

Abstract
We report the synthesis of a new class of recombinant elastin-mimetic triblock copolymer capable of both physical and chemical crosslinking. These investigations were motivated by a desire to capture features unique to both physical and chemical crosslinking schemes so as to exert optimal control over a wide range of potential properties afforded by protein-based mutiblock materials. We postulated that by chemically locking a multiblock protein assembly in place, functional responses that are linked to specific domain structures and morphologies may be preserved over a broader range of loading conditions that would otherwise disrupt microphase structure solely stabilized by physical crosslinking. Specifically, elastic modulus was enhanced and creep strain reduced through the addition of chemical crosslinking sites. Additionally, we have demonstrated excellent in vivo biocompatibility of glutaraldehyde treated multiblock systems.
Keywords: Recombinant elastin-mimetic, protein polymer, mechanical testing, in vivo biocompatibility
Introduction
Genetic engineering provides a facile route for the design of novel protein polymers composed of repetitive amino acid sequences or peptide blocks whose structural complexity imparts distinct mechanical, chemical or biological properties. To date, the majority of recombinant multiblock protein polymers have been designed with relatively short block sequences that limit structural polymorphism. As a consequence, opportunities to access diverse polymer morphologies are limited and the potential to tune a wide range of functional responses reduced [1, 2]. Recently, we have reported a new class of elastin-mimetic multiblock copolymer composed of identical endblocks derived from self-associating, hydrophobic sequences that display plastic-like mechanical responses (Ile-Pro-Ala-Val-Gly), separated by a central block that is both hydrophilic and elastomeric (Val-Pro-Gly-Glu-Gly) [3, 4]. Block sizes, typically, exceed 35 kDa, which has allowed us to explore the production of protein-based materials that are structurally polymorphic [3-8].
Significantly, multiblock systems afford the ability to form physical or non-covalent crosslinked networks through the self-association of chemically similar domains. In the case of elastin-mimetic proteins [3-8], repeat peptide sequences of self-associating blocks are chosen such that coacervation or phase separation of these domains occurs in water under physiologically relevant conditions (pH 7.4, 37°C), which maximizes hydrophobic interactions that drive self-assembly. In turn, the sequence of the non-crosslinking domain is selected in a manner that precludes coacervation. This typically has required the incorporation of hydrophilic residues in the fourth position of the pentapeptide repeat sequence (Val-Pro-Gly-Xaa-Gly), such as glutamic acid, which limits the tendency for block aggregation. Physically crosslinked protein-based materials possess a number of advantages over their chemically crosslinked counterparts, including ease of processability, the ability to avoid the addition or removal of reagents or unreacted intermediates needed for chemical crosslinking, and the capacity to incorporate biologically or chemically active agents or cells that might otherwise be sensitive to covalent crosslinking schemes. Moreover, if blocks are of sufficient size and chemical diversity the potential to access diverse polymer morphologies exists. This provides the capacity to tune a wide range of functional responses, such as mechanical behavior, permeability or drug elution characteristics, as well as the potential to design templated materials [1, 9, 10]. Notwithstanding these desirable features, physical crosslinks and the related domains so formed may be deformed or damaged at applied stresses lower than those required to disrupt covalent crosslinks.
Native elastin is enzymatically crosslinked upon proper alignment of two pairs of lysine residues between adjacent tropoelastin chains with formation of desmosine or isodesmosine linkages [11, 12]. Likewise, most recombinant elastin analogues that have been designed to date have relied on crosslinking through available amino groups, albeit with most reports describing the use of chemical crosslinkers, including isocyanates, NHS-esters, phosphines, aldehydes, or genipin [9, 13-23]. In this regard, we have previously reported the design of a synthetic elastin sequence, (Val-Pro-Gly-Val-Gly)4(Val-Pro-Gly-Lys-Gly), in which lysine residues were chemically crosslinked using bis(sulfosuccinimidyl) suberate and disuccinimidyl suberate [20]. Subsequent studies have reported the application of transglutaminase or lysyl oxidase for enzymatic crosslinking [24]. In addition, we have also explored solid-state crosslinking of recombinant elastin-mimetic proteins using both UV and visible light activated photoinitiators [25]. In tropoelastin, lysine residues are often interspersed among alanine repeats (eg. Ala-Ala-Ala-Lys-Ala-Ala-Lys-Ala-Ala), which has suggested that self-association of alanine-rich sequences facilitates crosslinking [26, 27]. Several elastin-like proteins have been designed in similar manner [9, 10, 28].
The capacity of chemical crosslinks to provide an independent mechanism for control of protein mechanical responses and biostability is well established. However, in this report we postulated that by chemically locking a multiblock protein assembly in place, functional responses that are linked to specific domain structures and morphologies may be preserved over a broader range of loading conditions that would otherwise disrupt microphase structure solely stabilized by physical crosslinking. We report herein the synthesis of a new class of recombinant elastin-mimetic triblock copolymer capable of both physical and chemical crosslinking. These investigations were motivated by a desire to capture features unique to both physical and chemical crosslinking schemes so as to exert optimal control over a wide range of potential properties afforded by protein-based mutiblock materials.
Materials and Methods
Synthetic gene construction of elastic- and plastic-like domains
Synthetic methods used to produce the DNA inserts that encode the various elastin-mimetic block copolymers have been described previously [3, 5, 7, 8]. Genes encoding two distinct chemically crosslinkable protein triblock copolymers were synthesized. Briefly, oligonucleotide cassettes encoding elastic- (E) and plastic-like (P) repeat units (Table 1) were independently synthesized and inserted into the BamH I and HinD III sites within the multiple cloning region of pZErO cloning vectors. Specifically, P1 and E1 encode the monomer repeat unit for plastic- and elastic-like domains designated for the triblock protein polymer, referred to as LysB10. A second set of oligonucleotide cassettes, P2 and E2, were designed to encode monomer repeat units for plastic- and elastic-like domains for a second protein triblock copolymer, designated R4. Recombinant clones were isolated after propagation in E. coli strain TOP10F′ and the identity of the DNA inserts E and P verified by double-stranded DNA sequence analysis. DNA monomers E and P were liberated from the respective plasmids via sequential restriction digestion with Bbs I and BsmB I, respectively. Multimerization, or self-ligation in a head-to-tail fashion of each DNA cassette afforded a population of multimers. This procedure was repeated separately for the four multimers synthesized in this report (P1, E1, P2, and E2).
Table 1.
Coding Sequences of Oligonucleotide Cassettes Employed for the Construction of Crosslinkable Protein Triblocks, LysB10 (AP1IE1IP1A) and R4 (AP2IE2IP2A)
| E1 Block (LysB10 Elastic-like Block) | ||||||||||||||
| Val | Pro | Gly | Ala | Gly | Val | Pro | Gly | Ala | Gly | Val | Pro | Gly | Glu | Gly |
| GTT | CCA | GGT | GCA | GGC | GTA | CCG | GGT | GCT | GGC | GTT | CCG | GGT | GAA | GGT |
| Val | Pro | Gly | Ala | Gly | Val | Pro | Gly | Ala | Gly | |||||
| GTT | CCA | GGC | GCA | GGT | GTA | CCG | GGT | GCG | GGT | |||||
| E2 Block (R4 Elastic-like Block) | ||||||||||||||
| Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly |
| GTT | CCA | GGT | ATT | GGC | GTT | CCG | GGT | ATC | GGT | GTG | CCA | GGC | ATC | GGT |
| Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly | |||||
| GTA | CCG | GGT | ATT | GGC | GTT | CCA | GGC | ATT | GGC | |||||
|
| ||||||||||||||
| P1 Block (LysB10 Plastic-like Block) | ||||||||||||||
| Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly |
| ATT | CCG | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGC |
| Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | |||||
| ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC | |||||
| P2 Block (R4 Plastic-like Block) | ||||||||||||||
| Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly |
| ATT | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGC |
| Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | |||||
| ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC | |||||
|
| ||||||||||||||
| I Block (Lysine Insert) | ||||||||||||||
| Ile | Pro | Ala | Val | Gly | Lys | Ala | Ala | Lys | Val | Pro | Gly | Ala | Gly | |
| ATT | CCA | GCT | GTT | GGT | AAG | GCG | GCC | AAG | GTT | CCA | GGT | GCA | GGC | |
|
| ||||||||||||||
| A Block (Modified Lysine Adaptor) | ||||||||||||||
| Val | Pro | Ala | Val | Gly | Lys | Val | Pro | Ala | ...... | Ile | Pro | Ala | Val | |
| GTT | CCA | GCT | GTT | GGT | AAG | GTT | CCA | GCT | ...... | ATT | CCA | GCT | GTT | |
| Gly | Lys | Ala | Ala | Lys | Ala | Stop | ||||||||
| GGT | AAG | GCG | GCC | AAG | GCG | TAA | ||||||||
Multimers derived from DNA monomers were inserted into the BsmB I site of their original plasmid containing the monomer cassette. Multimers encoding 33 repeats of the P1 monomer, 16 repeats of the P2 monomer, 28 repeats of the E1 monomer, and 15 repeats of the E2 monomer were isolated and identified via restriction cleavage with BamH I and HinD III. Double-stranded DNA sequence analysis confirmed the integrity of the concatemers within the recombinant plasmids, which were labeled pP1, pP2, pE1, and pE2, respectively. pP1 and pE1 were utilized in the construction of the LysB10 gene, pP2 and pE2 for the R4 gene.
Synthetic gene construction of chemical crosslinking domains
Single stranded oligonucleotides encoding the sense and anti-sense strands of the Lysine Insert (I) and Lysine Adaptor (A) were chemically synthesized (Sigma Genosys, Inc.) (Table 1). The Lysine Insert is a 60 bp DNA cassette encoding the crosslinking sequence, Lys-Ala-Ala-Lys, which was inserted between the plastic- and elastic-like domains. The Lysine Adaptor is a 50 bp DNA cassette designed with restriction enzyme cut sites midway through the cassette to allow for insertion of the assembled triblock gene. The Lysine Adaptor encodes for a single N-terminal lysine residue and two C-terminal lysine residues. Additionally, it allows for facile cloning into the pET24-a expression vector within the multiple cloning region. This ensures correct insertion of the gene in frame with the N-terminal polyhistidine tag.
The following procedure detailing the protocol to generate double stranded DNA was implemented for both the Lysine Insert and Lysine Adaptor. The DNA was suspended in 10 mM Tris buffer (pH 8) to a final concentration of 0.5 μg/uL. A solution of 10 μg of each corresponding oligonucleotide, 4 μL 5M NaCl, 4 μL 1M MgCl2, and 152 μL of sterile ddH2O was subjected to an annealing procedure initiated at a reaction temperature of 99°C with temperature decrements of 1°C every 5 minutes to a final reaction temperature of 30°C. The resultant double stranded DNA cassette was analyzed by agarose gel electrophoresis (4% GTG NuSieve agarose, 1X TBE buffer).
Double stranded synthetic DNA was phosphorylated through a 2-hour incubation with T4 Poynucleotide Kinase (New England Biolabs) in the presence of T4 DNA ligase buffer with 10 mM ATPs (New England Biolabs). The enzymes were removed with phenol/chloroform/isoamyl alcohol (25:24:1) and the double stranded DNA (dsDNA) was recovered through an ethanol precipitation.
The pZErO-1 acceptor plasmid (1 μg), was prepared via BamH I and HinD III double digestion, followed by heat inactivation of the enzymes at 65°C and a dilution of the digested plasmid to 10 ng/μL. The Lysine Insert and Lysine Adaptor were designed with BamH I and HinD III overhangs to enable cloning into pZErO-1 at these restriction sites.
The DNA cassette and respective acceptor plasmid were ligated together in the presence of T4 DNA ligase at 16°C for 30 minutes. A 2 μL aliquot of the ligation reaction mixture was used to transform 40 μL of electrocompetent TOP10F′ E. coli cells. A total of 100 μL of the transformation mixture was spread onto low salt Luria Broth (LSLB) agar supplemented with Zeocin (50 μg/μL). The plates were incubated for 12 hours at 37°C. Five transformants were selected from each plate to inoculate individual 7 mL cultures of LSLB/Zeocin. Cultures were rotary incubated for 12 hours at 37°C. Plasmid DNA was isolated following a Qiagen Spin Miniprep protocol (Qiagen, Inc.). DNA was initially screened by a BamH I and HinD III double digestion. Positive transformants were verified by agarose gel electrophoresis (4% GTG NuSieve agarose, 1X TBE buffer). Automated DNA sequencing utilizing the M13 forward and M13 reverse primers confirmed correct DNA products. Plasmids containing the correct sequence for the Lysine Insert and Lysine Adaptor are identified as pI and pA, respectively.
