

Abstract
While the therapeutic activity of the deoxycytidine analog decitabine is thought to reflect its ability to reactivate methylation-silenced genes, this agent is also known to trigger p53-dependent DNA damage responses. Here we report that p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a robust transcriptional target of decitabine. In cancer cells, decitabine treatment induces p53R2 mRNA expression, protein expression, and promoter activity in a p53-dependent fashion. The mechanism of p53R2 gene induction by decitabine does not appear to be promoter DNA hypomethylation, as the p53R2 5′ CpG island is hypomethylated before treatment. siRNA targeting of DNA methyltransferase 1 (DNMT1) in wild-type p53 cells leads to genomic DNA hypomethylation but does not induce p53R2, suggesting that DNMT/DNA adduct formation is the molecular trigger for p53R2 induction. Consistent with this idea, only nucleoside-based DNMT inhibitors that form covalent DNA adducts induce p53R2 expression. siRNA targeting of p53R2 reduces the extent of cell cycle arrest following decitabine treatment, supporting a functional role for p53R2 in decitabine mediated cellular responses. To determine the clinical relevance of p53R2 induction, we measured p53R2 expression in bone marrow samples from 15 myelodysplastic syndrome/acute myelogenous leukemia (MDS/AML) patients undergoing decitabine therapy. p53R2 mRNA and protein were induced in 7/13 (54%) and 6/9 (67%) of patients analyzed, respectively, despite a lack of methylation changes in the p53R2 promoter. Most notably, there was a significant association (P=0.0047) between p53R2 mRNA induction and clinical response in MDS/AML. These data establish p53R2 as a novel hypomethylation-independent decitabine gene target associated with clinical response.
Keywords: p53R2/RRM2B, decitabine, DNA methylation, myelodysplastic syndrome, epigenetic therapy, acute myelogenous leukemia
Introduction
Aberrant DNA hypermethylation is a key oncogenic mechanism that leads to epigenetic silencing of tumor suppressor genes (1). This fact has led to increasing interest in the development of pharmacological inhibitors of DNA methyltransferase enzymes (DNMTs) as anti-cancer agents (2, 3). The two DNMT inhibitors in current clinical use are the cytidine analog 5-azacytidine and the deoxycytidine analog decitabine (5-aza-2′-deoxycytidine; Dacogen). These agents are suicide substrates that incorporate into DNA following enzymatic conversion, and form covalent adducts with DNMT proteins present at the replication fork (4). Early studies of 5-azacytidine and decitabine utilized these drugs at high doses to achieve an anti-metabolite-driven cytotoxic mechanism of action (5). In contrast, recent clinical studies have utilized lower doses of these agents directed at reversing tumor-specific gene silencing and promoting tumor cell differentiation (6). The success of the latter strategy in Phase III clinical studies led to FDA approval of 5-azacytidine and decitabine for treatment of myelodysplastic syndrome (MDS) (6, 7).
Despite the success of these agents as treatments for MDS the basis for their activity, and to what extent it is related to reversal of epigenetic gene silencing, is unresolved (8). Gene expression profiling studies in cancer cell lines have revealed transcriptional responses to DNMT inhibitors that include activation of methylation-silenced tumor suppressor genes, as well as induction of distinct groups of genes not hypermethylated in tumor cells (9). As the result of these and other studies, at least three distinct mechanisms have been proposed to explain therapeutic responses to decitabine, including: 1) activation of methylation-silenced tumor suppressor genes such as p15INK4b (10, 11); 2) activation of cancer-testis or cancer-germline tumor antigen expression and MHC Class I expression, which may provoke anti-tumor immunity (12–14); and 3) activation of p53-mediated DNA damage responses (15, 16). Consistent with the last mechanism, MDS, the malignancy for which decitabine therapy is FDA-approved, rarely displays p53 mutations (17–19). Furthermore, decitabine treatment has been observed to induce p53-mediated DNA damage responses in vitro at clinically relevant concentrations (15, 16), and pifithrin-alpha, a selective p53 inhibitor (20), reverses the growth inhibitory and apoptotic effects of 5-azacytidine treatment in colon cancer cells (21). The mechanism by which 5-azacytdine and decitabine lead to DNA damage and p53 activation may be related to cellular recognition of DNMT/DNA adducts or to structural changes in chromatin caused by global genomic DNA hypomethylation (3, 22, 23). Further support of a key role for the activation of DNA damage pathways in the cellular response to decitabine comes from recent reports indicating that decitabine treatment activates classical DNA damage detection and signaling molecules, including ATM, CHK1 and γ-H2AX (24, 25).
Here we report the discovery of a novel transcriptional target of decitabine treatment, p53R2/RRM2B, which plays a key role in the resolution of cellular DNA damage by providing deoxynucleotides (dNTPs) to allow DNA repair to proceed in cells undergoing cell cycle arrest in response to p53 activation (26, 27). We show that decitabine treatment induces p53R2 expression in a p53-dependent manner, via a mechanism likely involving the formation of covalent DNMT/DNA adducts but not involving promoter DNA hypomethylation. In addition, we show that p53R2 induction occurs in response to treatment with other nucleoside analog-based DNMT inhibitors, and that its induction functionally contributes to cell cycle arrest in tumor cells responding to decitabine treatment. Most notably, we demonstrate that p53R2 induction occurs in vivo in the bone marrow of MDS/AML patients treated with decitabine, and that its induction directly correlates with clinical responses to decitabine.
Materials and Methods
Cell lines and drug treatments
HT29, HCT116, and RKO colorectal cancer (CRC) cells were obtained from American Type Culture Collection (ATCC) and were cultured as described previously (15). LOVO CRC cells were obtained from ATCC and were cultured using the same conditions as described for RKO (15). RKO cells expressing human papilloma virus E6 oncoprotein (RKO E6 cells) were a kind gift from Dr. Mark Meuth, Institute for Cancer Studies, University of Sheffield, United Kingdom, and were cultured as described for RKO (15). Decitabine (5-aza-2′-deoxycytidine), 5-azacytidine, zebularine [1-(β-D-ribofuranosyl)-1,2-dihydropyrimidin-2-one], and RG108 (N-Phthalyl-1-tryptophan) were obtained from Sigma Chemical Company, St. Louis, MO. The first three drugs were dissolved in 1× PBS, while RG108 was dissolved in DMSO. Drug concentrations and time points for specific experiments are described in the text and figure legends.
