

Abstract
Background and purpose:
The α1L-adrenoceptor has pharmacological properties that distinguish it from three classical α1-adrenoceptors (α1A, α1B and α1D). The purpose of this was to identify α1L-adrenoceptors in mice and to examine their relationship to classical α1-adrenoceptors.
Experimental approach:
Radioligand binding and functional bioassay experiments were performed on the cerebral cortex, vas deferens and prostate of wild-type (WT) and α1A-, α1B- and α1D-adrenoceptor gene knockout (AKO, BKO and DKO) mice.
Key results:
The radioligand [3H]-silodosin bound to intact segments of the cerebral cortex, vas deferens and prostate of WT, BKO and DKO but not of AKO mice. The binding sites were composed of two components with high and low affinities for prazosin or RS-17053, indicating the pharmacological profiles of α1A-adrenoceptors and α1L-adrenoceptors. In membrane preparations of WT mouse cortex, however, [3H]-silodosin bound to a single population of prazosin high-affinity sites, suggesting the presence of α1A-adrenoceptors alone. In contrast, [3H]-prazosin bound to two components having α1A-adrenoceptor and α1B-adrenoceptor profiles in intact segments of WT and DKO mouse cortices, but AKO mice lacked α1A-adrenoceptor profiles and BKO mice lacked α1B-adrenoceptor profiles. Noradrenaline produced contractions through α1L-adrenoceptors with low affinity for prazosin in the vas deferens and prostate of WT, BKO and DKO mice. However, the contractions were abolished or markedly attenuated in AKO mice.
Conclusions and implications:
α1L-Adrenoceptors were identified as binding and functional entities in WT, BKO and DKO mice but not in AKO mice, suggesting that the α1L-adrenoceptor is one phenotype derived from the α1A-adrenoceptor gene.
Keywords: α1L-adrenoceptor, α1A-adrenoceptor, vas deferens, prostate, cerebral cortex, knockout mouse
Introduction
In the α1-adrenoceptors, three distinct subtypes (α1A-, α1B- and α1D-adrenoceptors; nomenclature follows Alexander et al., 2008) have been cloned and are known to be involved in various physiological functions (Lomasney et al., 1991; Hieble et al., 1995; Zhong and Minneman, 1999; Michelotti et al., 2000). The three classical α1-adrenoceptors have high (subnanomolar) affinity for prazosin, a prototypic, selective α1-adrenoceptor antagonist, but show distinct pharmacological profiles for several antagonists. For example, silodosin, 5-methylurapidil and RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-α,α-dimethyl-1H-indole-3-ethamine hydrochloride) are selective for α1A-adrenoceptors, and BMY-7378 (8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4,5]decane-7,9-dione-dihydrochloride) is selective for α1D-adrenoceptors (Lomasney et al., 1991; Hieble et al., 1995; Ford et al., 1996; Murata et al., 1999; Piao et al., 2000). Tamsulosin is a potent antagonist for the three α1-adrenoceptor subtypes or shows slightly lower affinity for the α1B subtype than for the other two subtypes (Ford et al., 1996; Morishima et al., 2008).
In addition to these classical α1-adrenoceptors, the presence of another subtype (the putative α1L-adrenoceptor) has been proposed (Flavahan and Vanhoutte, 1986; Muramatsu et al., 1990; Ford et al., 1996). The α1L-adrenoceptor shows a unique pharmacological profile: low affinity for prazosin, 5-methylurapidil, RS-17053 and BMY 7378, but high affinity for silodosin and tamsulosin (Muramatsu et al., 1995, 1998; Ford et al., 1996; Murata et al., 1999). The α1L-adrenoceptors have been identified primarily by functional studies: rabbit thoracic aorta (Oshita et al., 1993), rabbit iris (Nakamura et al., 1999; Suzuki et al., 2002), rat small mesenteric arteries (Stam et al., 1999), canine subcutaneous arteries (Argyle and McGrath, 2000), rat and human vas deferens (Ohmura et al., 1992; Amobi et al., 2002), and rabbit, rat, mouse and human prostate (Ford et al., 1996; Van der Graaf et al., 1997; Hiraoka et al., 1999; Gray and Ventura, 2006; Morishima et al., 2007a; Su et al., 2008). However, the gene corresponding to the α1L-adrenoceptor has not yet been cloned, even though many trials of candidate genes have been carried out, including splicing variants of α1-adrenoceptor genes and heterodimeric expression of different subtypes (Ramsay et al., 2004). Subsequently, it has been considered that the α1L-adrenoceptor may be a functional phenotype of the α1A-adrenoceptor because the functional studies with recombinant α1A-adrenoceptors have revealed a relatively low affinity for prazosin (Ford et al., 1997; Daniels et al., 1999).