Assembly of elastin-mimetic triblock copolymers
The proteins, LysB10 and R4 were designed to contain the Lysine Insert between each plastic-like and elastic-like block and to be flanked by the Lysine Adaptor (-Lysine Adaptor-Plastic-like Domain-Lysine Insert-Elastic-like Domain-Lysine Insert-Plastic-like Domain-Lysine Adaptor-). All subcloning steps were performed in the pZErO-1 plasmid using LSLB media under Zeocin antibiotic selection.
Recombinant plasmids encoding the elastic-like (pE) (E1=2.1 kB, E2=1.1 kB) and plastic-like (pP) (P2=2.5 kB, P2=1.2 kB) domains were constructed, as described above. Each gene was isolated from its respective plasmid with Bbs I and BsmB I sequential digestion. The gene fragment was isolated via preparative gel electrophoresis (1% agarose, 0.5X TBE) and purified using Zymoclean Gel Recovery (ZymoResearch, Inc). Preparative amounts of pI DNA were isolated for two separate reactions. A total of 5 μg of pI was digested with restriction enzyme Bbs I and Shrimp Alkaline Phosphatase (SAP) dephosphorylated (1 U SAP per 1 pmol strand ends) to prevent re-ligation. A separate 5 μg of pI was digested with the restriction enzyme BsmB I and SAP dephosphorylated. The linearized plasmids were isolated via preparative gel electrophoresis (1% agarose, 0.5X TBE) and purified using Zymoclean Gel Recovery.
Similar protocols for ligation, transformation, and propagation were followed, as previously described. Two separate ligation reactions were performed between Bbs I digested pI and P and BsmB I digested pI and P. Isolated DNA from clones were screened by a BamH I and HinD III double digestion and cleavage fragments analyzed by agarose gel electrophoresis. Correct ligation was confirmed by automated DNA sequence analysis using M13 forward and reverse primers. Plasmids containing the correct sequences are identified as pIP and pPI.
Analogously, 5 μg of pPI was digested with restriction enzyme BsmB I and Shrimp Alkaline Phosphatase (SAP) dephosphorylated (1U SAP per 1 pmol strand ends) to prevent relegation. The linearized plasmid was isolated via preparative gel electrophoresis (1% agarose, 0.5X TBE) and purified using Zymoclean Gel Recovery. Linearized pPI was ligated with E, followed by transformation and propagation. Isolated DNA from transformants was screened by a BamH I and HinD III double digestion and cleavage fragments analyzed by agarose gel electrophoresis. Correct ligation was confirmed by automated DNA sequence analysis using M13 forward and reverse primers. The plasmid containing the correct sequence was termed pPIE.
Recombinant plasmids pIP and pPIE were digested with Bbs I / Xma I and BsmB I / Xma I, respectively. The gene fragment from each of these digestions was isolated via preparative gel electrophoresis (1% agarose, 0.5X TBE) and purified using Zymoclean Gel Recovery. IP and PIE fragments were ligated by T4 DNA ligase, transformed into TOP10F′ and plated on LSLB/Zeocin plates. As the Xma I site cuts within the Zeocin coding region, only clones containing the correctly assembled triblock (PIEIP), and thus, the correctly reassembled antibiotic coding region, were able to propagate. Transformants were confirmed by analysis of BamH I and HinD III restriction digest fragments with agarose gel electrophoresis (1% agarose, 0.5X TBE) and automated DNA sequence analysis using M13 forward and reverse primers. The plasmid containing the correct sequence for the triblock was termed, pPIEIP.
pA, containing the Lysine Adaptor, was assembled, as described above. This plasmid was digested with restriction enzyme BsmB I and SAP dephosphorylated. The triblock, PIEIP, was excised from the pZErO-1 plasmid via sequential digestion using restriction enzymes Bbs I and BsmB I and purified via gel isolation. A ligation reaction was performed to relocate the PIEIP gene from pPIEIP to pA. The ligation mixture was transformed into competent TOP10F′ cells and plated on LSLB media under Zeocin antibiotic selection. Isolated DNA from transformants was screened via agarose gel electrophoresis analysis of a BamH I and HinD III double digestion. Automated DNA sequence analysis using M13 forward and reverse primers confirmed correct insertion of the gene pAP1IE1IP1A and pAP2IE2IP2A. pAP1IE1IP1A was identified as the cloning plasmid, pLysB10 and pAP2IE2IP2A as the cloning plasmid, pR4.
The pET24-a plasmid (1 μg, Invitrogen) was prepared via BamH I and HinD III double digestion, followed by gel isolation and purification. The APIEIPA gene was released from the pZErO-1 vector at analogous sites. Adaptor and plasmid were ligated together in the presence of T4 DNA ligase at 16°C for 30 minutes. A 2 μL aliguot of the ligation reaction mixture was used to transform 40 μL of electrocompetent TOP10F′ E. coli cells. A 100 μL aliquot of the transformation mixture was spread onto LB agar supplemented with kanamycin (50 μg/μL). The plates were incubated for 12 hours at 37°C. Five transformants were selected from each plate to inoculate individual 7 mL cultures of LB/kanamyacin media. Cultures were rotary incubated for 12 hours at 37°C. Plasmid DNA was isolated following a Qiagen Spin Miniprep protocol (Qiagen, Inc.). DNA was screened by a BamH I and HinD III double digestion. Positive transformants were verified by agarose gel electrophoresis (4% GTG NuSieve agarose, 1X TBE buffer). Automated DNA sequencing utilizing the T7 promoter and T7 terminator primers confirmed the correct DNA product. The resultant plasmid was identified as expression plasmids, pLysB10 (AP1IE1IP1A) and pR4 (AP2IE2IP2A). DNA agarose gels in Figures 1 and 2 depict gene products at each subcloning step in the assembly of pLysB10 and pR4, respectively.
Figure 1.
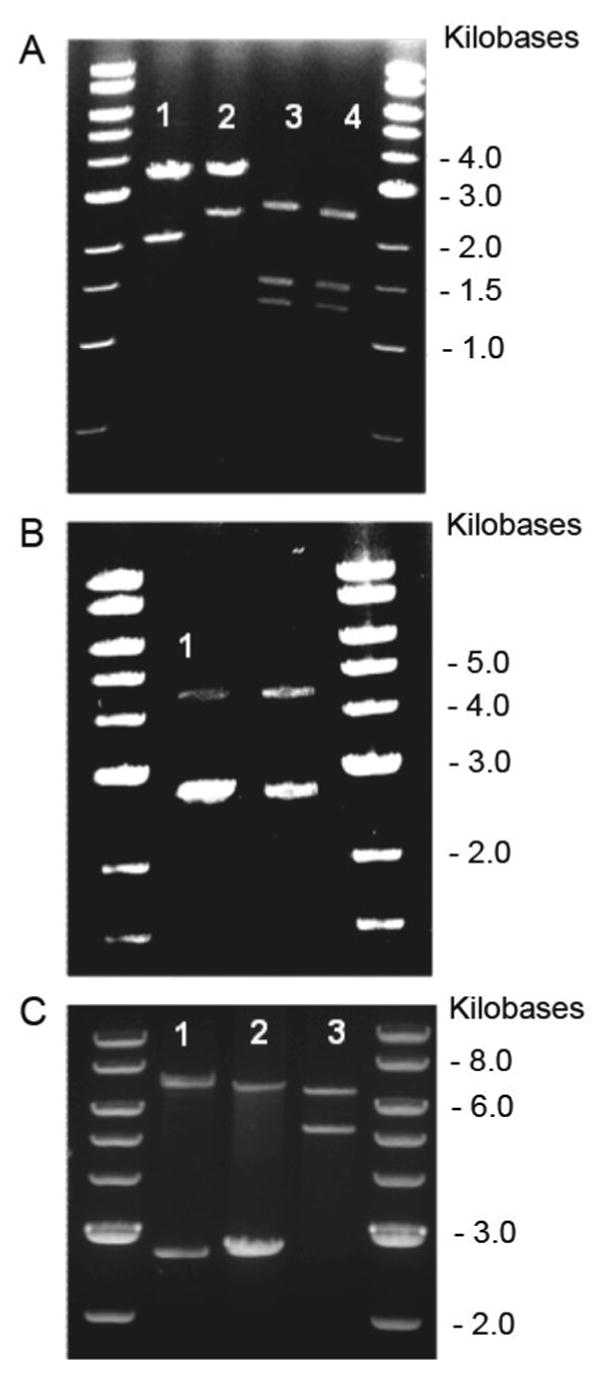
Analytical restriction digests, 1% TAE (Tris-acetate-EDTA) agarose gel, depicting gene and vector sizes at each stage of the LysB10 assembly process with corresponding digestion schemes. DNA standard used was a 1Kb DNA ladder (NEB). A. Lane 1: BamHI / HinDIII digest pE1 (2.1 Kb), pZerO-2 (3.3 Kb). Lane 2: BamHI / HinDIII digest pP1 (2.5 Kb), pZerO-2 (3.3Kb). Lane 3: Nsi I / Xma I digest pP1I (2.56 Kb), pZerO-1 (1.3, 1.5 Kb). Lane 4: Nsi I / Xma I digest pIP1 (2.56 Kb), pZerO-1 (1.3, 1.5 Kb). B. Lane 1: Nsi I digest pP1IE1 (4.66 Kb), pZErO-1 (2.8 Kb). C. Lane 1: Nsi I digest pP1IE1IP1 (7.22 Kb), pZErO-1 (2.8 Kb). Lane 2: Nsi I digest LysB10 (7.28 Kb), pZErO-1 (2.8 Kb). Lane 3: BamHI / HinDIII digest LysB10 (7.28 Kb), pET 24a (5.3 Kb).
Figure 2.
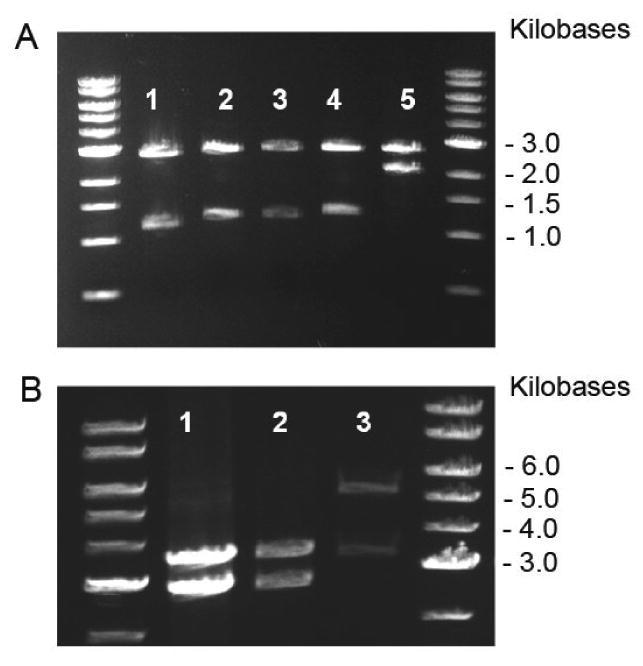
Analytical restriction digests, 1% TAE (Tris-acetate-EDTA) agarose gel, depicting gene and vector sizes at each stage of the R4 assembly process with corresponding digestion schemes. DNA standard used was a 1Kb DNA ladder (NEB). A. Lane 1: Nsi I digest pE2 (1.1 Kb), pZerO-1 (2.8 Kb). Lane 2: Nsi I digest pP2 (1.2 Kb), pZerO-1 (2.8Kb). Lane 3: Nsi I digest pE2I (1.16 Kb), pZerO-1 (2.8 Kb). Lane 4: Nsi I digest pP2I (1.26 Kb), pZerO-1 (2.8 Kb). Lane 5: Nsi I digest pP2IE2I (2.42 Kb), pZerO-1 (2.8 Kb). B. Lane 1: Nsi I digest pP2IE2IP2 (3.62 Kb), pZerO-1 (2.8 Kb). Lane 2: Nsi I digest pR4 (3.7 Kb), pZerO-1 (2.8Kb). Lane 3: BamH I / HinD III digest pR4 (3.7 Kb), pET24-a (5.3 Kb).
Isolation and purification of protein triblock copolymers
The plasmid, pLysB10, encoding the protein LysB10 as a single contiguous reading frame within plasmid pET24-a was used to transform the E. coli expression strain BL21(DE3). This afforded a protein triblock of sequence (Table 2):
Table 2.