Human tissue samples
Fifteen patients with MDS (n=10), AML (n=4), and CMML (n=1) by World Health Organization (WHO) criteria (28) were treated with decitabine administered as 15 mg/m2 intravenously over three hours every eight hours for three days, repeated every six weeks (29), or as 20 mg/m2 intravenously over one hour daily for five days, repeated every four weeks (30). Bone marrow aspirates were performed pre-treatment and at recovery from cycles 2, 4, 6, etc. and cells were cryopreserved under an Institutional Review Board (IRB)-approved protocol. Response to treatment was defined using 2006 International Working Group criteria (31). The laboratory study was approved by the IRB. Cryopreserved bone marrow samples were briefly thawed in a 37°C water bath, and contents were separated to obtain RNA, DNA, and cytosolic protein extracts. For RNA extractions, bone marrow cells were pelleted via centrifugation at 4°C, and the supernatant was removed. 500 μl Trizol (Invitrogen, Carlsbad, CA) was added to the pellet and the suspension was homogenized with a pestle motor (VWR International, Bridgeport, NJ). After complete homogenization, an additional 500 μl of Trizol was added, and the sample was mixed by pipetting up and down several times. Subsequent extraction steps were performed according to the manufacturer’s recommendations. For DNA isolation, genomic DNA was isolated using the Puregene kit (Gentra Systems, Minneapolis, MN). For protein isolations, cytosolic protein fractions were isolated using the NE-PER kit (Pierce Biochemical, Rockland, IL) according to the specifications of the manufacturer. A motor pestle was used in steps that required suspension of cell pellet in buffer. 25–30 μg of protein extracts were used for Western blot analysis.
Northern blot analysis
Northern blot analyses were performed as described previously (32). The p53R2 probe was generated by reverse transcriptase PCR (RT-PCR) of the p53R2 mRNA using the following primers: F: 5′ GACTTGAGAAGACAGTTTGATCC 3′ and R: 5′ ATGCAGGCTTGGCTTGATTGACC 3′.
Western blot analysis
Western blots were performed as described previously (33). p53R2 was detected using the N-16 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer’s instructions. DNMT1 was detected as described previously (34).
5′RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) mapping
RLM-RACE mapping for the 5′ end of the p53R2 mRNA was performed as described previously (35). Primer sequences are available upon request.
Quantitative reverse transcriptase PCR (qRT-PCR)
qRT-PCR was performed as described previously (33). Primer sequences for p53R2 and GAPDH are reported in Supplemental Table 1, and PCR conditions are available upon request.
DNA methylation analyses
Global DNA methylation was assessed by liquid chromatography-mass spectrometry measurement of 5-methyldeoxycytidine (5mdC), and methylation of the LINE-1 repetitive element was determined by quantitative sodium bisulfite pyrosequencing as described previously (36, 37). Methylation-specific PCR (MSP), and sodium bisulfite sequencing were performed as described previously (33). Primers for p53R2 methylation analysis by MSP and sodium bisulfite sequencing are listed in Supplemental Table 1. Human genomic DNA modified with SssI (New England Biolabs, Beverly, MA) served as a methylated DNA control, and testis DNA (BioChain Institute, Hayward, CA) served as an unmethylated DNA control.
siRNA knockdowns
Transient siRNA knockdown of DNMT1 in RKO cells was performed as described previously (33). Transient knockdown of p53R2 in RKO cells was accomplished by transfecting cells once with 50 nM p53R2si (sc-36158, Santa Cruz Biotechnology, Santa Cruz, CA) or 50 nM controlsi #2 (Dharmacon, Chicago, IL) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
p53R2 promoter luciferase assays
A 572 bp fragment of the p53R2 gene, containing the intron 1 p53 binding site (27), was amplified from RKO genomic DNA using the following primers: F: 5′-ATTAAATGGCATGCCCAGGA-3′ and R: 5′-TGACACTGAGCCACTGGTCT-3′. After gel purification, the PCR product was cloned into the pcDNA 2.1 vector (Invitrogen, Calsbad, CA, USA), according to the manufacturer’s instructions. Resulting clones were digested with Xho I and Hind III (Fermentas, Hanover, MD, USA), and ligated into the pGL3-basic vector (Promega, Madison, WI), generating pGL3-R2. A previously described p53 binding site point mutation (27) was introduced into the construct using the Phusion Site-Directed Mutagenesis kit (New England Biolabs, Ipswich, MA, USA), generating pGL3-R2mut. The Dual Luciferase Reporter Assay (Promega) was performed according to the manufacturer’s instructions.
For drug treatment experiments, cells were treated with decitabine at day 0 and again at day 3. 24 hours later (day 4), cells were co-transfected with 50 ng of a Renilla construct (to normalize for transfection efficiency) and 150 ng of pGL3-R2 or pGL3-R2mut using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cell extracts were harvested for luciferase assays 24 hours later (day 5). For DNMT1 siRNA experiments, RKO cells were treated as described above except that DNMT1-specific or control siRNAs (33) were transfected at day 0 and again at day 3 post-treatment.
Cell cycle analyses
Triplicate wells of RKO cells in 6-well plates were transfected with either 50 nM sip53R2 or sicontrol as described above. 24 hours later, cells were treated with 1×PBS, 0.5 μM, or 2.0 μM decitabine. After three days, cells were trypsinized and 2 × 106 cells were washed in PBS. Next, 100% ice cold EtOH was added dropwise to the cell pellet while vortexing. After a 30-minute incubation on ice, cells were spun down, and the cell pellet was washed in 0.5 % BSA. Cells were then pelleted and resuspended in propidium iodide staining solution (0.1% sodium citrate, 0.02 mg/ml RNAse, 0.2 % NP40, 0.05 mg/ml propidium iodide, 1 N HCL, in deionized water). Cells were then incubated in the dark for 4 hours, passed through an 18 gauge needle, and analyzed using a FacScan instrument (Becton Dickinson, Franklin Lakes, NJ) using standard methods. Data were analyzed using ModFit LT (Verity Software House, Topsham, ME).
