH gene family continues rapid evolution in the human lineage driven by the relaxation of selective constraints and/or directional selection towards subfunctionalization and/or population-specific diversification. The excess of rare SNPs, the lower than average intercontinental genetic differentiation, and the minimal variation of the CSH1 locus suggests species-wide directional selection. Consistently, a significantly higher evolutionary rate and the excess of nonsynonymous to synonymous substitutions in Old World Monkey and hominoid CSH genes, compared to the GH1 gene, has been reported [Ye et al., 2005]. In contrast, the human GH2 gene shows very strong intercontinental genetic structuring: European compared to Asian chromosomes, FST=0.829 and for African – Asian differentiation FST = 0.908. According to a large-scale study [Akey et al., 2002] (25,530 autosomal SNPs) the mean FST among human populations was estimated 0.123; 6% of SNPs had FST > 0.40 and a small fraction of autosomal SNPs exhibit FST >0.9. Only a few other genes have been reported with FST ≥0.5 (e.g. LCT associated with lactose intolerance and showing directional selection in Europeans [Bersaglieri et al., 2004]) and several of these represent members of duplicated gene families (e.g. loci for Duffy blood group [Hamblin et al., 2002]; ADH1B Arg47His, FST=0.478, East-Asian populations [Han et al., 2007]). Notably, in contrast to the pattern of higher African compared to non-African diversity observed in most studies of human variation, the Mandenkalu possessed only one common GH2 haplotype carried by >85% chromosomes. A similar observation has been published for the African Hausa GCK gene, which is also involved in regulation of glucose metabolism and associated with birth weight [Weedon et al., 2005]. As the variation patterns of the other linked genes within the 48 kb hGH/CSH cluster were strongly dissimilar to GH2, we would reject the scenario of population subdivision by drift and suggest the observed variation pattern of GH2 reflects regional population-specific selection. The third placental expressed protein-coding locus CSH2 differs from both CSH1 and GH2, as it is represented by 2-3 major gene variants in each studied population (each shared by at least two populations), a significant excess of common SNPs in non-African populations and genetic structuring among studied populations. This may indicate either the effect of genetic drift or balancing selection among human populations through interplay with local environmental factors.
Which biological processes regulated by hGH/CSH genes could be potential targets for selection? What do we know about the diverse functions of the placental genes that could explain the evolution under different constraints? Although the activation of GH2 and CSH gene expression in the human placenta occurs coordinately at 6-8 weeks of pregnancy [MacLeod et al., 1992], the expression level and patterns of individual placental genes differ, supporting their diverse functions. Chorionic somatomammotropin (CSH) is secreted into both the maternal and fetal circulations, whereas the placental growth hormone variant (PGH) coded by the GH2 gene is released only into maternal circulation. CSH1 and CSH2 are transcribed equally at 8 weeks of gestation, but during the mid- and late gestation placenta the CSH1 transcripts level rises and exceed CSH2 mRNA level 5-fold by term. Interestingly, although a lower level of CSH in maternal serum has been associated with intrauterine growth retardation (IUGR) [Chard et al., 1985], the complete deficiency of CSH does not necessarily cause a major clinical disorder of growth and development in utero or during infancy [Parks et al., 1985; Simon et al., 1986; Wurzel et al., 1982]. There are no data available, however, on how the reduction in the availability of CSH in utero would affect the metabolism and disease susceptibility in adult life. The two studies on the association of a microsatellite marker upstream CSH1 gene with adult fasting insulin levels have given contradictory results [Day et al., 2004; Freathy et al., 2006]. In contrast to the CSH genes, the complete deletion of GH2 leads to severe intrauterine growth retardation and the levels of PGH are significantly decreased in maternal circulation in pregnancies with IUGR [Alsat et al., 1997; McIntyre et al., 2000]. Birth-weight is a likely target for natural selection. Low birth-weight babies have a high level of neonatal mortality; a too high birth weight may lead to complications during birth, posing a danger to the mother and the child. During pregnancy, PGH suppresses maternal GH1 gene expression in the pituitary and, by the third trimester, becomes the major GH in maternal serum, playing an important role in maternal metabolism during pregnancy (reviewed by [Lacroix et al., 2002]). It appears to regulate the maternal levels of IGF-1, which is an important determinant of glucose and amino acid transport to the fetus [Zumkeller, 2000], and has been suggested as a major mediator of the insulin resistance observed during human pregnancy [Barbour et al., 2004]. As the secretion of PGH is inhibited by glucose in vitro and in vivo (reviewed by [Alsat et al., 1997]), maternal nutrition, food availability and diet most probably affect PGH synthesis. Different GH2 expressional variants may have had a selective advantage to guarantee the optimal birth weight for a given population environment and life-style. The functional impact of the different GH2 haplotypes identified in this study is still to be explored.