Recently, we have demonstrated in radioligand binding studies that α1L-adrenoceptors occur as a distinct entity from α1A-, α1B- and α1D-adrenoceptors (Hiraizumi-Hiraoka et al., 2004; Morishima et al., 2007a, 2008). However, the α1L-adrenoceptors were detected only under intact tissue conditions and the pharmacological profile converted to that of the α1A-adrenoceptor after homogenization (Morishima et al., 2008; Su et al., 2008). This line of evidence strongly suggests that the α1L-adrenoceptor may be a phenotypic subtype of α1-adrenoceptors (probably the α1A-adrenoceptor) rather than a genetically different subtype (Su et al., 2008).
The aims of this study were to identify α1L-adrenoceptors in mice by radioligand binding and bioassay approaches and to examine the effects of gene knockout of the classical α1-adrenoceptor subtypes. The present results show that binding and functional profiles of α1L-adrenoceptors can be clearly detected in wild-type (WT) mice, but that the α1L-adrenoceptor is selectively abolished in α1A-adrenoceptor gene knockout (AKO) mice. A part of this study was presented in the Satellite Meetings (Pharmacology of Adrenoceptors, Beijing, China, 2006) of the 15th International Congress of Pharmacology and the 80th Annual Meeting of The Japanese Pharmacological Society (Morishima et al., 2007b).
Materials and methods
Animals
All animal procedures and this study were performed according to the Guidelines for Animal Experiments, University of Fukui and University of California.
AKO (Rokosh and Simpson, 2002), α1B-adrenoceptor gene knockout (BKO) (Cavalli et al., 1997), α1D-adrenoceptor gene knockout (DKO) (Sadalge et al., 2003) and WT mice were all of the C57Bl/6J background. Mice of both sexes were used, aged 1–6 months (mean 3). Mice were housed under a 12-h light-dark cycle and had free access to standard laboratory food and tap water. The room temperature and relative humidity were regulated at 22 °C and 40–70%, respectively.
Mice were anaesthetized with isoflurane or halothane and killed by cervical dislocation or removal of the heart under deep anaesthesia. The brain cortex, vas deferens and prostate were rapidly excised and placed in a modified Krebs–Henseleit solution (120.7 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.0 mM CaCl2, 1.2 mM NaH2PO4, 25.5 mM NaHCO3 and 11.5 mM D-glucose). The modified Krebs–Henseleit solution was aerated with a mixture of 95% O2 and 5% CO2 and maintained at 4 °C (pH 7.4).