Results
Identification and characterization of p53R2 as a p53-dependent decitabine target gene
In a previous epigenomic reactivation microarray screen (9, 34), we identified p53R2 as a potential decitabine target gene in human cancer cells (data not shown). As p53R2 is a p53 target gene (26, 27), its induction in response to decitabine treatment was of interest based on observations that p53 status impacts cellular responses to pharmacologic or genetic inhibition of DNA methylation (15, 16, 22). To evaluate p53R2 as a decitabine target gene, we first measured p53R2 mRNA expression in human colorectal cancer cells following treatment with 0.5 or 5.0 μM decitabine, which are clinically relevant concentrations based on human pharmacokinetic studies (38, 39). Additionally, to examine the role of p53 in this response, we utilized cell lines with differing p53 status, including cells containing wildtype p53 (RKO, LOVO, and HCT116), cells containing mutant p53 (HT29), and RKO cells sustaining partial p53 disruption via the expression of a viral oncoprotein (RKOE6). Decitabine treatment led to a robust induction of p53R2 expression in LOVO and RKO cells, a lower level of induction in HCT116 and RKOE6, and no induction in HT29 (Fig. 1A). Thus, p53R2 induction by decitabine is drug concentration as well as p53 dependent. Very similar results were obtained when p53R2 protein expression was examined (Fig. 1B). Of the three wild-type p53 lines studied, p53R2 induction was least dramatic in HCT116 (Fig 1A and B). This may be due to a general impairment of p53R2 induction in this cell type as etoposide, a classical DNA damaging agent and p53 activator, robustly induces p53R2 expression in RKO but not HCT116 cells, despite activating p53 equally well in both cell types (Supplemental Figure 1). In addition, one allele of p53R2 is mutated in HCT116 (40), suggesting that impaired p53R2 function may have been selected for in the HCT116 tumor. Notably, activation of p53R2 by etoposide treatment in RKO cells suggested that decitabine-mediated p53R2 induction may not be a consequence of promoter DNA hypomethylation.
Fig. 1.
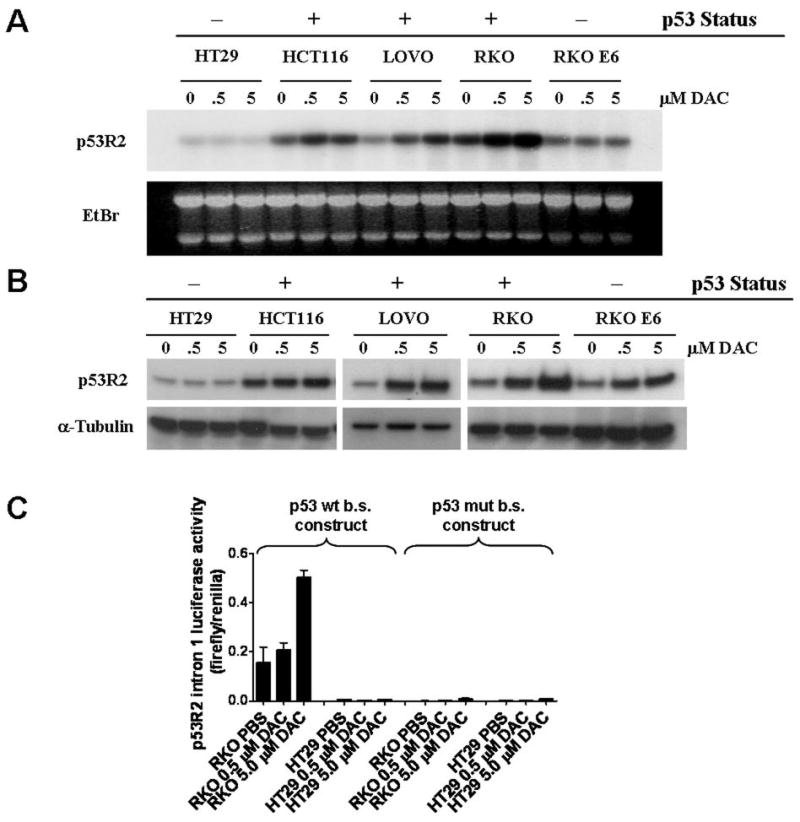
p53R2 is a p53-dependent decitabine gene target. A, p53R2 mRNA expression in cancer cell lines following decitabine treatment. p53R2 was measured by Northern blot analysis in the indicated colorectal cancer cell lines five days post decitabine treatment, using the indicated drug concentrations. The p53 status of each cell line is indicated and the cell lines are described in the text. Ethidium bromide staining of total RNA confirmed equivalent RNA loading. B, p53R2 protein expression in cancer cell lines following decitabine treatment. p53R2 was measured by Western blot analysis under the same experimental conditions described in A. C, p53-dependent p53R2 promoter activity in decitabine-treated cells. Left: The activity of a p53R2 luciferase reporter construct containing the intron 1 p53 binding site was determined before and after decitabine-treatment in RKO (p53 wildtype) and HT29 (p53 mutant) cells. Right: activity of the p53R2 intron 1 construct containing a mutated p53 binding site.
We hypothesized that decitabine-mediated p53R2 induction may be driven in part by transcriptional activation by p53. To test this, we cloned a region of the p53R2 intron 1, which contains the p53 binding site (27), into a luciferase reporter construct and measured its activity in decitabine treated RKO cells as well as p53 mutant HT29 cells (as a negative control). The activity of the reporter construct was induced by decitabine in a concentration-dependent fashion in RKO but not HT29 cells (Fig 1C). Furthermore, decitabine-mediated reporter activation in RKO cells is dependent on the presence of a wildtype p53 binding site (Fig. 1C). Taken together, these data suggest that decitabine-mediated activation of p53R2 involves direct transcriptional activation by p53.
The p53R2 5′ CpG island is hypomethylated
To further investigate the mechanism of p53R2 induction, we measured DNA methylation of the p53R2 gene. Examination of the p53R2 5′ region revealed that it contains a large and dense CpG island flanking the transcriptional start site (TSS) (Fig. 2A). RLM-RACE analysis revealed that the p53R2 TSS in human CRC cells is located within this 5′ CpG island and approximately 80 to 200 bp downstream of the NCBI-predicted TSS diagrammed in Fig 2A (data not shown). We initially utilized methylation-specific PCR (MSP) (41) to determine the methylation status of the p53R2 5′ CpG island in the cell lines described in Fig. 1. MSP revealed that two different regions of the p53R2 5′ CpG island, one upstream and one downstream of the TSS, are hypomethylated at baseline, before decitabine treatment, regardless of cellular p53 status (Fig. 2B). To confirm this finding, we performed sodium bisulfite sequencing of three different regions of the p53R2 5′ CpG island in HCT116, RKO, and LOVO cells. These analyses revealed that the p53R2 5′ CpG island region is fully hypomethylated at each region and in each cell type (Fig. 2C and Supplemental Figure 2). These data strongly suggest that decitabine-induced hypomethylation of the 5′ CpG island of p53R2 does not contribute to decitabine-mediated p53R2 gene activation in these cell lines. Also consistent with this idea, HCT116, LOVO, and RKO cells each express low levels of p53R2 in the absence of decitabine treatment (Fig. 1 A and B). We also note that there are no CpGs sites near the p53 binding site in intron 1 (the closest sites are 40 bp upstream and 128 bp downstream of the p53 binding site) (Fig. 2A and data not shown), suggesting that differential DNA methylation at the p53 binding site also does not account for the induction of p53R2 by decitabine.
Fig. 2.