Amino acid sequence of LysB10 and related nucleic acid coding sequence [VPAVGKVPAVG(IPAVG)4][(IPAVG)5]33 [IPAVGKAAKVPGAG][(VPGAG)2VPGEG(VPGAG)2]28 [VPAVGKAAKVPGAGVPAVG(IPAVG)4][(IPAVG)5]33[IPAVGKAAKA]
| Val | Pro | Ala | Val | Gly | Lys | Val | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val |
| GTT | CCA | GCT | GTT | GGT | AAG | GTT | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT |
| Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val |
| GGT | ATC | CCA | GCT | GTT | GGC | ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG |
| Gly | [Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val |
| GGC | ATT | CCG | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT |
| Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly]33 | Ile | Pro | Ala | Val |
| GGC | ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC]33 | ATT | CCA | GCT | GTT |
| Gly | Lys | Ala | Ala | Lys | Val | Pro | Gly | Ala | Gly | [Val | Pro | Gly | Ala | Gly |
| GGT | AAG | GCG | GCC | AAG | GTT | CCA | GGT | GCA | GGC | GTT | CCA | GGT | GCA | GGC |
| Val | Pro | Gly | Ala | Gly | Val | Pro | Gly | Glu | Gly | Val | Pro | Gly | Ala | Gly |
| GTA | CCG | GGT | GCT | GGC | GTT | CCG | GGT | GAA | GGT | GTT | CCA | GGC | GCA | GGT |
| Val | Pro | Gly | Ala | Gly]28 | Val | Pro | Ala | Val | Gly | Lys | Ala | Ala | Lys | Val |
| GTA | CCG | GGT | GCG | GGT]28 | GTT | CCA | GCT | GTT | GGT | AAG | GCG | GCC | AAG | GTT |
| Pro | Gly | Ala | Gly | Val | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile |
| CCA | GGT | GCA | GGC | GTT | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC |
| Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | [Ile |
| CCA | GCT | GTT | GGC | ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC | ATT |
| Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile |
| CCG | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGC | ATT |
| Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly]33 | Ile | Pro | Ala | Val | Gly | Lys |
| CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC]33 | ATT | CCA | GCT | GTT | GGT | AAG |
| Ala | Ala | Lys | Ala | Stop | ||||||||||
| GCG | GCC | AAG | GCG | TAA |
N-terminal endblock: VPAVGK[(VPAVG)(IPAVG)4][(IPAVG) 5]33
Midblock: (IPAVG)KAAK(VPGAG)[(VPGAG)2VPGEG(VPGAG)2]28(VPAVG)KAAK(VPGAG)
C-terminal endblock: [(VPAVG)(IPAVG)4][(IPAVG)5]33IPAVGKAAKA
The plasmid, pR4, encoding the protein R4 as a single contiguous reading frame within plasmid pET24-a was used to transform the E. coli expression strain BL21(DE3). This afforded a protein triblock of sequence (Table 3):
Table 3.
Amino acid sequence of R4 and related nucleic acid coding sequence [VPAVGKVPAVG[(IPAVG)5]16 (IPAVGIPAVG)KAAK(VPGAGVPGIG) [(VPGIG)5]15 (VPGIGVPAVG)KAAK(VPGAGVPAVG) [(IPAVG)5]16 IPAVGVPAVGKAAKA]
| Val | Pro | Ala | Val | Gly | Lys | Val | Pro | Ala | Val | Gly | [Ile | Pro | Ala | Val |
| GTT | CCA | GCT | GTT | GGT | AAG | GTT | CCA | GCT | GTT | GGT | [ATT | CCG | GCT | GTT |
| Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val |
| GGT | ATC | CCA | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGC | ATT | CCG | GCT | GTA |
| Gly | Ile | Pro | Ala | Val | Gly]16 | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val |
| GGT | ATC | CCG | GCA | GTG | GGC]16 | ATT | CCG | GCT | GTT | GGT | ATT | CCA | GCT | GTT |
| Gly | Lys | Ala | Ala | Lys | Val | Pro | Gly | Ala | Gly | Val | Pro | Gly | Ile | Gly |
| GGT | AAG | GCG | GCC | AAG | GTT | CCA | GGT | GCA | GGC | GTT | CCA | GGT | ATT | GGT |
| [Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly |
| [GTA | CCT | GGT | ATT | GGC | GTT | CCG | GGT | ATC | GGT | GTG | CCA | GGC | ATC | GGT |
| Val | Pro | Gly | Ile | Gly | Val | Pro | Gly | Ile | Gly]15 | Val | Pro | Gly | Ile | Gly |
| GTA | CCG | GGT | ATT | GGC | GTT | CCA | GGC | ATT | GGC]15 | GTA | CCT | GGT | ATT | GGT |
| Val | Pro | Ala | Val | Gly | Lys | Ala | Ala | Lys | Val | Pro | Gly | Ala | Gly | Val |
| GTT | CCA | GCT | GTT | GGT | AAG | GCG | GCC | AAG | GTT | CCA | GGT | GCA | GGC | GTT |
| Pro | Ala | Val | Gly | [Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile |
| CCA | GCT | GTT | GGT | [ATT | CCG | GCT | GTT | GGT | ATC | CCA | GCT | GTT | GGT | ATC |
| Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly]16 | Ile |
| CCA | GCT | GTT | GGC | ATT | CCG | GCT | GTA | GGT | ATC | CCG | GCA | GTG | GGC]16 | ATT |
| Pro | Ala | Val | Gly | Ile | Pro | Ala | Val | Gly | Lys | Ala | Ala | Lys | Ala | Stop |
| CCG | GCT | GTT | GGT | ATT | CCA | GCT | GTT | GGT | AAG | GCG | GCC | AAG | GCG | TAA |
N-terminal endblock: VPAVGKVPAVG[(IPAVG)5]16 (IPAVG)
Midblock: (IPAVG)KAAK(VPGAG)(VPGIG) [(VPGIG)5]15(VPGIG)(VPAVG)KAAK(VPGAG)
C-terminal endblock: (VPGAG) [(IPAVG)5]16 (IPAVG)VPAVGKAAKA
Large-scale fermentation (100 L) was performed at 37°C in Circle Grow (Q-BIOgene) medium supplemented with kanamycin (50 μg/mL) at the Bioexpression and Fermentation Facility, University of Georgia. The fermentation cultures were incubated under antibiotic selection for 24 hours at 37°C.
Cells were harvested through centrifugation in sterile tubes at 1660 RCF for 20 minutes at 4°C. The supernatant was carefully decanted, cell pellets were resuspended in cold, sterile PBS (phosphate buffered saline, 20 mL per large culture flask pellet) and frozen at -80°C. Three freeze (-80°C) / thaw (25°C) cycles were employed for the initial cell fracture with equilibration back to cold temperatures following the cycles. Once cells were completely resuspended, six cycles of sonication, consisting of 20 second bursts followed by a 20 second rests in an ice bath, was employed to thoroughly break the cells. To recover any unbroken cells, a preparative centrifugation step was used (1660 RCF for 10 minutes at 4°C). Unbroken cells, which pelleted out during the spin were resuspended in cold, sterile PBS and re-sonicated, as described above.
The cold cell lysate was centrifuged at 20,000 g for 40 minutes at 4°C. The supernatant was transferred to a cold, sterile tube and poly(ethyleneimine) (PEI) was added to a final concentration of 0.5%. This solution was centrifuged again at 20,000 g for 40 minutes at 4°C to remove all nucleic acids and contaminating cellular material precipitated by the PEI. The supernatant was transferred to new sterile 50 mL tubes and NaCl was added to a 2M final concentration. The elastin-mimetic protein was salted out of solution at 25°C for 30-45 minutes. This solution was centrifuged at 9500 g for 15 minutes at 25°C to recover the protein product (‘hot-spin’). The supernatant was discarded and the protein pellet was resuspended in cold, sterile PBS on ice for only 10-20 minutes to avoid solubilizing unwanted contaminates. The resuspended protein solution was then subjected to a ‘cold spin’ at 20,000 g for 40 minutes at 4°C. The supernatant was transferred to sterile 50 mL tubes and salting precipitation was repeated. The hot (25°C) / cold (4°C) spin cycles were repeated until no contaminating pellet was observed after the cold spin. The number of cycles ranged between 6 to 10 and ended with a hot spin. Dialysis and lyophillization afforded proteins LysB10 and R4 as fibrous solids in isolated yield of 150 mg/L and 200 mg/L of culture, respectively.
Lyophilized protein was resuspended in sterile molecular grade water at 1 mg/mL and endotoxin levels were assessed according to manufacturer instructions using the Limulus Amoebocyte Lysate (LAL) assay (Cambrex). Levels of 0.1 EU/mg were obtained (1 EU = 100 pg of endotoxin), which corresponds to endotoxin levels for clinically used alginate (Pronova sodium alginate, endotoxin ≤ 100 EU/gram).
Identification of elastin-mimetic proteins
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis revealed a single protein band at 250 kDa and 100 kDa corresponding to LysB10 (Figure 3A) and R4 (Figure 3B), respectively. A total of 10 μg of the elastin-mimetic polypeptide along with molecular weight markers (Precision Plus Protein Kaleidoscope, Bio-Rad) were run on a 7.5% gel and negatively stained with a Copper stain (Bio-Rad). As previously reported, molecular weights observed by SDS-PAGE for elastin-mimetic proteins are approximately 20% greater than calculated molecular weights [18, 29].
Figure 3.
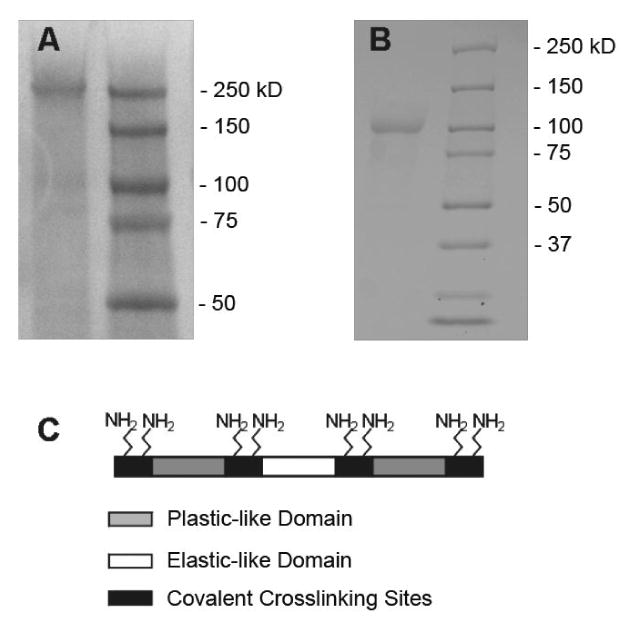
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of crosslinkable elastin-mimetic triblock copolymers. A. LysB10, run on 7.5% SDS-PAGE stained with Copper Stain (BioRad). Expected molecular weight: 209 KDa. B. R4 run on 7.5% SDS-PAGE stained with Copper Stain. Expected molecular weight: 108 KDa. Marker lane: Precision Plus Protein Kaleidoscope (Bio-Rad). C. Assembly scheme for crosslinkable elastin-mimetic proteins, LysB10 and R4. Both proteins are triblock copolymers with lysine-containing crosslinking domains flanking each plastic-like and elastic-like domain. Together, there are eight possible sites for chemical crosslinking afforded by the free amine of the lysine residues and the N-terminal amine of the peptide chain. Plastic-like domain (grey), elastic-like domain (white), crosslinking domain (black).
Amino acid composition analysis was performed by the Microchemical Facility at Harvard University. Lyophilized protein was resuspended in HPLC grade water and dialyzed against the same water at 4°C with buffer changes. Solutions of 1 mg/mL were submitted for analysis. Amino acid compositional analysis of LysB10. Calc. (mol.-%): Ala, 19; Glu, 1; Gly, 25.7; Ile, 13.9; Pro, 19.8; Val, 19.9. Obs. (mol.-%): Ala, 18.7; Glu, 2.3; Gly, 23; Ile, 13.5; Pro, 18.1; Val, 18. R4. Calc. (mol.-%): Ala, 14.2; Gly, 26.1; Ile, 19.2; Pro, 19.8; Val, 20.2. Obs. (mol.-%): Ala, 15.6; Gly, 24.3; Ile, 17.5; Pro, 19.4; Val, 18.9.