Implications
The contribution of genes expressed during intrauterine development to the susceptibility for adult diseases has been under-explored. The human GH/CSH genes, which regulate growth in utero and in childhood and are involved in feto-maternal and adult glucose metabolism, are good candidate targets for association studies of IUGR, gestational diabetes, and adult diseases associated with insulin-resistance. This study, which uncovers the major worldwide hGH/CSH gene variants and explores the roles of regional selection and gene conversion, will pave the way for successful case-control studies to follow. The results imply that resequencing of the study population may be a prerequisite for an association study targeted to duplicated genes in order to determine normal population variation and functionally relevant candidate polymorphisms.
Supplementary Material
ACKNOWLEDGEMENTS
We thank dr. Howard Cann for providing Mandenka and Chinese Han DNA samples from the CEPH Human Genome Diversity Cell Line Panel, Tõnu Margus for bioinformatics help, Bob Zimmermann, Drs. Mait Metspalu, Jaana Männik, Kristiina Rull and Pille Vaas for discussions on the project and comments to the manuscript. M.L. is a HHMI International Scholar (grant #55005617) and Wellcome Trust International Senior Research Fellow (grant no. 070191/Z/03/Z) in Biomedical Science in Central Europe. Additionally, the study has been supported by Estonian Ministry of Education and Science core grant no. 0182721s06 and Estonian Science Foundation grant no. 5796 as well as personal scholarships for L.S. from the Kristjan Jaak Stipend Program and Tartu University Raefond. B. P. was supported by NSF grant DBI-0606461 to Dr. Susan McCouch and Dr. Carlos D.Bustamante and as well as by National Institutes of Health Center for Excellence in Genomic Sciences grant (1P50 HG002790-01A1) to Dr. Paul Marjoram and Dr. Magnus Nordborg.
REFERENCES
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12(12):1805–14. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers T, Lichten M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell. 2001;106(1):47–57. doi: 10.1016/s0092-8674(01)00416-0. [DOI] [PubMed] [Google Scholar]
- Alsat E, Guibourdenche J, Luton D, Frankenne F, Evain-Brion D. Human placental growth hormone. Am J Obstet Gynecol. 1997;177(6):1526–34. doi: 10.1016/s0002-9378(97)70103-0. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297(5583):1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology. 2004;145(3):1144–50. doi: 10.1210/en.2003-1297. [DOI] [PubMed] [Google Scholar]
- Barbour LA, Shao J, Qiao L, Pulawa LK, Jensen DR, Bartke A, Garrity M, Draznin B, Friedman JE. Human placental growth hormone causes severe insulin resistance in transgenic mice. Am J Obstet Gynecol. 2002;186(3):512–7. doi: 10.1067/mob.2002.121256. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. Bmj. 1990;301(6746):259–62. doi: 10.1136/bmj.301.6746.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bennett AJ, Sovio U, Ruokonen A, Martikainen H, Pouta A, Taponen S, Hartikainen AL, King VJ, Elliott P, Järvelin MR, McCarthy MI. Variation at the insulin gene VNTR (variable number tandem repeat) polymorphism and early growth: studies in a large Finnish birth cohort. Diabetes. 2004;53(8):2126–31. doi: 10.2337/diabetes.53.8.2126. [DOI] [PubMed] [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74(6):1111–20. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296(5566):261–2. doi: 10.1126/science.296.5566.261b. and others. [DOI] [PubMed] [Google Scholar]
- Chard T, Sturdee J, Cockrill B, Obiekwe BC. Which is the best placental function test? A comparison of placental lactogen and unconjugated oestriol in the prediction of intrauterine growth retardation. Eur J Obstet Gynecol Reprod Biol. 1985;19(1):13–7. doi: 10.1016/0028-2243(85)90159-5. [DOI] [PubMed] [Google Scholar]
- Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE, Seeburg PH. The human growth hormone locus: nucleotide sequence, biology, and evolution. Genomics. 1989;4(4):479–97. doi: 10.1016/0888-7543(89)90271-1. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet. 2004;36(7):700–6. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- Day IN, Chen XH, Gaunt TR, King TH, Voropanov A, Ye S, Rodriguez S, Syddall HE, Sayer AA, Dennison EM. Late life metabolic syndrome, early growth, and common polymorphism in the growth hormone and placental lactogen gene cluster. J Clin Endocrinol Metab. 2004;89(11):5569–76. doi: 10.1210/jc.2004-0152. and others. [DOI] [PubMed] [Google Scholar]
- Dunger DB, Ong KK, Huxtable SJ, Sherriff A, Woods KA, Ahmed ML, Golding J, Pembrey ME, Ring S, Bennett ST. Association of the INS VNTR with size at birth. ALSPAC Study Team. Avon Longitudinal Study of Pregnancy and Childhood. Nat Genet. 1998;19(1):98–100. doi: 10.1038/ng0598-98. and others. [DOI] [PubMed] [Google Scholar]
- Esteban C, Audi L, Carrascosa A, Fernandez-Cancio M, Perez-Arroyo A, Ulied A, Andaluz P, Arjona R, Albisu M, Clemente M, et al. Human growth hormone (GH1) gene polymorphism map in a normal-statured adult population. Clin Endocrinol (Oxf) 2007;66(2):258–68. doi: 10.1111/j.1365-2265.2006.02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Fleenor D, Oden J, Kelly PA, Mohan S, Alliouachene S, Pende M, Wentz S, Kerr J, Freemark M. Roles of the lactogens and somatogens in perinatal and postnatal metabolism and growth: studies of a novel mouse model combining lactogen resistance and growth hormone deficiency. Endocrinology. 2005;146(1):103–12. doi: 10.1210/en.2004-0744. [DOI] [PubMed] [Google Scholar]
- Frayling TM, Hattersley AT, McCarthy A, Holly J, Mitchell SM, Gloyn AL, Owen K, Davies D, Smith GD, Ben-Shlomo Y. A putative functional polymorphism in the IGF-I gene: association studies with type 2 diabetes, adult height, glucose tolerance, and fetal growth in U.K. populations. Diabetes. 2002;51(7):2313–6. doi: 10.2337/diabetes.51.7.2313. [DOI] [PubMed] [Google Scholar]
- Freathy RM, Mitchell SM, Knight B, Shields B, Weedon MN, Hattersley AT, Frayling TM. A study of association between common variation in the growth hormone-chorionic somatomammotropin hormone gene cluster and adult fasting insulin in a UK Caucasian population. J Negat Results Biomed. 2006;5:18. doi: 10.1186/1477-5751-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freathy RM, Weedon MN, Bennett A, Hypponen E, Relton CL, Knight B, Shields B, Parnell KS, Groves CJ, Ring SM, Pembrey ME, Ben-Shlomo Y, Strachan DP, Power C, Jarvelin MR, McCarthy MI, Davey Smith G, Hattersley AT, Frayling TM. Type 2 diabetes TCF7L2 risk genotypes alter birth weight: a study of 24,053 individuals. Am J Hum Genet. 2007;80(6):1150–61. doi: 10.1086/518517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano M, Marchetti C, Chiorboli E, Bona G, Momigliano Richiardi P. Evidence for gene conversion in the generation of extensive polymorphism in the promoter of the growth hormone gene. Hum Genet. 1997;100(2):249–55. doi: 10.1007/s004390050500. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. Bmj. 1991;303(6809):1019–22. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallast P, Nagirnaja L, Margus T, Laan M. Segmental duplications and gene conversion: Human luteinizing hormone/chorionic gonadotropin beta gene cluster. Genome Res. 2005;15(11):1535–46. doi: 10.1101/gr.4270505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet. 2002;70(2):369–83. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Gu S, Oota H, Osier MV, Pakstis AJ, Speed WC, Kidd JR, Kidd KK. Evidence of positive selection on a class I ADH locus. Am J Hum Genet. 2007;80(3):441–56. doi: 10.1086/512485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handwerger S, Freemark M. The roles of placental growth hormone and placental lactogen in the regulation of human fetal growth and development. J Pediatr Endocrinol Metab. 2000;13(4):343–56. doi: 10.1515/jpem.2000.13.4.343. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Fujii K, Yamada M, Igarashi Y, Tachibana K, Tanaka T, Onigata K, Nishi Y, Kato S, Hasegawa T. Identification of novel human GH-1 gene polymorphisms that are associated with growth hormone secretion and height. J Clin Endocrinol Metab. 2000;85(3):1290–5. doi: 10.1210/jcem.85.3.6468. [DOI] [PubMed] [Google Scholar]
- Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet. 1999;353(9166):1789–92. doi: 10.1016/S0140-6736(98)07546-1. [DOI] [PubMed] [Google Scholar]
- Horan M, Millar DS, Hedderich J, Lewis G, Newsway V, Mo N, Fryklund L, Procter AM, Krawczak M, Cooper DN. Human growth hormone 1 (GH1) gene expression: complex haplotype-dependent influence of polymorphic variation in the proximal promoter and locus control region. Hum Mutat. 2003;21(4):408–23. doi: 10.1002/humu.10167. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159(4):1805–17. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, May CA. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat Genet. 2004;36(2):151–6. doi: 10.1038/ng1287. [DOI] [PubMed] [Google Scholar]
- Jorgensen JO, Krag M, Jessen N, Norrelund H, Vestergaard ET, Moller N, Christiansen JS. Growth hormone and glucose homeostasis. Horm Res. 2004;62(Suppl 3):51–5. doi: 10.1159/000080499. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Chuzhanova NA, Cooper DN. Evolution of the proximal promoter region of the mammalian growth hormone gene. Gene. 1999;237(1):143–51. doi: 10.1016/s0378-1119(99)00313-3. [DOI] [PubMed] [Google Scholar]
- Lacroix MC, Guibourdenche J, Frendo JL, Muller F, Evain-Brion D. Human placental growth hormone--a review. Placenta. 2002;23(Suppl A):S87–94. doi: 10.1053/plac.2002.0811. [DOI] [PubMed] [Google Scholar]
- Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165(4):2213–33. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ye C, Shi P, Zou XJ, Xiao R, Gong YY, Zhang YP. Independent origin of the growth hormone gene family in New World monkeys and Old World monkeys/hominoids. J Mol Endocrinol. 2005;35(2):399–409. doi: 10.1677/jme.1.01778. [DOI] [PubMed] [Google Scholar]
- MacLeod JN, Lee AK, Liebhaber SA, Cooke NE. Developmental control and alternative splicing of the placentally expressed transcripts from the human growth hormone gene cluster. J Biol Chem. 1992;267(20):14219–26. [PubMed] [Google Scholar]
- McIntyre HD, Serek R, Crane DI, Veveris-Lowe T, Parry A, Johnson S, Leung KC, Ho KK, Bougoussa M, Hennen G. Placental growth hormone (GH), GH-binding protein, and insulin-like growth factor axis in normal, growth-retarded, and diabetic pregnancies: correlations with fetal growth. J Clin Endocrinol Metab. 2000;85(3):1143–50. doi: 10.1210/jcem.85.3.6480. and others. [DOI] [PubMed] [Google Scholar]
- Millar DS, Lewis MD, Horan M, Newsway V, Easter TE, Gregory JW, Fryklund L, Norin M, Crowne EC, Davies SJ. Novel mutations of the growth hormone 1 (GH1) gene disclosed by modulation of the clinical selection criteria for individuals with short stature. Hum Mutat. 2003;21(4):424–40. doi: 10.1002/humu.10168. and others. [DOI] [PubMed] [Google Scholar]
- Morrell PL, Toleno DM, Lundy KE, Clegg MT. Estimating the contribution of mutation, recombination and gene conversion in the generation of haplotypic diversity. Genetics. 2006;173(3):1705–23. doi: 10.1534/genetics.105.054502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JC, Lohmussaar E, Magi R, Remm M, Bettecken T, Lichtner P, Biskup S, Illig T, Pfeufer A, Luedemann J. Linkage disequilibrium patterns and tagSNP transferability among European populations. Am J Hum Genet. 2005;76(3):387–98. doi: 10.1086/427925. and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310(5746):321–4. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Newsome CA, Shiell AW, Fall CH, Phillips DI, Shier R, Law CM. Is birth weight related to later glucose and insulin metabolism?--A systematic review. Diabet Med. 2003;20(5):339–48. doi: 10.1046/j.1464-5491.2003.00871.x. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25(14):2745–51. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Pattern of nucleotide substitutions in growth hormone-prolactin gene family: a paradigm for evolution by gene duplication. Genetics. 1993;134(4):1271–6. doi: 10.1093/genetics/134.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhukasahasram B, Wall JD, Marjoram P, Nordborg M. Estimating recombination rates from single-nucleotide polymorphisms using summary statistics. Genetics. 2006;174(3):1517–28. doi: 10.1534/genetics.106.060723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks JS, Nielsen PV, Sexton LA, Jorgensen EH. An effect of gene dosage on production of human chorionic somatomammotropin. J Clin Endocrinol Metab. 1985;60(5):994–7. doi: 10.1210/jcem-60-5-994. [DOI] [PubMed] [Google Scholar]