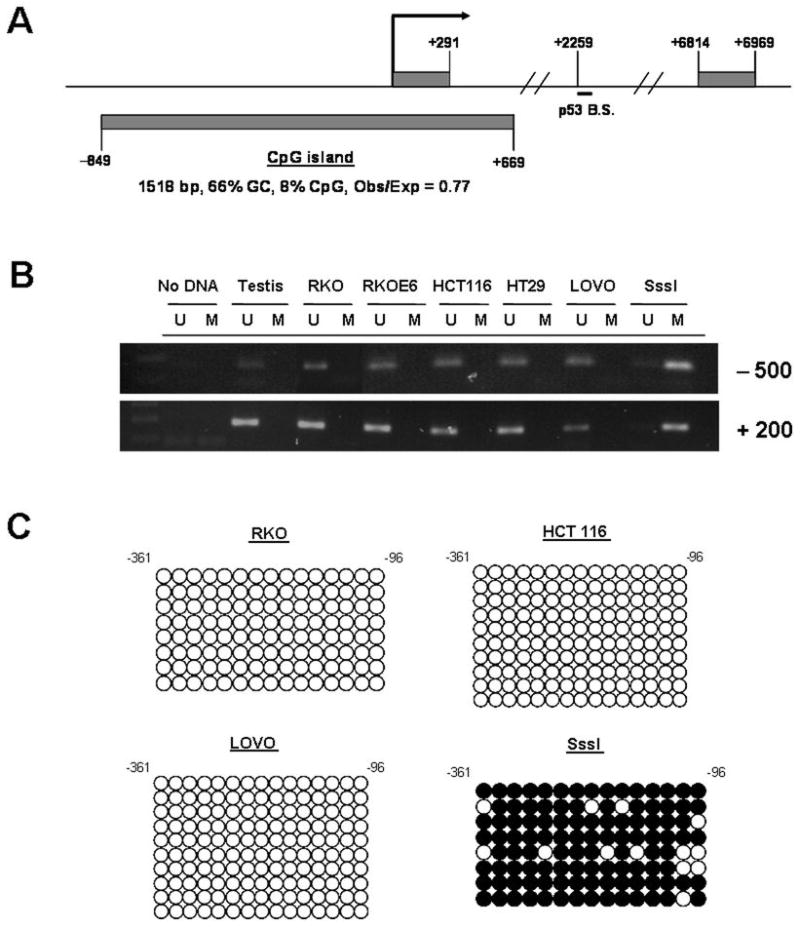
The p53R2 5′ CpG island is hypomethylated in colon cancer cells in the absence of decitabine treatment. A, Diagram of the 5′ region of the p53R2 gene, showing the CpG island (lower rectangle), intron 1, and exons 1 and 2 (upper rectangles). The position of the p53 binding site (p53 B.S.) in intron 1 is also shown. The bent arrow corresponds to the NCBI-predicted transcriptional start site (TSS). B, MSP analysis of two different regions of the p53R2 5′ CpG island. Testis DNA served as an unmethylated control, and SssI modified DNA served as a methylated control. Cell lines are the same as in Fig. 1A–B. C, Sodium bisulfite sequencing analysis of the central region of the p53R2 5′ CpG island in RKO, HCT116, and LOVO colorectal cancer cell lines. Open and filled circles represent unmethylated and methylated CpG sites, respectively, and rows correspond to individually sequenced clones. SssI modified DNA was analyzed as a positive control for DNA methylation. Numbers indicate nucleotide positions relative to the NCBI-predicted p53R2 TSS.
p53R2 expression is induced by nucleoside analog-based DNMT inhibitors but not by genetic targeting of DNMT1
We next used a genetic approach to investigate the mechanism of p53R2 induction by decitabine. We targeted DNMT1 using siRNA to test whether global genomic DNA hypomethylation, or hypomethylation of other loci caused by DNMT1 loss, leads to p53R2 activation in decitabine-treated cells. We efficiently knocked down DNMT1 expression in RKO cells using direct transfection of siRNAs as described previously (33) (Supplemental Fig. 3). DNMT1 knockdown in RKO cells leads to global genomic DNA hypomethylation, as assessed by analysis of genomic 5-methyldeoxycytidine (5mdC), to a similar extent as is seen following decitabine treatment (Fig. 3A). However, p53R2 mRNA expression was unaltered in RKO cells following DNMT1 knockdown (Fig. 3B), and p53R2 protein expression was similarly unaffected (Supplemental Fig. 3). In addition, unlike decitabine, DNMT1 knockdown did not induce p53R2 promoter activity (Fig. 3C). Taken together, these data suggest that genomic DNA hypomethylation does not induce p53R2, but rather that this effect is linked to a different characteristic of decitabine treatment. We speculate that p53R2 induction emanates from the formation of DNMT/DNA adducts (42, 43). Supporting this idea, the loss of DNMT1 from the soluble nuclear protein compartment of RKO cells following decitabine treatment (which is a surrogate measure of DNA adduct formation) parallels the drug concentrations at which p53R2 induction occurs (Supplemental Fig 4).
Fig. 3.
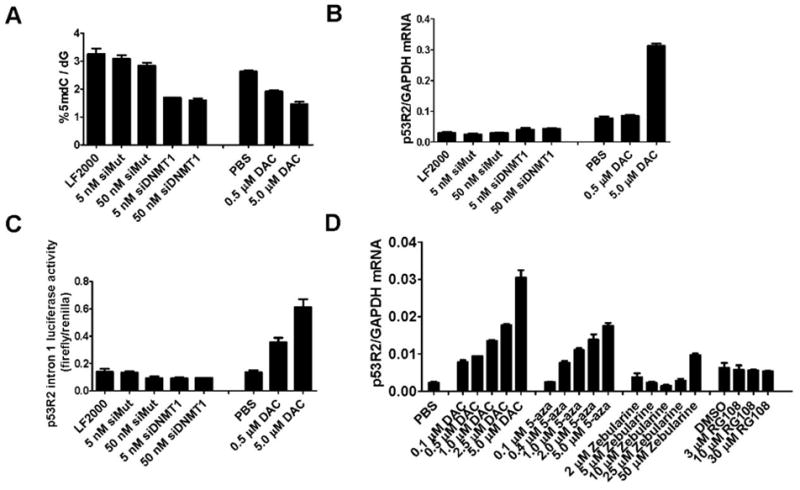
The effect of DNMT1 siRNA knockdown and treatment with other DNMT inhibitors on p53R2 expression in RKO cells. A, RKO cells were treated with the indicated concentrations of wildtype or mutant DNMT1 siRNA for five days. LF2000 indicates cells treated with only the transfection reagent. Alternatively, RKO cells treated with the indicated concentrations of decitabine (DAC) and harvested five days post treatment. DNA extracts were obtained, and genomic 5-methyldeoxycytidine (5mdC) levels were determined as described in Materials and Methods. B, p53R2 expression was measured by qRT-PCR under the same set of conditions described in A. C, The activity of the p53R2 intron 1 construct was measured under the same set of conditions described in A. D, p53R2 expression in RKO cells was measured by qRT-PCR five days post-treatment with the indicated concentrations of decitabine (DAC), 5-azacytidine (5-aza), zebularine, or RG108. RKO cells were treated with the indicated concentrations of drugs at day 0 and day 3 and cells were harvested at day 5 post treatment. PBS serves as the vehicle control for the 1st three drugs, while DMSO serves as the vehicle control for RG108.