Tissue segment binding experiments with mouse cerebral cortex, vas deferens and prostate
Tissue segment binding experiments with mouse cerebral cortex, vas deferens and prostate were performed as described earlier (Muramatsu et al., 2005; Morishima et al., 2008). The cerebral cortex was cleaned from the pia mater and substantia alba, and cut into small pieces (approximately 2 × 2 × 1 mm). The whole vas deferens was cleared of connective tissue and the epithelium, and then the muscle layer was cut into small pieces (approximately 3 mm long and 1–2 mm wide, 16–20 segments from one mouse). The prostate was cleared of connective tissue and separated into four to five fragments (approximately 1–2 mm long and 1 mm wide) without damaging the lobules. Each segment of cerebral cortex, vas deferens and prostate was incubated with [3H]-silodosin for 16 h at 4 °C in 1 mL of a Krebs incubation buffer containing 135.7 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.0 mM CaCl2, 1.2 mM NaH2PO4, 10.5 mM NaHCO3 and 11.5 mM D-glucose (pH 7.4). On samples of the cerebral cortex, [3H]-prazosin was also used. In binding saturation experiments, [3H]-silodosin or [3H]-prazosin at concentrations 50–1000 pM were used. Binding competition experiments were performed with [3H]-silodosin and [3H]-prazosin at 500 pM. After incubation, the pieces were gently washed and then solubilized in 1 mL of 0.3 M NaOH solution to estimate the radioactivity and protein content. The specific binding was determined by subtracting the nonspecific binding estimated in the presence of 30 μM phentolamine from the total radioactivity bound per milligram protein. Experiments with the cerebral cortex were carried out in duplicate at each radioligand concentration for saturation experiments or with a competing ligand for competition binding experiments. The abundance of α1-adrenoceptors in mouse cerebral cortex was represented as maximum binding capacity per milligram of total tissue protein (fmol per mg of total tissue protein). In the case of vas deferens, each segment was used for a single concentration of [3H]-silodosin in saturation experiments or with a competing ligand for competition binding experiments. In the experiments with prostate segments, binding of only 500 pM [3H]-silodosin was examined because of the small number of segments.
Membrane binding experiments with mouse cerebral cortex
Mouse cortex was minced with scissors and homogenized in 40 volumes (v/w) of Krebs incubation buffer containing proteinase inhibitors (Complete, EDTA-free tablet, Roche, Penzberg, Germany) using a Polytron homogenizer at 4 °C. The tissue homogenate was subjected to centrifugation at 1000 g for 10 min at 4 °C, and then the supernatant was centrifuged at 80 000 g for 30 min at 4 °C. The resulting pellet was resuspended in ice-cold Krebs incubation buffer without proteinase inhibitors and used as the crude membrane preparations for conventional membrane binding experiments. The incubation was carried out for 4 h at 4 °C in 1 mL Krebs incubation buffer. For saturation binding experiments, 30–1000 pM [3H]-silodosin was used. For competition binding experiments, the membranes were incubated with 500 pM [3H]-silodosin in the absence or presence of unlabelled competing ligands. Nonspecific binding of [3H]-silodosin was defined as the binding in the presence of 30 μM phentolamine. Experiments were carried out in duplicate. The protein contents of the homogenates before centrifugation and of the crude membrane fractions were measured using a Bio-Rad Protein Assay kit (Bio-Rad Japan, Tokyo, Japan). The abundance of α1-adrenoceptors in the crude membranes was represented as maximum binding capacity per milligram of total tissue protein (fmol per mg of total tissue protein), but not of crude membrane protein after centrifugation.
Functional studies with mouse vas deferens and prostate
Functional studies were performed as described earlier (Ohmura et al., 1992). Briefly, mouse vas deferens (epididymal portion) and prostate strips were placed at 37 °C in organ baths containing a modified Krebs–Henseleit solution composed of 120.7 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.0 mM CaCl2, 1.2 mM NaH2PO4, 25.5 mM NaHCO3 and 11.5 mM D-glucose. Noradrenaline was applied non-cumulatively to the vas deferens and cumulatively to the prostate, and the isometric tension changes were recorded through a force transducer. Desipramine (0.3 μM), deoxycorticosterone acetate (5 μM) and propranolol (1 μM) were added to inhibit neural and extraneural reuptake of noradrenaline and to block β-adrenoceptors, as described elsewhere (Muramatsu et al., 1995). Antagonists were used 60 min before and during the evaluation of contractile responses to noradrenaline. The tissue responsiveness to α,β-methylene ATP was also examined. The concentrations of α,β-methylene ATP were 1 μM in vas deferens, which produced roughly the same amplitude of contraction as 100 μM noradrenaline, and 10 μM in prostate, which corresponded to the maximum concentration.
Data analysis
Data are presented as the mean±s.e.mean with the number of experiments or mice. Data from WT, AKO, BKO and DKO mice were statistically analysed by ANOVA followed by Scheffe's post hoc test.