- Procter AM, Phillips JA, 3rd, Cooper DN. The molecular genetics of growth hormone deficiency. Hum Genet. 1998;103(3):255–72. doi: 10.1007/s004390050815. [DOI] [PubMed] [Google Scholar]
- Rosa A, Ornelas C, Jobling MA, Brehm A, Villems R. Y-chromosomal diversity in the population of Guinea-Bissau: a multiethnic perspective. BMC Evol Biol. 2007;7(1):124. doi: 10.1186/1471-2148-7-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld RG. Insulin-like growth factors and the basis of growth. N Engl J Med. 2003;349(23):2184–6. doi: 10.1056/NEJMp038156. [DOI] [PubMed] [Google Scholar]
- Rousset F, Raymond M. Testing heterozygote excess and deficiency. Genetics. 1995;140(4):1413–9. doi: 10.1093/genetics/140.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6(5):526–38. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- Simon P, Decoster C, Brocas H, Schwers J, Vassart G. Absence of human chorionic somatomammotropin during pregnancy associated with two types of gene deletion. Hum Genet. 1986;74(3):235–8. doi: 10.1007/BF00282540. [DOI] [PubMed] [Google Scholar]
- Smith GR. Homologous recombination in prokaryotes. Microbiol Rev. 1988;52(1):1–28. doi: 10.1128/mr.52.1.1-28.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH, et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001a;293(5529):489–93. doi: 10.1126/science.1059431. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001b;68(4):978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker CJ, Arch JR, Cawthorne MA. Fetal origins of insulin resistance and obesity. Proc Nutr Soc. 2005;64(2):143–51. doi: 10.1079/pns2005417. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser G, Hermann M, Rumpold H, Pfister G, Berger P. An unusual member of the human growth hormone/placental lactogen (GH/PL) family, the testicular alternative splicing variant hPL-A2: recombinant expression revealed a membrane-associated growth factor molecule. Mol Cell Endocrinol. 2000;167(1-2):117–25. doi: 10.1016/s0303-7207(00)00287-2. [DOI] [PubMed] [Google Scholar]
- Vaessen N, Janssen JA, Heutink P, Hofman A, Lamberts SW, Oostra BA, Pols HA, van Duijn CM. Association between genetic variation in the gene for insulin-like growth factor-I and low birthweight. Lancet. 2002;359(9311):1036–7. doi: 10.1016/s0140-6736(02)08067-4. [DOI] [PubMed] [Google Scholar]
- Wallis M. Variable evolutionary rates in the molecular evolution of mammalian growth hormones. J Mol Evol. 1994;38(6):619–27. doi: 10.1007/BF00175882. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Frayling TM, Shields B, Knight B, Turner T, Metcalf BS, Voss L, Wilkin TJ, McCarthy A, Ben-Shlomo Y. Genetic regulation of birth weight and fasting glucose by a common polymorphism in the islet cell promoter of the glucokinase gene. Diabetes. 2005;54(2):576–81. doi: 10.2337/diabetes.54.2.576. and others. [DOI] [PubMed] [Google Scholar]
- Wurzel JM, Parks JS, Herd JE, Nielsen PV. A gene deletion is responsible for absence of human chorionic somatomammotropin. DNA. 1982;1(3):251–7. doi: 10.1089/dna.1.1982.1.251. [DOI] [PubMed] [Google Scholar]
- Ye C, Li Y, Shi P, Zhang YP. Molecular evolution of growth hormone gene family in old world monkeys and hominoids. Gene. 2005;350(2):183–92. doi: 10.1016/j.gene.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Zumkeller W. Current topic: the role of growth hormone and insulin-like growth factors for placental growth and development. Placenta. 2000;21(5-6):451–67. doi: 10.1053/plac.2000.0505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.