Rheological analysis of concentrated protein polymer solutions
Rheological data were acquired on an Advanced Rheological Expansion System III rheometer (ARES III, TA instrument, NJ) in parallel plate geometry with a plate diameter of 25 mm. The testing protocol for rheological analysis has been detailed elsewhere [4]. In brief, 100 mg/mL protein solutions were prepared by adding distilled, deionized water to lyophilized protein at 4°C, shaking the solution for 48 hours, and then allowing the solution to equilibrate for 72 hours. The gap between parallel plates was adjusted between 0.2 and 0.35 mm and dynamic mechanical experiments were performed in shear deformation mode. An initial strain amplitude sweep was performed at 4°C and 37°C at a frequency of 1 Hz to confirm the linear viscoelastic range for the protein polymer.
The gelation temperature was determined by heating samples from 4°C to 37°C at a rate of 1°C per minute. Following temperature equilibration at 37°C, viscoelastic properties were examined by a strain sweep at a fixed frequency of 1Hz and a frequency sweep at fixed strain amplitude of 2%. Experiments were repeated on six samples and representative data presented.
Fabrication of water cast protein films
For mechanical property analysis, films were cast from protein solutions in water at room temperature. In brief, lyophilized proteins were dissolved at a concentration of 100 mg/mL in water at 4°C. The protein solution was then poured into Teflon casting molds and regulated solvent evaporation performed at 23°C for 48 hours. After complete solvent evaporation, films were subjected to glutaraldehyde (GTA) vapor phase crosslinking. Specifically, films were enclosed in a 2 L chamber containing a 10 mL of 25% glutaraldehyde (GTA) solution. Films were placed on a platform 4 cm above the GTA solution and exposed to GTA vapor for 24 hours. Subsequently, the films were rinsed in PBS for 48 hours with a change of PBS at 24 hours. Test samples were referred to as crosslinked or non-crosslinked indicating whether GTA treatment was used. Prior to testing, films were hydrated in PBS at 37°C, which contained NaN3 at 0.2 mg/mL to prevent biological contamination. Samples were cut into a dumbbell shape using a stainless steel die with gauge dimensions of 13 mm x 4.75 mm. Hydrated film thickness, as measured by optical microscopy, was typically 0.07 mm for non-crosslinked and crosslinked LysB10 films and 0.1 mm for R4 films.
Evaluation of water content in protein films
For evaluation of water content, 200 μL of a 10 wt% protein solution was cast as a disk measuring 1 cm in diameter. Dried films were vapor phase crosslinked with a 25% GTA solution for 24 hours, fully dehydrated under vacuum, and the dehydrated weight obtained using a Mettler balance. Films were subsequently incubated in PBS at 37°C for 24 hours and fully hydrated weights were obtained. A total of six films were evaluated for each protein. The equilibrium water content and equilibrium swelling ratio were determined according to equations (1) and (2), respectively, and expressed as mean ± standard deviation [30].
| (1) |
| (2) |
Characterization of non-crosslinked extractables
To determine the percent of potentially extractable protein polymer, 200 μL of a 10-wt% protein solution was cast as a disk measuring 1 cm in diameter. Dried films were vapor phase crosslinked with a 25% GTA solution for 24 hours, rinsed in PBS and incubated at 4°C, below the inverse transition temperature of the protein, for a period of seven days. Every 48 hours films were dehydrated and dry weight was monitored for material losses. Six films were investigated for each protein. The percent extractable was determined by equation (3) and expressed as a mean ± standard deviation.
| (3) |
Mechanical analysis of hydrated protein films
A preconditioning protocol was employed for LysB10 samples that consisted of a single cyclic stretch to 50% strain for one cycle followed by 20 cycles of 30% strain with off-loading periods of 5 minutes between each cycle. Due to the plasticity of R4 protein films, preconditioning was not conducted. Uniaxial stress-strain properties of protein films were determined on at least five to six individual specimens using a dynamic mechanical thermal analyzer (DMTA V, Rheometric Scientific Inc., Newcastle, DE) with a 15 N load cell in the inverted orientation, so that samples could be immersed in a jacketed beaker filled with PBS at 37°C. The maximum travel distance of the drive shaft was 23 mm, which limited maximum strain to 70% of engineering strain. Given the extensibility of these materials, uniaxial stress-strain responses were also characterized using a miniature materials tester (Minimat 2000, Rheometric Scientific) in tensile deformation mode at a rate of 5 mm/min conducted in air at room temperature. All samples were coated with a thin layer of mineral oil to prevent dehydration. For both DMTA and Minimat testing, samples were cut into a dumbbell shape using a stainless steel die with gauge dimensions of 13 mm x 4.75 mm. In addition, to calculating Young's modulus (E), ultimate tensile stress (UTS), and strain at failure (ε), resilience was determined from equation (4).
| (4) |
Creep analysis was performed on 6 to 12 specimens for each film type subjected to varying levels of constant engineering stress for periods of up to 11 hours.
In vivo evaluation of crosslinked protein gels
Syringe casting method for creating cylindrical implants
In order to minimize sample manipulation and the risk of cross-contamination, the following protocol was used for preparation of samples for in vivo implant studies. A 10 wt% cold protein solution was drawn into a chilled sterile 1 mL syringe (Becton Dickenson) and subsequently gelled at 37°C. The tip of the syringe was removed with a sterile scalpel and the molded protein gel extruded into room temperature PBS. Gel samples, 4.75 mm in diameter by 8 mm in length, were incubated in 0.5% GTA for 24 hours. Following crosslinking, samples were washed in sterile PBS for 48 hours with 20 buffer exchanges.
Subcutaneous and peritoneal implant models
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University. Eight-week-old inbred male C57BL/6 mice weighing 25–30 g were obtained from the Jackson Laboratory (Bar Harbor, ME). Under ketamine (95 mg/kg, IM) and xylazine (5 mg/kg, IM) anesthesia, a 1 cm dorsal midline incision was performed and a single test sample implanted in the subcutaneous space, parallel to the longitudinal body axis of each mouse. Each implant type was implanted in at least three separate mice. Three weeks after implantation, animals were sacrificed and samples explanted with overlying skin. For peritoneal implants, a 1 cm long midline incision was made along the linea alba of the abdominal wall and a single implant placed into the peritoneal cavity. After closing the abdominal muscle with 4-0 absorbable surgical suture (Vicryl, Ethicon, Inc, NJ), the skin incision was closed with wound clips. Each recombinant protein was implanted in at least five mice. Mice were sacrificed one week later and, prior to sample removal, a peritoneal saline lavage was performed to harvest cells for FACS analysis.
Histological examination
Retrieved samples were processed for histological and immunohistochemical evaluation to characterize the local cellular response and to determine the extent and thickness of fibrous capsule formation. All samples were fixed in 10% neutral buffered formalin overnight and processed for paraffin embedding. Sections were prepared at a thickness of 5 μm and stained with hematoxylin and eosin (H&E) or rat anti-mouse monoclonal F4/80 (CI:A3-1, Abcam) to identify infiltrating macrophages. In all cases, multiple sections were examined in three to five separate samples for each protein polymer type.
Fluorescent-activated cell sorting (FACS) of peritoneal lavage
Prior to harvesting implants, each peritoneal cavity was initially lavaged with 10 mL of cold Hank's Balanced Salt Solution containing 10 U/mL heparin and 1% BSA (Mediatech, Inc). Typically, 6 to 7 mL of lavage solution was retrieved and cells immunostained for flow cytometry with PE-conjugated rat monoclonal anti-mouse CD11b for macrophages, FITC-conjugated hamster anti-mouse CD3 for total T cells, FITC-conjugated rat monoclonal anti-mouse CD4 for helper T cells, FITC-conjugated rat monoclonal anti-mouse CD8 for cytotoxic T cells, FITC-conjugated rat monoclonal anti-mouse CD19 for B cells, and FITC-conjugated rat monoclonal anti-mouse Gr-1 for neutrophils (BD Biosciences Pharmingen). Typically, antibodies were diluted to 1 μg/50 μL/106 cells in PBS containing 1% BSA and 0.1% sodium azide. Cells were incubated in the dark for 30 minutes on ice, then washed three times in staining buffer, and fixed in 1% paraformaldehyde. Analysis was performed on a FACScan using Cellquest (Becton Dickinson) and FlowJo software (Tree Star) [31]. Comparison between groups was analyzed via a Student's t-test and p < 0.05 were considered to be significant. Results are presented as mean ± SEM. Two control groups were employed; one that did not undergo surgery and another in which surgery was performed without sample implantation. At least five mice were enrolled in each experimental and control group.
Results and Discussion
Synthesis of triblock protein copolymers capable of both chemical and physical crosslinking
We have recently reported the design of a new class of recombinant elastin-mimetic triblock copolymer that has the capacity to form physical or virtual crosslinks, which stabilize protein network structure [3-5]. Moreover, through selective engineering of block structure, including the design of block size or sequence, and choice of film casting conditions, microphase structure can be manipulated and, as a consequence, material properties, such as drug elution characteristics and mechanical behavior tailored over a wide range of responses [4, 7, 8]. However, physical crosslinks formed as a result of hydrophobic aggregation may be deformed or disrupted under external stresses lower than that required to disrupt covalent crosslinks. This feature may limit the capacity of physically crosslinked protein-based materials to retain material integrity under loading conditions operative for a number of potential applications in tissue engineering or regenerative medicine. Given these considerations, we have postulated that chemically locking a multiblock protein assembly in place may provide a strategy to preserve functional responses that are linked to specific domain structures and morphologies over a broader range of externally applied loads. Further, it would also provide an additional approach for altering material strength and compliance, as well as stress induced creep behavior.
In this report, elastin-mimetic triblock copolymers were designed with endblock sequences, encoded by a hydrophobic repeat sequence, (IPAVG), that exhibits plastic-like mechanical responses, and a central elastomeric block of varying amino acid structure. The endblock sequence was selected to display an inverse temperature transition in water below 37°C, thereby mediating protein self-assembly due to endblock coacervation at or above this temperature. Sites for covalent crosslinking were engineered at positions flanking each block. Two target proteins were genetically engineered. A 209 kDa triblock, LysB10, was designed with hydrophobic endblocks with a mass of approximately 75 kDa each, which contained 33 repeats of the pentapeptide sequence [IPAVG]5, separated by a 58 kDa hydrophilic midblock comprised of 28 repeats of the pentapeptide sequence [(VPGAG)2VPGEG(VPGAG)2] (Table 2). Crosslinking sites that contained a pair of lysine residues (KAAK) flanked each block, such that a total of eight crosslinkable residues were potentially accessible. The presence of glutamic acid residues (E) was responsible for the hydrophilic character of the midblock.
In addition, a 108 kDa triblock protein polymer, designated as R4, was synthesized with flanking hydrophobic plastic-like endblocks, each with a mass of approximately 37 kDa that contained 16 repeats of [IPAVG]5, separated by a 35 kD midblock comprised of 15 repeats of [VPGIG]5 (Table 3). Likewise, a total of eight potential crosslinking sites were engineered into the protein sequence; positioned predominantly as lysine pairs (KAAK) that flank each block. The substitution of isoleucine for glutamic acid in the midblock yielded a protein that was largely hydrophobic with little difference in block polarity. While VPGEG and VPGIG are both reported to form β-spiral structures that display elastic responses when crosslinked as gels, polypeptides from each sequence differ significantly in their inverse transition temperature. Specifically, by incorporating the hydrophobic residue, isoleucine, into the fourth position of the repeat sequence, the inverse transition temperature of VPGIG is much lower than that associated with VPGEG [32-36].
Lysine crosslinking domains were engineered with an appreciation of the structure of similar crosslinking sites in native elastin and a consideration of the ‘N-end rule’, such that the identity of the N-terminal residue of a recombinant protein may influence degradation in bacterial expression systems. In native elastin, crosslinking domains contain paired lysines within polyalanine repears (eg. Ala-Ala-Ala-Lys-Ala-Ala-Lys-Ala-Ala) [26, 27], which promotes formation of an alpha-helix that has been reported to facilitate intermolecular crosslinking [37-40]. Thus, for both protein triblock copolymers, a lysine containing insert was designed encoding two lysine residues separated by two alanine residues (Lys-Ala-Ala-Lys) that was inserted between component blocks. Additionally, lysine containing adaptor sequences were designed to encode for two C-terminal, as well as a single lysine residue near the N-terminus. Lysine was not incorporated as an N-terminal residue, as previous efforts to encode lysine in this position have lead to a 10-fold decrease in protein yield [1, 10, 41, 42]. This design afforded eight free amines for crosslinking, seven from lysine and one from the N-terminal amine (Figure 3C).