Binding data in saturation and competition experiments were analysed using PRISM software (version 3, GraphPad, San Diego, CA, USA). The number of α1-adrenoceptors was presented as the maximal binding capacity per milligram of total tissue protein (Bmax: fmol per mg of total tissue protein). That is, in the case of conventional binding experiments with membrane fractions of cerebral cortex, the proteins in the homogenates before fractionation were measured as total protein. In saturation binding studies, data were fitted by a one-site saturation-binding isotherm. In competition studies, the data were first fitted to a one- and then a two-site model, and if the residual square sums were significantly less for a two-site fit of the data than for a one-site fit (P-value <0.05 as determined by F-test), then a two-site model was accepted. Slopes of pseudo-Hill plots were also determined for some competitors to validate one- or two-site fitting. For pseudo-Hill plot analyses, Origin (version 7.5, Origin Lab Co., Northampton, MA, USA) was used.
In functional studies, antagonist affinity estimates (pKB values) were obtained by plotting the data according to the Schild analysis. When the straight lines had a slope of unity, the pA2 value estimated was taken as the pKB value. When a single concentration of an antagonist was tested, the pKB value was also determined for a single concentration of the antagonist by the concentration ratio method (Furchgott, 1972).
Drugs
Silodosin (KMD-3213) and tamsulosin were donated from Kissei Pharmaceutical Co., Ltd. (Matsumoto, Japan). [3H]-silodosin (specific activity 1.92 TBq mmol−1) was manufactured by GE Healthcare UK (Buckinghamshire, UK), and provided by Kissei Pharmaceutical Co., Ltd. The other drugs were obtained from commercially available sources.
Results
[3H]-silodosin bindings in the mouse cerebral cortex
[3H]-silodosin (50–1000 pM) bound to tissue segments and membrane preparations of the WT mouse cerebral cortex in a concentration-dependent manner (Figure 1a). The Hill coefficients were 1.00±0.04 for tissue segments and 1.05±0.05 for membrane preparations, and it was assumed that [3H]-silodosin bound to a single class of sites. The dissociation constants and maximal binding capacities were 470±35 pM and 228±12 fmol per mg total tissue protein in the segments (n=6) and 390±20 pM and 193±15 fmol per mg total tissue protein in the membrane preparations (n=6), respectively. Thus, there was no significant difference in the abundance of [3H]-silodosin binding sites between tissue segments and membrane preparations of the WT mouse cerebral cortex (Figure 1b).
Figure 1.

Binding of [3H]-silodosin and [3H]-prazosin to mouse cerebral cortex. (a) Saturation curves for specific binding of [3H]-silodosin in tissue segments and membrane preparations of WT mouse cortex and in tissue segments of AKO mouse cortex. (b) Binding density of [3H]-silodosin estimated from saturation experiments with WT, AKO, BKO and DKO mouse cortices. The saturation experiments were conducted with intact tissue segments (S) and membranes (M) of cerebral cortex. High- and low-affinity sites for prazosin at [3H]-silodosin binding sites were represented as α1A and α1L, respectively. (c) Binding density of [3H]-prazosin estimated from saturation experiments with intact segments of WT, AKO, BKO and DKO mouse cortices. High- and low-affinity sites for silodosin at [3H]-prazosin-binding sites were represented as α1A and α1B, respectively. The number of experiments was six in WT, five in AKO, four in BKO and three in DKO. *Significantly different from WT mouse (P<0.01). AKO, α1A-adrenoceptor gene knockout; BKO, α1B-adrenoceptor gene knockout; DKO, α1D-adrenoceptor gene knockout; WT, wild type.
In contrast, [3H]-silodosin binding was negligible in tissue segments and membrane preparations of the AKO mouse cortex (Bmax<20 fmol per mg protein, n=5; Figures 1a and b), whereas the same Bmax values as that of WT mouse were estimated in tissue segments of BKO and DKO mice (Figure 1b).