Decitabine is one of three nucleoside analog-based DNMT inhibitors, each of which acts by trapping DNMT enzymes to DNA, following their incorporation into DNA (2). The other two compounds are 5-azacytidine, which is FDA-approved for treatment of MDS, and zebularine, which is in preclinical testing (2). In addition, a non-nucleoside and non-competitive inhibitor of DNMT1, RG108, has recently been described (44). To determine whether p53R2 induction is characteristic of treatment with these other DNMT inhibitors, we measured p53R2 expression in RKO cells following treatment with each of these drugs (Fig. 3D). Both decitabine and 5-azacytidine caused dose-dependent induction of p53R2 expression, while zebularine treatment induced p53R2 only at the highest drug concentration, consistent with the lower potency of this agent (25) (Fig. 3D). In contrast, RG108 did not induce p53R2 expression at any concentration (Fig. 3D). We did note a slight effect of RG108 on global 5mdC levels, but not LINE-1 methylation, suggesting that this agent is capable of inhibiting DNA methylation (Supplemental Fig. 5). Taken together, these data suggest that p53R2 induction is a cellular response to covalent DNMT/DNA adducts and not to genomic DNA hypomethylation per se.
siRNA knockdown of p53R2 reduces decitabine-mediated G1 arrest
Decitabine treatment induces growth arrest in cancer cell lines, characterized by G1 arrest and a reduction of cells in S phase (15, 45). To determine whether p53R2 is functionally involved in this response, we utilized siRNA to downregulate p53R2 expression in RKO cells responding to decitabine treatment. (Supplemental Fig. 6). We utilized flow cytometry in conjunction with propidium iodide staining to quantify the proportion of RKO cells in G1, S, and G2 following decitabine treatment. Notably, p53R2 knockdown led to a significant reduction in the proportion of cells in G1 and an increased proportion of cells in S and G2 following treatment of RKO cells with either 0.5 or 2.0 μM decitabine (Table 1). In contrast, p53R2 knockdown had little effect on the cell cycle profile of untreated RKO cells (Table 1). These data reveal that p53R2 induction functionally contributes to cell cycle arrest elicited by decitabine treatment, primarily through activation of the G1/S checkpoint.
Table 1.
Alteration of RKO cell cycle response to decitabine (DAC) following p53R2 knockdown
| Treatment1 | % of cells | ||
|---|---|---|---|
| G1 | S | G2 | |
| PBS, control siRNA2 | 39.5 ± 0.9 | 40.3 ± 1.0 | 20.2 ± 0.3 |
| PBS, p53R2 siRNA | 36.1 ± 1.7 | 40.9 ± 4.9 | 23.0 ± 3.2 |
| 0.5 μM DAC, control siRNA | 52.6 ± 1.2 | 27.3 ± 0.3 | 20.1 ± 1.0 |
| 0.5 μM DAC, p53R2 siRNA | 43.4 ± 0.9 | 31.2 ± 1.4 | 25.4 ± 0.5 |
| 2.0 μM DAC, control siRNA | 57.2 ± 1.1 | 17.9 ± 0.6 | 24.9 ± 1.2 |
| 2.0 μM DAC, p53R2 siRNA | 46.0 ± 1.7 | 22.3 ± 1.2 | 31.7 ± 0.9 |
Cells were treated with siRNA for 24 hours, and then treated with DAC. Three days later, cells were harvested for flow cytometry analysis as described in the Materials and Methods.
All siRNA treatments (control and p53R2 siRNA) utilized 50nM concentrations of siRNA.
p53R2 expression is induced by decitabine therapy in vivo and correlates with clinical response in MDS/AML
To determine whether p53R2 induction is a characteristic of decitabine therapy in vivo, we focused our attention on MDS and AML, diseases for which decitabine is in current clinical use (6). We obtained bone marrow samples from MDS (n=10), AML (n=4), and CMML (n=1) patients before and after a variable number of courses of decitabine treatment. Fig. 4A shows the results of p53R2 expression analysis in 13 patients from whom sufficient RNA was obtained for analysis. Notably, seven patients (54%) showed significant induction of p53R2 (ranging from 4- to 38-fold) at a minimum of one time point following drug treatment (Fig. 4A). Overall, there was good concordance of p53R2 induction levels at different treatment times within individual patients, with the exception of patient #1, who showed a high level of p53R2 induction only after the fourth cycle of therapy (Fig. 4A). Considering all data points, 13/28 (46%) of post-treatment samples displayed > 4-fold p53R2 induction, and the difference in p53R2 expression pre- and post-treatment over the entire sample group was statistically significant (Supplemental Fig. 7A). We measured p53R2 protein expression by Western blot analyses in the nine patients whose samples yielded sufficient protein for analysis (Fig. 4B). Six of nine patients (67%) analyzed showed significantly increased p53R2 protein expression (> 1.5-fold induction) at one or more time point post-drug treatment (Fig. 4B). As with p53R2 mRNA induction, the level of p53R2 protein induction in patient samples was in many instances similar to or greater than that observed in decitabine-treated RKO cells (Fig. 4B). Interestingly, a number of post-treatment samples displayed decreased migration of p53R2 protein coincident with its induction (Fig. 4B), suggesting that the induced protein could have altered post-translational modifications, or that a novel form of p53R2 protein is induced. We determined the methylation status of the central region of the 5′ CpG island of p53R2 in pre- and post-treatment samples from two patients showing robust p53R2 induction (pt. 8, 8c; pt. 14, 8c) (Supplemental Fig 7B). As shown, p53R2 was hypomethylated both before and after decitabine treatment, suggesting that, similar to what was observed in vitro, hypomethylation does not account for p53R2 induction in vivo. In addition, the 3′ region of the 5′ CpG island of p53R2 was also hypomethylated in these clinical samples (data not shown).
Fig. 4.