Rheological analysis confirms formation of viscoelastic protein gels
The gelation point of protein solutions can be determined by measurement of G′ and G″ as a function of temperature at a fixed frequency. Above 13°C, the shear storage (G′) and loss (G″) modulus of concentrated aqueous solutions of LysB10 increased by a factor of approximately 103 and 10 (Pa), respectively, while tan δ (G′/G″) decreased, consistent with the formation of a viscoelastic gel (Figure 4A). Above 15 °C, R4 solutions displayed an increase in shear storage (G′) and loss (G″) modulus to 104 and 103 (Pa), respectively, with only a modest reduction in tan δ (Figure 4C). For both LysB10 and R4 protein solutions at 37°C, G′ and G″ were independent of frequency between 1 to 10 rad/s at a fixed strain amplitude of 2% (Figure 4B, 4D). In addition, the complex viscosity (η*) was a linear function of the logarithm of frequency with a slope of -1. However, as evident by a significantly higher tan δ and complex viscosity, aqueous gels of R4 were more viscous than those of LysB10. This difference highlights the significance of the midblock structure in triblock design. Despite similar endblock structure and the presence of an elastomeric midblock sequence in both R4 and LysB10 triblocks, the R4 midblock is considerably more hydrophobic and coacervates along with the endblock at 37°C. Indeed, when expressed as single blocks the inverse transition temperatures of the R4 endblock and midblock proteins were 26°C and 16°C, respectively, while the comparable transition temperatures for LysB10 blocks were 21°C and >80°C (data not shown). Thus, significant mixing of the elastomeric and plastic-like blocks occurs only in the case of gels produced from the R4 protein polymer, which limits its elastomeric response. We have previously demonstrated that selected changes in midblock size and amino acid sequence result in significant changes in viscoelastic mechanical properties. Indeed, a more viscous response, similar to that observed for R4, was displayed by a protein composed entirely of hydrophobic plastic-like endblock sequences [4].
Figure 4.
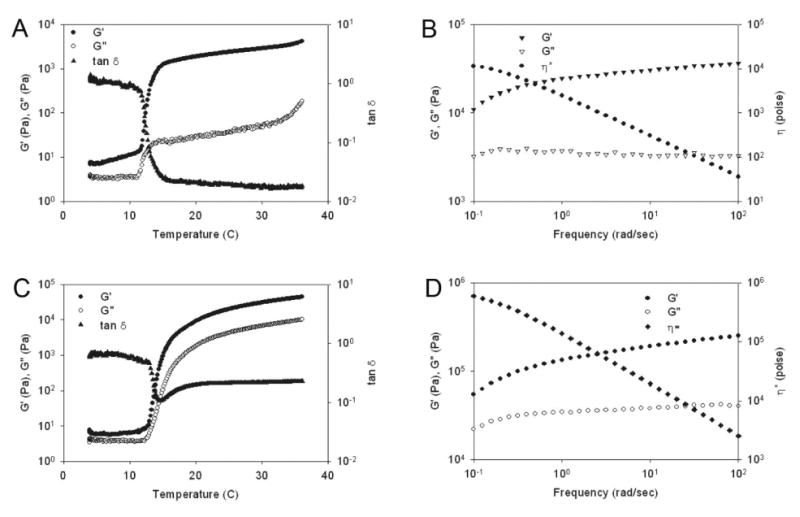
Rheological behavior of triblock protein polymers in water. (A) LysB10 dynamic shear storage (G′) and loss modulus (G″) are plotted as a function of temperature (γ 2%, ω 1Hz). (B) LysB10 dynamic shear storage (G′), loss modulus (G″), and complex viscosity (η*) are plotted as a function of frequency (γ 2%, 37°C). (C) R4 dynamic shear storage (G′) and loss modulus (G″) are plotted as a function of temperature (γ 2%, ω 1Hz). (D) R4 dynamic shear storage (G′), loss modulus (G″), and complex viscosity (η*) are plotted as a function of frequency (γ 2%, 37°C).
Glutaraldehyde crosslinking of elastin-mimetic triblock copolymer films
Investigations by our group and others have demonstrated that covalent crosslinks can enhance the mechanical stability of a variety of elastin analogues (Table 4). Aldehyde crosslinking agents, such as glutaraldehyde, have been commonly used to process implanted tissues and proteins because of their capacity for efficient crosslink formation with an associated reduction in tissue antigenicity and enhanced mechanical strength [43, 44]. While limitations of glutaraldehyde exist [17, 24], since it remains an industry standard and its effects are otherwise well established, it was used in this study as a model crosslinking system.
Table 4.
Review of Chemically Crosslinkable Elastin-Mimetic Systems and Chemical Crosslinking Strategies
| Crosslinkable Recombinant Elastin-Mimetic Proteins | Molecular Weight (kDa) | Crosslinking Agent | Fabrication Technique | Mechanical Evaluation | Biocompatibility Evaluation | Reference |
|---|---|---|---|---|---|---|
| Poly(KGGVG) (polydisperse) | 6.5-14 (polydisperse) | GTA and DSG | hydrogel | - | - | [15] |
| VPGIG2VPGKGVPGIG2-CS5-VPGIG2VPGKGVPGIG2 | 80.7 | GTA | film | - | - | [14] |
| CS5-(VPGIG)20
[CS5-(VPGIG)20]3 [CS5-(VPGIG)20]5 |
14, 36.6, 59 | GTA | film | E = 0.099-0.32 MPa | - | [10] |
| σ = 0.5-0.9 MPa | ||||||
| ε = 251-393 % | ||||||
| [VPGVG4VPGKG]25 | 80.6 | BS3 and DSS | hydrogel | - | - | [19] |
| CS5-(VPGIG)25
[CS5-(VPGIG)25]3 [CS5-(VPGIG)25]5 |
14, 36.6, 59 | Isocyanate | hydrogel | E = 0.4-0.9 MPa | - | [17] |
| G = 0.13-0.31 MPa | ||||||
| E = 340-570 % | ||||||
| ELP[KV6]-102, ELP[KV16]-112 | 42.7, 47 | TSAT | hydrogel | G* = 1.6-15 kPa | - | [18] |
| ELP[KV6-112] | 47.1 | TSAT | Hydrogel + cells | G* = 0.28-1.7 kPa | non-cytotoxic in vitro | [24] |
| KV7F-72 AND KV2F-64 | 31, 28.3 | THPP | Hydrogel | G* = 5.8-45.8 kPa | non-cytotoxic in vitro, 3 days | [22, 23] |
| Exons 20-24 | 10 | Genipin | Film | E = 1.8 MPa | - | [13, 37] |
| ε = 0.68 mm/mm | ||||||
| PQQ | Film | E = 0.4 MPa | - | |||
| ε = 0.90 mm/mm | ||||||
| CS5-[(VPGIG)2(VPGKG)(VPGIG)2] 4 | 37 | BS3 | Film | E = 0.08-0.7 MPa | - | [21] |
| G = 0.022-0.060 MPa | ||||||
| DSS | Film | E = 0.3-0.97 MPa | - | |||
| G = 0.12-0.32 MPa |
Abbreviations: CS5-RGD cell binding domain, GTA-glutaraldehyde, DSG-disuccinimidyl glutarate, HMDI-hexamethylene diisocyanate, BS3-bis(sulfosuccinimidyl) suberate, DSS-disuccinimidyl suberate, TSAT-tris-succinimidyl aminotriacetrate, THPP-tris(hydroxymethyl)phosphino-propionic acid, PQQ-pyrroloquinoline quinine, E= elastic modulus, σ = ultimate tensile strength, ε = elongation at break, G* = complex modulus, G = shear moduli
As a measure of the extent of crosslinking, the percent of extractable protein was examined after incubating samples at 4°C for seven days. Below the inverse transition temperature, non-crosslinked films dissolved immediately due to disruption of physical crosslinks. After vapor phase GTA crosslinking, films retained approximately 86 ± 7% (LysB10) and 88 ± 5% (R4) of their mass consistent with a high degree of chemical crosslinking, despite a relatively limited number of crosslink sites per protein species.
The equilibrium water content observed for R4 and LysB10 films was 32.0 ± 3.7% and 54.3 ± 3.8%, respectively; values that are similar to the 32% water content reported for hydrated elastin at 36°C [45]. Likewise, water swelling ratios were 1.5 ± 0.1 for R4 and 2.4 ± 0.2 for LysB10, consistent with the higher proportion of hydrophobic amino acids in R4.
Mechanical responses of LysB10 copolymer films
Most biomolecular constructs represent dynamic systems of hydrated biopolymer chains whose entanglements and structural interrelationships may be altered in response to an external load. As a consequence, initial mechanical properties may change in response to repetitive loading forces until stable behavior is observed. Thus, mechanical preconditioning is presumed to induce changes in microstructure that lead to fixed structural rearrangements of the constituent polymer chains and, as a consequence, stable material properties under a given loading environment [46-49]. Preconditioned, non-crosslinked LysB10 films were robust and elastomeric; displaying an elastic modulus of 0.49 ± 0.03 MPa, an ultimate tensile strength of 2.88 ± 0.91 MPa, a strain at failure of 430 ± 34%, and a resilience of 53 ± 2%. In contrast, preconditioned crosslinked samples exhibited a two- to three-fold increase in Young's modulus and a 50% decrease in strain at failure with a modest increase in ultimate tensile strength, as compared to their non-crosslinked counterparts (Table 5, Figures 5, 6). We speculate that these differences are largely related to the stabilization of semi-rigid endblocks by chemical crosslinking. While crosslinking enhanced strength and modulus, a small reduction in resilience was noted. Since crosslinking was performed prior to preconditioning, the capacity of chain entanglements between midblock and endblock sequences to structurally rearrange in response to the conditioning protocol may have been restricted. In other words, in addition to restricting the mobility of the elastomeric phase, crosslinked-fixed mixing between rigid and elastomeric domains may have contributed to this effect.
Table 5.
Summary of Mechanical Parameters in Crosslinked and Non-Crosslinked Films
| Protein | Treatment | Resilience at 30% strain (%) | Young's Modulus DMTA (MPa) | Young's Modulus Minimat (MPa) | UTS (MPa) | Strain at Failure (%) |
|---|---|---|---|---|---|---|
| LysB10 | GTA Xlinked | 39 ± 1* | 1.10 ± 0.45* | 1.60 ± 0.48* | 3.62 ± 0.98 | 223 ± 30* |
| LysB10 | Non-Xlinked | 52 ± 2* | 0.49 ± 0.03* | 0.53 ± 0.02* | 2.88 ± 0.91 | 430 ± 34* |
|
| ||||||
| R4 | GTA Xlinked | - | 67.4 ± 5.14* | - | 4.49 ± 0.27* | 8 ± 2* |
| R4 | Non-Xlinked | - | 48.6 ± 0.90* | - | 1.72 ± 0.30* | 4.9 ± 1* |
|
| ||||||
| B10§ | Water-25 | 67± 1 | 0.71 ± 0.12 | - | - | - |
Average values obtained from 3-10 replicates. Resilience and Young's Modulus determined from DMTA testing. Young's modulus, UTS, and % Strain determined from Minimat testing.
p < 0.05 between crosslinked and non-crosslinked samples
B10 Data obtained from Wu et al, Biomacromolecules, In press [72].
Figure 5.
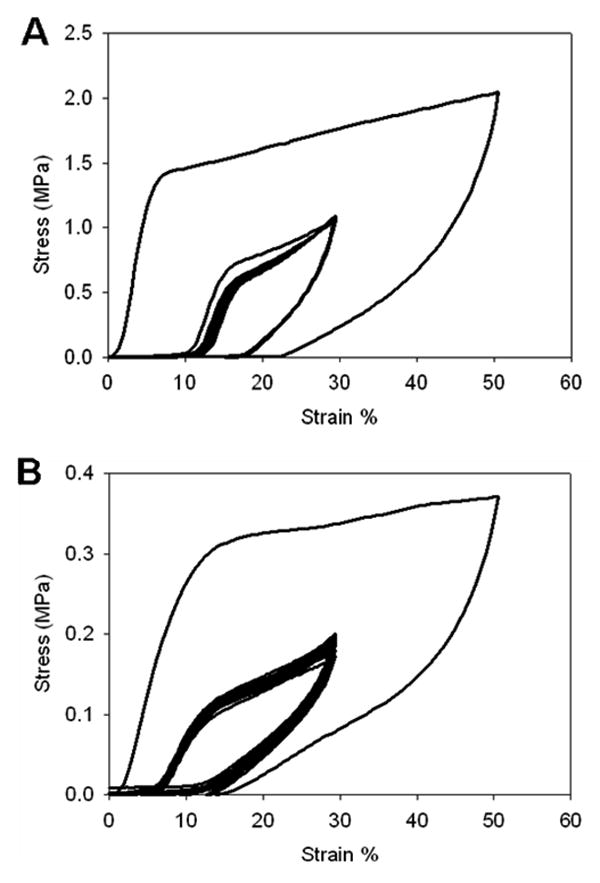
Mechanical preconditioing of glutaraldehyde crosslinked (A) and non-crosslinked (B) LysB10 films. Sample were cyclically stretched to 50% strain and then to 30% strain for 20 cycles.