The pharmacological profiles of [3H]-silodosin binding sites in the tissue segments and the membrane preparations of WT mouse cortex were examined in competition binding studies using several drugs. Competition curves for prazosin and RS-17053 in the tissue segments were shallow, better fitting a two-site model in computer analysis (Figures 2a and b). The slope factors in the pseudo-Hill plot analyses were −0.67±0.03 for prazosin and −0.56±0.07 for RS-17053, which also supported the existence of two affinity sites for these antagonists. The proportion of high-affinity sites was approximately 55% (Table 1). However, in the membrane preparations, prazosin and RS-17053 showed monophasic competition curves with high affinity (Figures 2a and b, Table 1). Tamsulosin, silodosin and BMY 7378 competed monotonically for the [3H]-silodosin binding in either tissue segments or membrane preparations (Table 1).
Figure 2.

Competition curves for various antagonists in mouse cerebral cortex. (a and b) Competition curves for prazosin and RS-17053 at 500 pM [3H]-silodosin-binding sites in the intact segments and membranes of the WT mouse cortex. (c) Competition curves for silodosin and BMY 7378 at 500 pM [3H]-prazosin-binding sites in the intact segments of the WT mouse cortex. (d) Competition of silodosin in the intact segments of AKO, BKO, and DKO mouse cortices at 500 pM [3H]-prazosin-binding sites. Each curve is representative of similar results obtained in 2–5 experiments. AKO, α1A-adrenoceptor gene knockout; BKO, α1B-adrenoceptor gene knockout; DKO, α1D-adrenoceptor gene knockout; WT, wild type.
Table 1.
Pharmacological characteristics of [3H]-silodosin and [3H]-prazosin-binding sites in the WT mouse cerebral cortex
| Antagonist |
[3H]-silodosin binding |
[3H]-prazosin binding |
|||
|---|---|---|---|---|---|
|
Segments |
Membranes |
Segments |
|||
 |
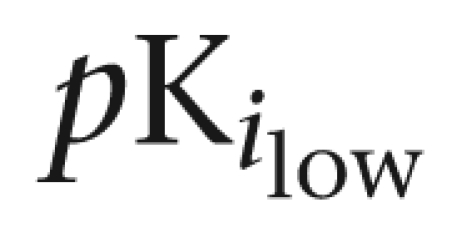 |
pKi |  |
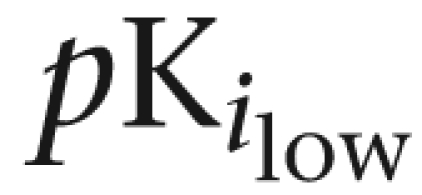 |
|
| Prazosin | 10.1±0.2 (55) | 8.5±0.2 | 9.9±0.1 | — | |
| Silodosin | 9.7±0.2 | 9.5±0.1 | 9.9±0.3 (38) | 7.4±0.2 | |
| Tamsulosin | 9.9±0.1 | 10.3±0.1 | — | ||
| BMY 7378 | 6.3±0.2 | 6.4±0.1 | 5.7±0.1 | ||
| RS-17053 | 8.4±0.1 (55) | 6.3±0.1 | 9.3±0.1 | 8.8±0.3 (40) | 7.7±0.2 |
Abbreviations: RS-17053, N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-α,α-dimethyl-1H-indole-3-ethamine hydrochloride; WT, wild type.
% indicates proportion of high-affinity sites.
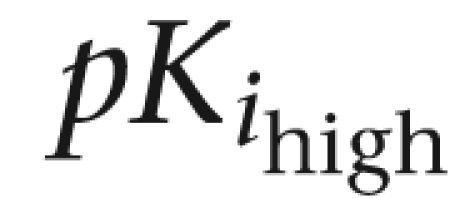 and
and 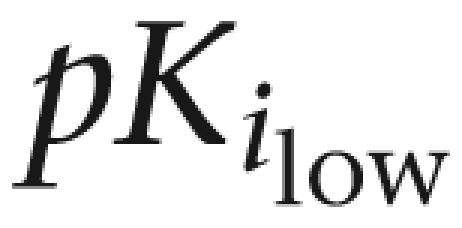 : negative logarithm of the equilibrium constants (pKi) at high- and low-affinity sites for tested drugs.
: negative logarithm of the equilibrium constants (pKi) at high- and low-affinity sites for tested drugs.
Competitive binding experiments with intact tissue segments and crude membrane preparations were carried out at 500 pM [3H]-silodosin or [3H]-prazosin. Data represent mean±s.e.mean of 4–6 experiments.