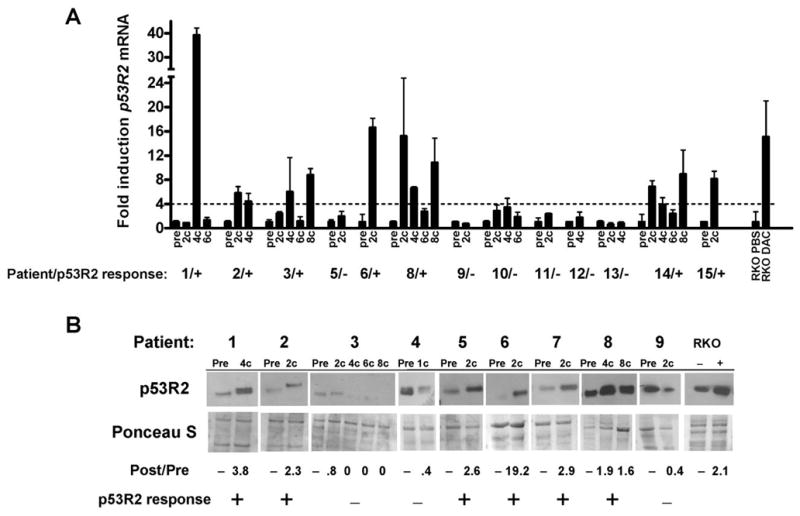
Decitabine treatment induces p53R2 mRNA and protein expression in MDS/AML patient bone marrow. A, p53R2 expression in bone marrow samples obtained from 13 MDS/AML patients undergoing decitabine therapy. Pre- and post-treatment samples are shown, and the number of cycles of therapy (#c) is listed for post-treatment samples. p53R2 expression was measured by qRT-PCR. For each patient, p53R2 expression was normalized to GAPDH, and pre-treatment samples were each set at one to normalize between patients. Plus signs (+) indicate patients in whom at least one post-treatment sample showed biologically significant (defined as > 4 fold) p53R2 induction compared to the pre-treatment sample. Dashes (−) indicate patients for whom no post-treatment sample showed significant p53R2 induction. p53R2 expression in RKO cells treated with decitabine (DAC) for five days is shown as a positive control. Note that patients #4 and #7 did not yield sufficient quality RNA for analysis and are not shown. B, p53R2 Western blot analysis of cytosolic protein extracts harvested from bone marrow samples from nine MDS/AML patients undergoing decitabine therapy. Pre- and post-treatment samples are shown, and the number of cycles of therapy (#c) is listed for post-treatment samples. Fold induction (or decline) relative to the pretreatment sample is shown beneath the Western blot. Plus signs (+) indicate patients in whom the post-treatment samples showed biologically significant (defined as > 1.5 fold) p53R2 induction. Dashes (−) indicate patients in whom p53R2 induction was not observed. RKO cells treated with decitabine (DAC) for five days are shown as a positive control. Ponceau S staining confirmed equivalent protein loading. Note that patients #10–15 did not yield sufficient quality protein for analysis and are not shown.
Finally, we investigated the potential relationship between clinical response and p53R2 induction in the 15 MDS/AML patients under study. Clinical responses included marrow complete remission and partial remission in one patient each and stable disease in six patients (Table 2). Strikingly, 7/8 patients (88%) showing a clinical response displayed p53R2 mRNA induction, while 0/5 (0%) non-responding patients displayed p53R2 mRNA induction, a difference that was highly significant (Fisher’s exact test, P = 0.0047) (Table 2). In addition, 4/5 (80%) patients showing a clinical response displayed p53R2 protein induction, while 2/4 (50%) non-responding patients displayed p53R2 protein induction, which was not significant (Fisher’s exact test, P = 0.5238). Taken together, these data reveal that p53R2 mRNA induction is associated with clinical responses to decitabine.
Table 2.
Clinical response data and correlation with p53R2 molecular response
| Patient | Age/Sex | Diagnosis | Clinical Response | p53R2 mRNA induction1 | p53R2 protein induction2 |
|---|---|---|---|---|---|
| 1 | 83F | MDS | Stable disease | Yes | Yes |
| 2 | 82M | MDS | Stable disease | Yes | Yes |
| 3 | 73M | MDS | Stable disease | Yes | No |
| 4 | 77F | MDS | Failure | n/d | No |
| 5 | 65M | MDS | Failure | No | Yes |
| 6 | 70M | MDS | Stable disease | Yes | Yes |
| 7 | 79M | AML | Failure | n/d | Yes |
| 8 | 70M | MDS | Partial remission | Yes | Yes |
| 9 | 69F | AML | Failure | No | No |
| 10 | 72F | MDS | Failure | No | n/d |
| 11 | 75M | MDS | Failure | No | n/d |
| 12 | 68M | AML | Failure | No | n/d |
| 13 | 70M | MDS | Stable disease | No | n/d |
| 14 | 72F | AML | Marrow complete remission | Yes | n/d |
| 15 | 47F | CMML | Stable disease | Yes | n/d |
| Correlation with Clinical Response (Fisher’s exact test) | P=0.0047 | P=0.5238 | |||
4-fold or greater p53R2 induction in bone marrow cells over pre-treatment at ≥1 post-treatment time point
1.5-fold or greater p53R2 induction in bone marrow cells over pre-treatment at ≥1 post-treatment time point
n/d = no data
Discussion
We have identified and characterized p53R2 as a novel decitabine gene target in human cancer. p53R2 activation links decitabine activity to p53-mediated DNA damage responses, as its induction is p53-dependent. This distinguishes p53R2 from p21/CDKN1A, which we have previously shown is induced by decitabine treatment by both p53-dependent and p53-independent mechanisms (15). p53R2 induction may thus be an appropriate pharmacodynamic marker for p53 activation by decitabine in vivo. In MDS, p53 mutations are infrequent, ranging from 6% to 14% in adult MDS patients (17–19). Thus, activation of p53R2 and other p53 target genes may be important in mediating therapeutic responses in this setting, consistent with the data presented here. This hypothesis is also supported by our finding that p53R2 contributes to growth arrest in vitro in tumor cells responding to decitabine treatment. Given the complex nature of cellular gene expression changes following decitabine treatment (34), it is remarkable that specific knockdown of p53R2 significantly alters decitabine-mediated growth arrest, and suggests that this protein may play a key role in decitabine responses in tumors in which wild-type p53 is retained.