Figure 6.
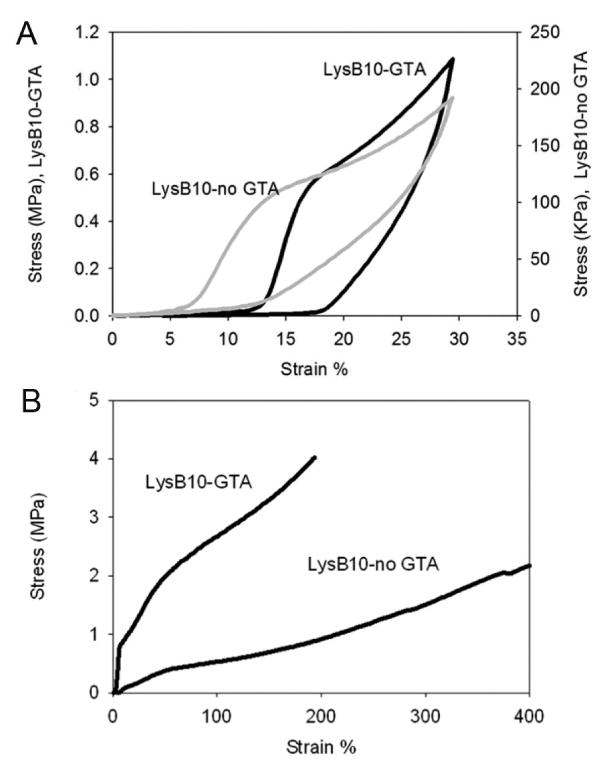
(A) Resilience of glutaraldehyde crosslinked and non-crosslinked LysB10 films after mechanical preconditioning. The response is representative of multiple data sets and illustrates cycle 20 of 30% stretch. (B) Uniaxial stress-strain response for preconditioned glutaraldehyde crosslinked and non-crosslinked LysB10 films. Films were strained to failure.
The approximate hoop stress of a blood vessel with an inner diameter of 4.5 mm and wall thickness of 0.8 mm is 45 kPa, when subjected to an intraluminal pressure of 16 kPa (120 mmHg). Crosslinked LysB10 films demonstrated limited creep (<10%) over 11 hours at stress levels at or below 45 kPa. Increasing the applied stress by a factor of ten increased creep to ∼30% (Figure 7A). In contrast, non-crosslinked films showed a four-fold greater level of creep strain in response to 45 kPa loading stress (Figure 7B).
Figure 7.
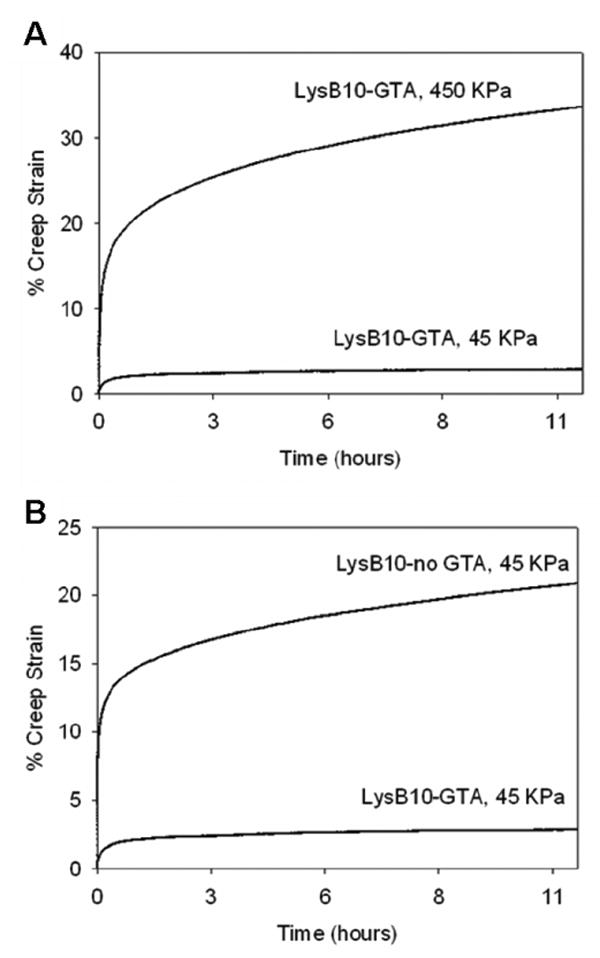
(A) Representative creep response of glutaraldehyde crosslinked LysB10 films subjected to tensile stress at 45kPa and 450kPa. (B) Creep behavior of glutaraldehyde crosslinked and non-crosslinked LysB10 films at a stress of 45 kPa.
Mechanical responses of R4 copolymer films
Hydrated R4 films revealed plastic-like deformation behavior. Specifically, stress increased linearly with increasing strain until a yield point was observed at 1.72 MPa and 4.49 MPa in non-crosslinked and crosslinked films, respectively (Figure 8A). Corresponding values of Young's modulus were 48.6 ± 0.9 MPa and 67 ± 5.14 MPa for non-crosslinked and crosslinked samples, respectively. These values are four- to ten-fold greater than those noted for LysB10 (Table 5). As compared to LysB10, the more hydrophobic character of R4 is associated with reduced water uptake, which contributes to an increase in material rigidity and tensile strength. Moreover, given the similarity of endblock and midblock polarity, size, and transition temperature, we speculate that a greater degree of block mixing occurs in films composed of R4 with rigid, plastic-like domains sustaining a higher level of the external load. Crosslinking appears to have a relatively greater effect on the yield point (2.6-fold increase) than Young's modulus (1.4-fold increase). Likely, the effect of block mixing has a more profound effect on the mobility of the elastomeric phase than crosslinking, which predominantly stabilizes the semi-rigid endblocks. Consistent with these observed uniaxial stress-strain properties, crosslinked R4 films demonstrated limited creep strain (<10%) over an 11-hour period, despite applied stresses as high as 400 kPa. Substantial deformation, however, was noted at stresses of nearly 1 MPa (Figure 8B).
Figure 8.
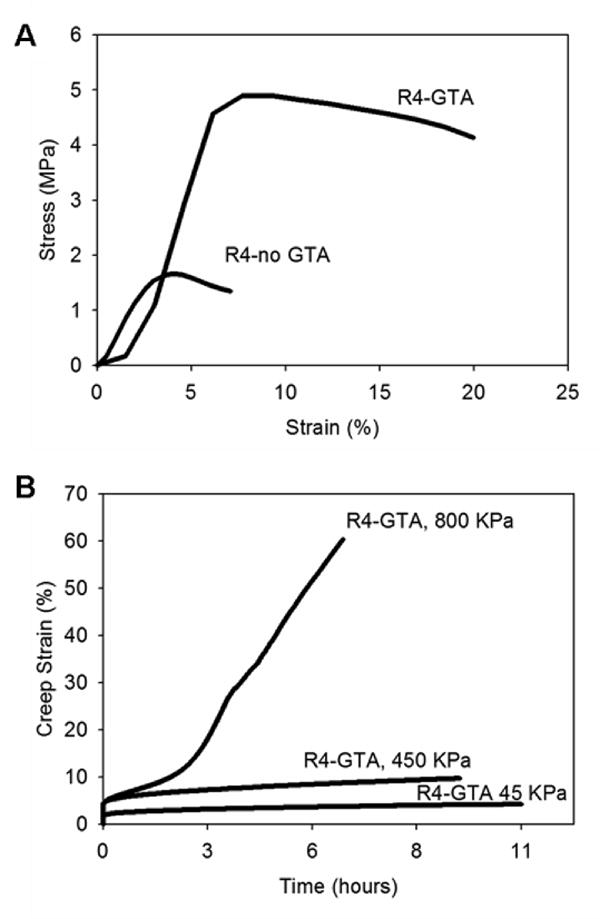
(A) Uniaxial stress-strain behavior for glutaraldehyde crosslinked and non-crosslinked hydrated R4 films. (B) Representative creep response of glutaraldehyde crosslinked R4 films subjected tensile stress at 45kPa, 450kPa, or 800kPa.
In vivo responses to crosslinked elastin-mimetic protein hydrogels
Using a murine model, crosslinked LysB10 and R4 hydrogels were implanted into either the subcutaneous space or the peritoneal cavity. FACS analysis demonstrated no difference in either the cell number or cell type identified within peritoneal lavage fluid, harvested one week after either sham surgery or protein polymer implantation (Figure 9). Samples implanted within the peritoneal cavity and subcutaneous tissue were explanted one and three weeks after implantation, respectively, without evidence of protein gel dissolution (Figures 10, 11). Macrophages were identified along the periphery of the fibrous capsule and in the surrounding tissue without infiltration into the implant. The fibrous capsule thickness of LysB10 samples within the subcutaneous space was 25.3 ± 16.1 μm, while the capsule measured 14.5 ± 5.3 μm for those placed in the peritoneal cavity. R4 implants displayed a similar response, with a fibrous capsule thickness of 24.2 ± 6.0 μm and 8.4 ± 1.2 μm for subcutaneous and peritoneal cavity implants, respectively.
Figure 9.
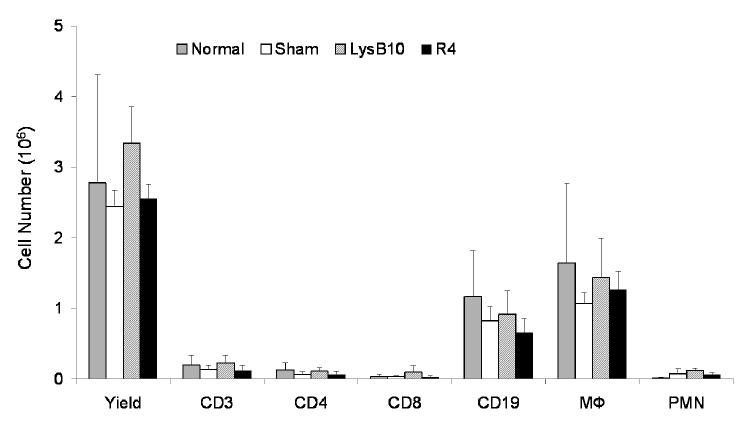
FACS analysis of peritoneal cells one week after implantation of LysB10 and R4 cylindrical hydrogels (n=5 for each group). Experimental groups displayed an identical cell profile to non-operated and sham surgery groups.
Figure 10.
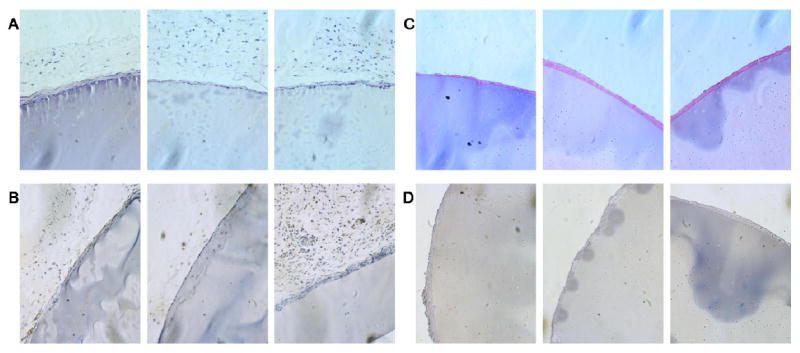
(A) Hematoxylin and eosin staining of subcutaneous LysB10 implants demonstrates the presence of a mild foreign body reaction along the periphery. (B) F4/80 staining of subcutaneous LysB10 implants demonstrate the presence of macrophages along the periphery of the fibrous capsule. (C) H&E staining of peritoneal LysB10 implants demonstrates the presence of a mild foreign body reaction along the periphery. (D) F4/80 staining of peritoneal LysB10 implants demonstrates the presence of macrophages along the periphery. Images are oriented so that the LysB10 gel is located in the bottom right corner, 20x magnification.
Figure 11.

(A) H&E staining of subcutaneous R4 implants demonstrates the presence of a mild foreign body reaction along the periphery of the sample. (B) F4/80 staining of subcutaneous R4 implants demonstrates the presence of macrophages along the periphery of the fibrous capsule. (C) H&E staining of peritoneal R4 implants demonstrates the presence of a mild foreign body reaction along the periphery of the implant. (D) F4/80 staining of peritoneal R4 implants demonstrates the presence of macrophages along the periphery of the fibrous capsule. Images are oriented so that the R4 gel is located in the bottom right corner, 20x magnification.
Tissue-material interactions, including biopolymer stability, are integral to assessing the suitability of elastin-like protein polymers for implant related applications. To date, reports documenting in vivo responses to elastin-mimetic protein implants have been limited; largely confined to studies performed 15 to 20 years ago on proteins synthesized chemically and subject to radiation crosslinking [36, 50]. In these investigations, homopolymers or copolymers composed of VPGVG, VPGKG, VPGEG, IPAVG, and VPAVG reportedly did not induce significant inflammatory or allergic reactions [50-53]. The most thoroughly characterized elastin variant, chemically synthesized poly(GVGVP), was subjected to in vitro toxicity and mutagenicity assays and was administered via intravenous, intraocular, intramuscular, intraperitoneal, and subcutaneous routes without toxic effect [36, 50]. A fibrous capsule was noted three weeks after intramuscular implantation of a radiation crosslinked sample [36]. In a more recent report, elastin microparticles prepared from chemically synthesized poly(VPAVG) were evaluated following subcutaneous and intravitreal injection. No inflammatory response was observed after 28 days. However, tractional retinal detachment was noted [51]. The failure to detect an immune mediated reaction to these polymers is consistent with other studies that have sought to identify potentially immunogenic epitopes on native elastin. While polyclonal and monoclonal antibodies can be raised against peptides derived from the hydrolysis of native elastin, neither VPGVG nor VPAVG peptides have been among the recognized sequences [53]. Moreover, VPGVG peptides were unable to competitively inhibit the binding of any of the antibodies raised against native elastin, which further supports the notion that this pentapeptide is not present among antigenic elastin epitopes [53].
Recently, genetically engineered elastin-mimetic protein polymers have been investigated in vivo as non-thrombogenic coatings [54, 55], targeted drug delivery vehicles [56-58], and as an implantable material [59]. In the latter instance, after a 13 week implant period in the subcutaneous space, recombinant human tropoelastin ‘sponges’, chemically crosslinked with bis(sulfosuccinimidyl) suberate, were surrounded by a fibrous capsule with a minimal to moderate inflammatory response [59]. Non-chemically crosslinked recombinant elastin-like proteins have also been administered within the intra-articular space as a 650 μM protein solution [59]. Although the biological response was not evaluated, this study revealed a three hour half-life for non-aggregating VPGVG proteins and a three day half-life for aggregating VPGXG proteins, where X = V:G:A at a ratio of 1:7:8. As a related material, a 47 kDa recombinant silk-elastin-like protein (SELP), comprised of GAGAS silk-like [S] and GVGVP elastin-like [E] amino acid sequences ([S]4[E]4[EK][E]3) has been studied after injection into the subcutaneous space. Histological analysis revealed minimal fibrous encapsulation after four weeks with a mild degree of inflammation that included the presence of macrophages in the surrounding tissue [60]. SELPs have been also used for adenoviral gene delivery and demonstrate prolonged and localized expression of adenoviruses for up to 15 days [61, 62]. A summary of in vivo biocompatibility studies conducted on elastin-mimetic proteins is presented in Table 6.
The evaluation of in vivo biocompatibility is largely based on characterizing local tissue responses to subcutaneously implanted materials where the intensity and duration of inflammation and wound healing, including capsule formation, is evaluated histologically [63, 64]. Although histological studies of biopolymers containing elastin-mimetic sequences have previously noted the presence of ‘mild inflammation’ [50, 51] and ‘a reaction that was limited to a typical cell mediated response to the presence of a foreign body’, the extent of fibrous capsule formation has not been reported [59]. Fibrous capsule thickness has been investigated for a variety of polymeric and ceramic implants designed for tissue repair, cell encapsulation, or as drug delivery systems [65-68]. Capsule thicknesses are dependent on implant site and material type and typically varies between 2 and 150 μm over implantation periods of one to three months. For example, greater capsule thickness has been observed for materials within intraperitoneal sites compared to those in subcutaneous sites over identical implant durations [66]. As an illustration of the effects of surface chemistry, implants comprised of poly(alkyl methacrylate) (PAMA) with short alkyl side chains exhibited a thicker fibrous capsule than those with long side chains (140 μm vs 120 μm) [65]. Additionally, self-assembled monolayers (SAMs) of alkanethiols on gold with different terminal functional groups displayed surface dependent inflammatory responses after one week with extremely hydrophobic methyl terminated surfaces inducing thick fibrous capsules (130 μm) and higher recruitment of inflammatory cells compared to hydrophilic COOH- and OH- terminated surfaces (80 μm and 70 μm, respectively) [69]. Likewise, functionalized polypropylene implants revealed similar foreign body reactions to surface modifications, with −OH surfaces triggering a stronger response (∼100 μm) compared to −COOH rich surfaces (37 μm) [70]. In contrast, surface topography does not appear to have a significant effect on capsule thickness, although it may influence local inflammatory responses [71]. In this report, there were no observable differences in biological responses for either chemically crosslinked triblock elastin-mimetic protein polymer. Both R4 and LysB10 implants initiated limited local inflammatory activity and displayed relatively thin fibrous capsules.
Conclusions
A new class of recombinant elastin-mimetic protein polymer has been designed that is capable of both physical and chemical crosslinking. We have demonstrated that chemical crosslinking provides an independent mechanism for control of protein mechanical responses. Specifically, elastic modulus can be enhanced and creep strain reduced through the addition of chemical crosslinking sites. Additionally, we have demonstrated exceptional biocompatibility of glutaraldehyde treated multiblock systems. By chemically locking a multiblock protein assembly in place, unique structures and morphologies are preserved and stabilized, which provides the capacity to modulate a wide range of functional responses, such as mechanical behaviors, permeability, and drug elution characteristics. We anticipate that these materials will find utility in a number of vascular and non-vascular biomedical applications.
Acknowledgments
This work was supported by NIH grants HL60464 and HL083867 (E.L.C. and V.P.C.). We thank the Bioexpression and Fermentation Facility at the University of Georgia for expression of the LysB10 protein. Additionally, we thank Wensheng Li of Emory University for the purification of the LysB10 protein materials used in these experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Reversible hydrogels from self-assembling artificial proteins. Science. 1998;281:389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 2.Rodríguez-Cabello JC, Reguera J, Girotti A, Arias FJ, Alonso M. Genetic engineering of protein-based polymers: The example of elastinlike polymers. Adv Polym Sci. 2006;200:119–167. [Google Scholar]
- 3.Wright ER, Conticello VP. Self-assembly of block copolymers derived from elastin-mimetic polypeptide sequences. Adv Drug Deliv Rev. 2002;54:1057–1073. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 4.Nagapudi K, Brinkman WT, Thomas BS, Park JO, Srinivasarao M, Wright E, et al. Viscoelastic and mechanical behavior of recombinant protein elastomers. Biomaterials. 2005;26(23):4695–4706. doi: 10.1016/j.biomaterials.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 5.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Thermoplastic elastomer hydrogels via self-assembly of an elastin-mimetic triblock polypeptide. Advanced Functional Materials. 2002;12(2):1–6. [Google Scholar]
- 6.Sallach RE, Wei M, Biswas N, Conticello VP, Lecommandoux S, Dluhy RA, et al. Micelle density regulated by a reversible switch of protein secondary structure. J Am Chem Soc. 2006;128(36):12014–12019. doi: 10.1021/ja0638509. [DOI] [PubMed] [Google Scholar]
- 7.Nagapudi K, Brinkman WT, Thomas BS, Wright ER, Conticello VP, Chaikof EL. Protein-based thermoplastic elastomers. Macromolecules. 2005;38:345–354. [Google Scholar]
- 8.Wu X, Sallach R, Haller CA, Caves JA, Nagapudi K, Conticello VP, et al. Alterations in physical cross-linking modulate mechanical properties of two-phase protein polymer networks. Biomacromolecules. 2005;6(6):3037–3044. doi: 10.1021/bm0503468. [DOI] [PubMed] [Google Scholar]
- 9.Bellingham CM, Lillie MA, Gosline JM, Wright GM, Starcher BC, Bailey AJ, et al. Recombinant human elastin polypeptides self-assemble into biomaterials with elastin-like properties. Biopolymers. 2003;70(4):445–455. doi: 10.1002/bip.10512. [DOI] [PubMed] [Google Scholar]
- 10.Welsh ER, Tirrell DA. Engineering the extracellular matrix: A novel approach to polymeric biomaterials. I. Control of the physical properties of artificial protein matrices designed to support adhesion of vascular endothelial cells. Biomacromolecules. 2000;1(1):23–30. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- 11.Urry DW, Parker TM. Mechanics of elastin: Molecular mechanism of biological elasticity and its relationship to contraction. J Muscle Res Cell Motil. 2002;23(56):543–559. [PubMed] [Google Scholar]
- 12.Vrhovski B, Weiss AS. Biochemistry of tropoelastin. Eur J Biochem. 1998;258(1):1–18. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 13.Vieth S, Bellingham CM, Keeley FW, Hodge SM, Rousseau D. Microstructural and tensile properties of elastin-based polypeptides crosslinked with genipin and pyrroloquinoline quinone. Biopolymers. 2007;85(3):199–206. doi: 10.1002/bip.20619. [DOI] [PubMed] [Google Scholar]
- 14.Girotti A, Reguera J, Rodriguez-Cabello JC, Arias FJ, Alonso M, Matestera A. Design and bioproduction of a recombinant multi(bio)functional elastin-like protein polymer containing cell adhesion sequences for tissue engineering purposes. J Mater Sci Mater Med. 2004;15(4):479–484. doi: 10.1023/b:jmsm.0000021124.58688.7a. [DOI] [PubMed] [Google Scholar]
- 15.Martino M, Tamburro AM. Chemical synthesis of cross-linked poly(KGGVG), An elastin-like biopolymer. Biopolymers. 2001;59(1):29–37. doi: 10.1002/1097-0282(200107)59:1<29::AID-BIP1003>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Macoscko CW, Urry DW. Elastomeric polypentapeptides cross-linked into matrixes and fibers. Biomacromolecules. 2001;2:170–179. doi: 10.1021/bm0000900. [DOI] [PubMed] [Google Scholar]
- 17.Nowatzki PJ, Tirrell DA. Physcial properties of artificial extracellular matrix protein films prepared by isocyanate crosslinking. Biomaterials. 2004;25:1261–1267. doi: 10.1016/s0142-9612(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 18.Trabbic-Carlson K, Setton LA, Chilkoti A. Swelling and mechanical behaviors of chemically cross-linked hydrogels of elastin-like polypeptides. Biomacromolecules. 2003;4:572–580. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 19.McMillan RA, Lee TAT, Conticello VP. Rapid assembly of synthetic genes encoding protein polymers. Macromolecules. 1999;32:3643–3648. [Google Scholar]
- 20.McMillan RA, Conticello VP. Synthesis and characterization of elastin-mimetic protein gels derived from a well-defined polypeptide precursor. Macromolecules. 2000;33:4809–4821. [Google Scholar]