Decitabine treatment induces p53R2 expression in vivo and its induction correlates with clinical response in MDS/AML. It is unclear whether p53R2 directly contributes to the therapeutic effect of decitabine, or alternatively whether its induction is a surrogate marker for p53 activation, which may contribute to the therapeutic response by a diverse set of mechanisms. Our observation that p53R2 mRNA but not p53R2 protein induction correlates with clinical response seems to support the latter model; however, analysis of a greater number of patient samples is required to adequately address this question. If p53R2 does contribute to therapeutic responses, we hypothesize that it may do so by promoting tumor cell differentiation, a long-established effect of DNMT inhibitors (46). p53R2, by catalyzing dNTP formation required for DNA repair in growth arrested cells, may allow for enhanced cell survival in the face of genomic DNA hypomethylation. This could in turn promote cellular differentiation by allowing the re-expression of epigenetically silenced genes in surviving hypomethylated cells.
p53R2 is not induced by genomic DNA hypomethylation mediated by genetic knockdown of DNMT1; furthermore, the 5′ CpG island of p53R2 does not display methylation changes during its activation by decitabine either in cell lines or in vivo. Moreover, etoposide treatment induces p53R2, suggesting that p53R2 is not silenced by DNA methylation in these cancer cell lines, even at potentially cryptic regulatory sites. Taken together, these data unlink DNA hypomethylation from p53R2 induction. Instead, we hypothesize that covalent adduct formation between decitabine-incorporated DNA molecules and DNMT enzymes leads to p53R2 induction. Supporting this model, dose response experiments revealed a tight correlation between the loss of soluble DNMT1 from nuclear extracts and the induction of p53R2 following decitabine treatment. In addition, two other DNMT inhibitors that form covalent adducts, 5-azacytidine and zebularine, also induce p53R2 expression while RG108, which does not form covalent adducts, does not. Despite these data, it is possible that p53R2 is silenced by DNA hypermethylation in certain tumors, although this has yet to been reported. More generally, the fact that p53R2 induction in vivo appears to be independent of promoter DNA hypomethylation emphasizes the role of hypomethylation-independent gene activation in the activity of decitabine in myeloid malignancies. Additional support for the role of hypomethylation-independent effects comes from an earlier study that found that the majority of decitabine-activated genes in MDS/AML do not show induced promoter DNA hypomethylation following drug treatment (47).
p53R2 is a homolog of the R2 subunit of ribonucleotide reductase that pairs with the catalytic R1 subunit to form a competent enzyme complex in non-proliferative cells (48). In addition to its function in nuclear DNA repair, p53R2 has recently been demonstrated to provide dNTPs for mtDNA replication (49, 50). Interestingly, p53R2 gene mutations were identified in individuals with severe mtDNA depletion in muscle (49). Based on these data, it has been suggested that the main function of p53R2 may not be to mediate nuclear DNA repair but rather for mtDNA synthesis (48). Thus it is of interest to determine whether decitabine treatment alters mtDNA function and whether this could impact cellular responses to DNMT inhibitors.
The original reports on p53R2 used overexpression studies to show that p53R2 induced cell cycle arrest via a G2 phase block (26, 27). In agreement, our overexpression studies indicate that p53R2 induces G2 arrest in RKO cells (data not shown). However, our siRNA knockdown studies revealed that p53R2 primarily contributes to decitabine-mediated growth arrest by mediating a G1 phase block. Consistent with this observation, it was recently shown that p53R2 contributes to G1 arrest in UV irradiated cells by disassociating from a direct interaction with p21 and facilitating the accumulation of nuclear p21 (51). It is currently unknown whether p53R2/p21 interactions are involved in cellular growth arrest in response to decitabine; however, it is clear from our data that overexpression studies may not accurately predict the functional role of endogenous p53R2 in mediating cell growth arrest.
In summary, we report that p53R2, a ribonucleotide reductase gene linked to p53-dependent DNA repair, is a robust gene target of decitabine therapy in vitro and in vivo. In addition, we show that p53R2 functionally contributes to cellular responses to decitabine and that its induction directly correlates with clinical responses in MDS/AML. These data open up a number of new lines of investigation relevant for understanding the molecular pharmacology of decitabine and other epigenetic therapies as treatments for human cancer.
Supplementary Material
Acknowledgments
We thank Ted Ririe and Dr. Pamela Cassidy of the Huntsman Cancer Institute and Stacy Fricke, Eun Ju-Lee, Anna Woloszynska-Read, and Wa Zhang of RPCI for contributions to this project. We also thank Laurie A. Ford of RPCI for expert clinical data management, and Dr. Jihnhee Yu of RPCI for assistance with statistical analysis.
Financial Support: PF-99-151-01-CDD, American Cancer Society (ARK), NIH grants CA11674 (ARK), CA73992 (DAJ), and CA16056 (Roswell Park Cancer Institute).
Abbreviations
- MDS
myelodysplastic syndrome
- AML
acute myelogenous leukemia
- CMML
chronic myelomonocytic leukemia
- p53R2/RRM2B
p53-inducible ribonucleotide reductase
- DNMT
DNA methyltransferase
- mtDNA
mitochondrial DNA
- dNTPs
deoxyribonucleotides
- DAC
decitabine, 5-aza-2′-deoxycytidine
- TSS
transcriptional start site
- CRC
colorectal cancer
References
- 1.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 3.Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002;21(35):5496–503. doi: 10.1038/sj.onc.1205602. [DOI] [PubMed] [Google Scholar]
- 4.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–95. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 5.Momparler RL, Rivard GE, Gyger M. Clinical trial on 5-aza-2′-deoxycytidine in patients with acute leukemia. Pharmacology & Therapeutics. 1985;30(3):277–86. doi: 10.1016/0163-7258(85)90052-x. [DOI] [PubMed] [Google Scholar]
- 6.Plimack ER, Kantarjian HM, Issa JP. Decitabine and its role in the treatment of hematopoietic malignancies. Leukemia & Lymphoma. 2007;48(8):1472–81. doi: 10.1080/10428190701471981. [DOI] [PubMed] [Google Scholar]
- 7.Kaminskas E, Farrell A, Abraham S, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clinical Cancer Research. 2005;11(10):3604–8. doi: 10.1158/1078-0432.CCR-04-2135. [DOI] [PubMed] [Google Scholar]
- 8.Oki Y, Aoki E, Issa JP. Decitabine--bedside to bench. Critical Reviews in Oncology-Hematology. 2007;61(2):140–52. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Karpf AR. Epigenomic reactivation screening to identify genes silenced by DNA hypermethylation in human cancer. Current opinion in molecular therapeutics. 2007;9(3):231–41. [PubMed] [Google Scholar]
- 10.Daskalakis M, Nguyen TT, Nguyen C, et al. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. [see comment] Blood. 2002;100(8):2957–64. doi: 10.1182/blood.V100.8.2957. [DOI] [PubMed] [Google Scholar]