- 21.Zio KD, Tirrell DA. Mechanical properties of artificial protein matrices engineered for control of cell and tissue behavior. Macromolecules. 2003;36:1553–1558. [Google Scholar]
- 22.Lim DW, Nettles DL, Setton LA, Chilkoti A. In situ cross-linking of elastin-like polypeptide block copolymers for tissue repair. Biomacromolecules. 2008;9(1):222–230. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim DW, Nettles DL, Setton LA, Chilkoti A. Rapid cross-linking of elastin-like polypeptides with (hydroxymethyl) phosphines in aqueous solution. Biomacromolecules. 2007;8(5):1463–1470. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McHale MK, Setton LA, Chilkoti A. Synthesis and in vitro evaluation of enzymatically cross-linked elastin-like polypeptide gels for cartilaginous tissue repair. Tissue Eng. 2005;11(1112):1768–1779. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- 25.Nagapudi K, Huang L, McMillan RA, Brinkman W, Conticello VP, Chaikof EL. Photomediated solid-state crosslinking of an elastin-mimetic recombinant protein polymer. Macromolecules. 2002;35:1730–1737. [Google Scholar]
- 26.Narayanan AS, Page RC, Kuzan F, Cooper CG. Elastin cross-linking in vitro. Studies on factors influencing the formation of desmosines by lysyl oxidase action on tropoelastin. Biochem J. 1978;173(3):857–862. doi: 10.1042/bj1730857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bressan GM, Pasquali-Ronchetti I, Fornieri C, Mattioli F, Castellani I, Volpin D. Relevance of aggregation properties of tropoelastin to the assembly and structure of elastic fibers. J Ultrastruct Mol Struct Res. 1986;94(3):209–216. doi: 10.1016/0889-1605(86)90068-6. [DOI] [PubMed] [Google Scholar]
- 28.Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA. Endothelial cell adhesion to the fibronectin CS5 domain in artificial extracellular matrix proteins. Biomaterials. 2003;24(23):4245–4252. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 29.Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: Examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3(2):357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 30.Dinerman AA, Cappello J, Ghandehari H, Hoag SW. Swelling behavior of a genetically engineered silk-elastinlike protein polymer hydrogel. Biomaterials. 2002;23(21):4203–4210. doi: 10.1016/s0142-9612(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 31.Safley SA, Kapp LM, Tucker-Burden C, Hering B, Kapp JA, Weber CJ. Inhibition of cellular immune responses to encapsulated porcine islet xenografts by simultaneous blockade of two different costimulatory pathways. Transplantation. 2005;79(4):409–418. doi: 10.1097/01.tp.0000150021.06027.dc. [DOI] [PubMed] [Google Scholar]
- 32.van Hest JC, Tirrell DA. Protein-based materials, toward a new level of structural control. Chem Commun. 2001;(19):1897–1904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 33.Chang DK, Urry DW. Molecular dynamics calculations on relaxed and extended states of the polypentapeptide of elastin. Chem Phys Letters. 1988;147(4):395–400. [Google Scholar]
- 34.Urry DW, Gowda DC, Parker TM, Luan CH, Reid MC, Harris CM, et al. Hydrophobicity scale for proteins based on inverse transition temperature. Biopolymers. 1992;32:1243–1250. doi: 10.1002/bip.360320913. [DOI] [PubMed] [Google Scholar]
- 35.Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, et al. Temperature of polypeptide inverse temperature transition depends on mean residue hydrophobicity. J Am Chem Soc. 1991;113:4346–4348. [Google Scholar]
- 36.Wood SA, Lemons JE, Prasad KU, Urry DW. In vitro calcification and in vivo biocompatibility of the crosslinked polypentapeptide of elastin. J Biomed Mater Res. 1986;20(3):315–335. doi: 10.1002/jbm.820200305. [DOI] [PubMed] [Google Scholar]
- 37.Bellingham CM, Woodhouse KA, Robson P, Rothstein SJ, Keeley FW. Self-aggregation characteristics of recombinantly expressed human elastin polypeptides. Biochim Biophys Acta. 2001;1550(1):6–19. doi: 10.1016/s0167-4838(01)00262-x. [DOI] [PubMed] [Google Scholar]
- 38.Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: The elastic fiber. FASEB J. 1993;7:1208–1218. [PubMed] [Google Scholar]
- 39.Cox BA, Starcher BC, Urry DW. Communication: Coacervation of tropoelastin results in fiber formation. J Biol Chem. 1974;249(3):997–998. [PubMed] [Google Scholar]
- 40.Wu WJ, Vrhovski B, Weiss AS. Glycosaminoglycans mediate the coacervation of human tropoelastin through dominant charge interactions involving lysine side chains. J Biol Chem. 1999;274(31):21719–21724. doi: 10.1074/jbc.274.31.21719. [DOI] [PubMed] [Google Scholar]
- 41.Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-end rule in bacteria. Science. 1991;254:1324–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- 42.Panitch A, Yamaoka T, Fournier MJ, Mason TL, Tirrell DA. Design and biosynthesis of elastin-like artificial extracellular matrix proteins containing periodically spaced fibronectin CS5 domains. Macromolecules. 1999;32:1701–1703. [Google Scholar]
- 43.Chang Y, Tsai CC, Liang HC, Sung HW. In vivo evaluation of cellular and acellular bovine pericardia fixed with a naturally occurring crosslinking agent (genipin) Biomaterials. 2002;23(12):2447–2457. doi: 10.1016/s0142-9612(01)00379-9. [DOI] [PubMed] [Google Scholar]
- 44.Jayakrishnan A, Jameela SR. Glutaraldehyde as a fixative in bioprostheses and drug delivery matrices. Biomaterials. 1996;17(5):471–484. doi: 10.1016/0142-9612(96)82721-9. [DOI] [PubMed] [Google Scholar]
- 45.Gosline JM, French CJ. Dynamic mechanical properties of elastin. Biopolymers. 1979;18(8):2091–2103. doi: 10.1002/bip.1979.360180818. [DOI] [PubMed] [Google Scholar]
- 46.Humphrey JD. Cardiovascular solid mechanics. New York: Springer; 2002. [Google Scholar]
- 47.Sacks MS. Biaxial mechanical evaluation of planar biological materials. J Elasticity. 2000;61:199–246. [Google Scholar]
- 48.Carew EO, Garg A, Barber JE, Vesely I. Stress relaxation preconditioning of porcine aortic valves. Ann Biomed Eng. 2004;32(4):563–572. doi: 10.1023/b:abme.0000019176.49650.19. [DOI] [PubMed] [Google Scholar]
- 49.Carew EO, Barber JE, Vesely I. Role of preconditioning and recovery time in repeated testing of aortic valve tissues: validation through quasilinear viscoelastic theory. Ann Biomed Eng. 2000;28(9):1093–1100. doi: 10.1114/1.1310221. [DOI] [PubMed] [Google Scholar]
- 50.Urry DW, Parker TM. Biocompatibility of the bioelastic materials, poly(GVGVP) and its gamma-irradiation cross-linked matrix: Summary of generic biological test results. J Bioactive Compatible Polym. 1991;6:263–282. [Google Scholar]
- 51.Rincon AC, Molina-Martinez IT, de Las Heras B, Alonso M, Bailez C, Rodriguez-Cabello JC, et al. Biocompatibility of elastin-like polymer poly(VPAVG) microparticles: In vitro and in vivo studies. J Biomed Mater Res A. 2006;78(2):343–351. doi: 10.1002/jbm.a.30702. [DOI] [PubMed] [Google Scholar]
- 52.Hoban LD, Pierce M, Quance J, Hayward I, McKee A, Gowda DC, et al. Use of polypentapeptides of elastin to prevent postoperative adhesions: Efficacy in a contaminated peritoneal model. J Surg Res. 1994;56(2):179–183. doi: 10.1006/jsre.1994.1029. [DOI] [PubMed] [Google Scholar]
- 53.Wei SM, Katona E, Fachet J, Fulop T, Jr, Robert L, Jacob MP. Epitope specificity of monoclonal and polyclonal antibodies to human elastin. Int Arch Allergy Immunol. 1998;115(1):33–41. doi: 10.1159/000023827. [DOI] [PubMed] [Google Scholar]
- 54.Jordan SW, Haller CA, Sallach RE, Apkarian RP, Hanson SR, Chaikof EL. The effect of a recombinant elastin-mimetic coating of an ePTFE prosthesis on acute thrombogenicity in a baboon arteriovenous shunt. Biomaterials. 2007;28(6):1191–1197. doi: 10.1016/j.biomaterials.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 55.Woodhouse KA, Klement P, Chen V, Gorbet MB, Keeley FW, Stahl R, et al. Investigation of recombinant human elastin polypeptides as non-thrombogenic coatings. Biomaterials. 2004;25(19):4543–4553. doi: 10.1016/j.biomaterials.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 56.Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, C A. Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hypothermia. Cancer Res. 2001;61:1548–1554. [PubMed] [Google Scholar]
- 57.Liu W, Dreher MR, Chow DC, Zalutsky MR, C A. Tracking the in vivo fate of recombinant polypeptides by isotopic labeling. J Control Release. 2006;114:184–192. doi: 10.1016/j.jconrel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. A thermally responsive biopolymer for intra-articular drug delivery. J Control Release. 2006;115(2):175–182. doi: 10.1016/j.jconrel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 59.Mithieux SM, Rasko JEJ, Weiss AS. Synthetic elastin hydrogels derived from massive elastic assemblies of self-organized human protein monomers. Biomaterials. 2004;25:4921–4927. doi: 10.1016/j.biomaterials.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 60.Cappello J, Crissman JW, Crissman M, Ferrari FA, Textor G, Wallis O, et al. In-situ self-assembling protein polymer gel systems for administration, delivery, and release of drugs. J Control Release. 1998;53(13):105–117. doi: 10.1016/s0168-3659(97)00243-5. [DOI] [PubMed] [Google Scholar]
- 61.Hatefi A, Cappello J, Ghandehari H. Adenoviral gene delivery to solid tumors by recombinant silk-elastinlike protein polymers. Pharm Res. 2007;24(4):773–779. doi: 10.1007/s11095-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 62.Megeed Z, Haider M, Li D, O'Malley BW, Jr, Cappello J, Ghandehari H. In vitro and in vivo evaluation of recombinant silk-elastinlike hydrogels for cancer gene therapy. J Control Release. 2004;94(23):433–445. doi: 10.1016/j.jconrel.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 63.Anderson JM. In vivo biocompatibility of implantable delivery systems and biomaterials. Eur J Pharm Biopharm. 1994;40:1–8. [Google Scholar]
- 64.Hunt JA, Vince DG, Williams DF. Image analysis in the evaluation of biomaterials. J Biomed Eng. 1993;15:39–45. doi: 10.1016/0141-5425(93)90091-c. [DOI] [PubMed] [Google Scholar]
- 65.Andersson M, Suska F, Johansson A, Berglin M, Emanuelsson L, Elwing H, et al. Effect of molecular mobility of polymeric implants on soft tissue reactions: An in vivo study in rats. J Biomed Mater Res A. 2008;84(3):652–660. doi: 10.1002/jbm.a.31389. [DOI] [PubMed] [Google Scholar]
- 66.Butler K, Benghuzzui H, Tucci M, Cason Z. A comparsion of fibrous tissue formation surrounding intraperitoneal and subcutaneous implantation of ALCAP, HA, and TCP ceramic devices. Biomed Sci Instrum. 1997;34:18–23. [PubMed] [Google Scholar]
- 67.Shin H, Quinten Ruhe P, Mikos AG, Jansen JA. In vivo bone and soft tissue response to injectable, biodegradable oligo(poly(ethylene glycol) fumarate) hydrogels. Biomaterials. 2003;24(19):3201–3211. doi: 10.1016/s0142-9612(03)00168-6. [DOI] [PubMed] [Google Scholar]
- 68.Giavaresi G, Torricelli P, Fornasari PM, Giardino R, Barbucci R, Leone G. Blood vessel formation after soft-tissue implantation of hyaluronan-based hydrogel supplemented with copper ions. Biomaterials. 2005;26(16):3001–3008. doi: 10.1016/j.biomaterials.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 69.Barbosa JN, Madureira P, Barbosa MA, Aguas AP. The influence of functional groups of self-assembled monolayers on fibrous capsule formation and cell recruitment. J Biomed Mater Res A. 2006;76(4):737–743. doi: 10.1002/jbm.a.30602. [DOI] [PubMed] [Google Scholar]
- 70.Nair A, Zou L, Bhattacharyya D, Timmons RB, Tang L. Species and density of implant surface chemistry affect the extent of foreign body reactions. Langmuir. 2008;24(5):2015–2024. doi: 10.1021/la7025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Voskerician G, Gingras PH, Anderson JM. Macroporous condensed poly(tetrafluoroethylene). I. In vivo inflammatory response and healing characteristics. J Biomed Mater Res A. 2006;76(2):234–242. doi: 10.1002/jbm.a.30481. [DOI] [PubMed] [Google Scholar]
- 72.Wu X, Sallach RE, Caves JM, Conticello VP, Chaikof EL. Deformation responses of a physically cross-linked high molecular weight elastin-like protein polymer. Biomacromolecules In Review. doi: 10.1021/bm800012x. [DOI] [PMC free article] [PubMed] [Google Scholar]