- 11.Issa JP, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103(5):1635–40. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 12.Karpf AR. A potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy. Epigenetics. 2006;1(3):116–20. doi: 10.4161/epi.1.3.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigalotti L, Altomonte M, Colizzi F, et al. 5-Aza-2′-deoxycytidine (decitabine) treatment of hematopoietic malignancies: a multimechanism therapeutic approach? Blood. 2003;101(11):4644–6. doi: 10.1182/blood-2002-11-3458. discussion 5–6. [DOI] [PubMed] [Google Scholar]
- 14.Maio M, Coral S, Fratta E, Altomonte M, Sigalotti L. Epigenetic targets for immune intervention in human malignancies. Oncogene. 2003;22(42):6484–8. doi: 10.1038/sj.onc.1206956. [DOI] [PubMed] [Google Scholar]
- 15.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Molecular Pharmacology. 2001;59(4):751–7. [PubMed] [Google Scholar]
- 16.Zhu WG, Hileman T, Ke Y, et al. 5-aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. Journal of Biological Chemistry. 2004;279(15):15161–6. doi: 10.1074/jbc.M311703200. [DOI] [PubMed] [Google Scholar]
- 17.Jekic B, Novakovic I, Lukovic L, et al. Lack of TP53 and FMS gene mutations in children with myelodysplastic syndrome. Cancer Genet Cytogenet. 2006;166(2):163–5. doi: 10.1016/j.cancergencyto.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Misawa S, Horiike S. TP53 mutations in myelodysplastic syndrome. Leuk Lymphoma. 1996;23(5–6):417–22. doi: 10.3109/10428199609054848. [DOI] [PubMed] [Google Scholar]
- 19.Sugimoto K, Hirano N, Toyoshima H, et al. Mutations of the p53 gene in myelodysplastic syndrome (MDS) and MDS-derived leukemia. Blood. 1993;81(11):3022–6. [PubMed] [Google Scholar]
- 20.Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 21.Schneider-Stock R, Diab-Assef M, Rohrbeck A, et al. 5-Aza-cytidine is a potent inhibitor of DNA methyltransferase 3a and induces apoptosis in HCT-116 colon cancer cells via Gadd45- and p53-dependent mechanisms. The Journal of pharmacology and experimental therapeutics. 2005;312(2):525–36. doi: 10.1124/jpet.104.074195. [DOI] [PubMed] [Google Scholar]
- 22.Jackson-Grusby L, Beard C, Possemato R, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. [see comment] Nature Genetics. 2001;27(1):31–9. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 23.Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994;91(25):11797–801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Zhao Y, Li L, et al. An ATM- and Rad3-related (ATR) signaling pathway and a phosphorylation-acetylation cascade are involved in activation of p53/p21Waf1/Cip1 in response to 5-aza-2′-deoxycytidine treatment. J Biol Chem. 2008;283(5):2564–74. doi: 10.1074/jbc.M702454200. [DOI] [PubMed] [Google Scholar]
- 25.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28(2):752–71. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano K, Balint E, Ashcroft M, Vousden KH. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19(37):4283–9. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. [see comment] Nature. 2000;404(6773):42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 28.Jaffee ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001. World Health Organization Classification of Tumors. [Google Scholar]
- 29.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 30.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–7. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 31.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419–25. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 32.Karpf AR, Peterson PW, Rawlins JT, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci U S A. 1999;96(24):14007–12. doi: 10.1073/pnas.96.24.14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James SR, Link PA, Karpf AR. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene. 2006;25(52):6975–85. doi: 10.1038/sj.onc.1209678. [DOI] [PubMed] [Google Scholar]
- 34.Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Mol Pharmacol. 2004;65(1):18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- 35.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, Karpf AR. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 36.Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77(2):504–10. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- 37.Woloszynska-Read A, Mhawech-Fauceglia P, Yu J, Odunsi K, Karpf AR. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin Cancer Res. 2008;14(11):3283–90. doi: 10.1158/1078-0432.CCR-07-5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aparicio A, Eads CA, Leong LA, et al. Phase I trial of continuous infusion 5-aza-2′-deoxycytidine. Cancer Chemother Pharmacol. 2003;51(3):231–9. doi: 10.1007/s00280-002-0563-y. [DOI] [PubMed] [Google Scholar]
- 39.van Groeningen CJ, Leyva A, O’Brien AM, Gall HE, Pinedo HM. Phase I and pharmacokinetic study of 5-aza-2′-deoxycytidine (NSC 127716) in cancer patients. Cancer Res. 1986;46(9):4831–6. [PubMed] [Google Scholar]
- 40.Yamaguchi T, Matsuda K, Sagiya Y, et al. p53R2-dependent pathway for DNA synthesis in a p53-regulated cell cycle checkpoint. Cancer Res. 2001;61(22):8256–62. [PubMed] [Google Scholar]
- 41.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christman JK, Schneiderman N, Acs G. Formation of highly stable complexes between 5-azacytosine-substituted DNA and specific non-histone nuclear proteins. Implications for 5-azacytidine-mediated effects on DNA methylation and gene expression. Journal of Biological Chemistry. 1985;260(7):4059–68. [PubMed] [Google Scholar]
- 43.Ferguson AT, Vertino PM, Spitzner JR, Baylin SB, Muller MT, Davidson NE. Role of estrogen receptor gene demethylation and DNA methyltransferase. DNA adduct formation in 5-aza-2′deoxycytidine-induced cytotoxicity in human breast cancer cells. Journal of Biological Chemistry. 1997;272(51):32260–6. doi: 10.1074/jbc.272.51.32260. [DOI] [PubMed] [Google Scholar]
- 44.Brueckner B, Boy RG, Siedlecki P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65(14):6305–11. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- 45.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58(1):95–101. [PubMed] [Google Scholar]
- 46.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 47.Schmelz K, Sattler N, Wagner M, Lubbert M, Dorken B, Tamm I. Induction of gene expression by 5-Aza-2′-deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia. 2005;19(1):103–11. doi: 10.1038/sj.leu.2403552. [DOI] [PubMed] [Google Scholar]
- 48.Thelander L. Ribonucleotide reductase and mitochondrial DNA synthesis. Nat Genet. 2007;39(6):703–4. doi: 10.1038/ng0607-703. [DOI] [PubMed] [Google Scholar]
- 49.Bourdon A, Minai L, Serre V, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39(6):776–80. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 50.Pontarin G, Ferraro P, Hakansson P, Thelander L, Reichard P, Bianchi V. p53R2-dependent ribonucleotide reduction provides deoxyribonucleotides in quiescent human fibroblasts in the absence of induced DNA damage. J Biol Chem. 2007;282(23):16820–8. doi: 10.1074/jbc.M701310200. [DOI] [PubMed] [Google Scholar]
- 51.Xue L, Zhou B, Liu X, et al. Ribonucleotide reductase small subunit p53R2 facilitates p21 induction of G1 arrest under UV irradiation. Cancer Res. 2007;67(1):16–21. doi: 10.1158/0008-5472.CAN-06-3200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.