From these results, it was suggested that [3H]-silodosin binding sites in WT mouse cortex are composed of two components having different affinities for prazosin and RS-17053 in the intact tissue segments, but that both components change the pharmacological profiles to that of a single component having high affinity for prazosin and RS-17053 after homogenization. These results are identical to those recently obtained in rat cerebral cortex (Morishima et al., 2008), suggesting the presence of α1A and α1L subtypes (see Introduction and Discussion for receptor subclassification). From the estimated proportions of high- and low-affinity sites for prazosin in the intact tissue segments, the densities of both α1A and α1L subtypes were extrapolated. Figure 1b shows that, in addition to total density, the density of high- or low-affinity sites for prazosin (α1A or α1L subtypes) is roughly the same among WT, BKO and DKO mouse cortex samples.
[3H]-prazosin bindings in the mouse cerebral cortex
As [3H]-silodosin recognized two distinct components of α1-adrenoceptors in tissue segments of the cerebral cortex, binding of [3H]-prazosin was also examined in the tissue segments of WT and KO mouse cortices. In saturation binding experiments, [3H]-prazosin bound to α1-adrenoceptors (KD=455±56 pM and Bmax=237±32 fmol per mg of total tissue protein, n=5). The same density as that in WT mice was estimated in DKO mice, but the abundance in AKO and BKO mice was significantly lower than in WT mice (Figure 1c).
In competition experiments with WT mouse cortex segments, silodosin biphasically inhibited 500 pM [3H]-prazosin binding; approximately 40% were high-affinity sites for silodosin (Figure 2c, Table 1). A similar biphasic competition curve for silodosin was observed in DKO mice (Figure 2d). However, the bindings in AKO and BKO mouse cortices showed monotonic competition for silodosin with low and high affinities, respectively (pKi=7.7±0.1 in AKO mice and 9.2±0.2 in BKO mice, n=3; Figure 2d). On the basis of the proportions of these different affinity sites for silodosin, the densities of both components were extrapolated in [3H]-prazosin binding sites and are shown in Figure 1c, where high- and low-affinity sites for silodosin are represented as α1A and α1B subtypes. Both α1A and α1B subtypes were identified in WT and DKO mice, but the α1B subtype only occurred in the AKO mouse cortex and α1A subtype only occurred in the BKO mouse cortex. BMY 7378 competed with [3H]-prazosin binding monotonically with low affinity in WT mouse cortex (Figure 2c, Table 1). These results suggest that α1A and α1B subtypes showing different affinities for silodosin occurred in the [3H]-prazosin binding sites of WT and DKO mouse cortex segments, but that each α1A or α1B subtype was selectively abolished by AKO or BKO mice. The prazosin low-affinity sites corresponding to α1L subtype were not detected in the binding study with [3H]-prazosin.
[3H]-silodosin bindings in the mouse vas deferens and prostate
As [3H]-silodosin recognized low-affinity sites for prazosin (α1L subtype) in mouse cerebral cortex segments, the binding approach with this radioligand was applied to tissue segments of vas deferens and prostate. Because both tissues were too small to provide a large number of tissue segments, very limited experiments were conducted. [3H]-silodosin specifically bound to tissue segments of the WT mouse vas deferens (KD=130±60 pM and Bmax=153±18 fmol per mg protein, n=5) (Figure 3a). However, specific binding of [3H]-silodosin was not detected in AKO mice. The binding capacities of 500 pM [3H]-silodosin in the vas deferens of BKO and DKO mice were not significantly different from that of WT mice (Figure 3c).
Figure 3.

Binding of [3H]-silodosin to mouse vas deferens and prostate. (a) Representative saturation curves for specific binding of [3H]-silodosin in tissue segments of vas deferens isolated from WT and AKO mice. (b) Representative competition curve for prazosin at 500 pM [3H]-silodosin-binding sites in tissue segments of the WT mouse vas deferens. (c and d) Binding capacities of 500 pM [3H]-silodosin in the intact segments of WT, AKO, BKO and DKO mouse vas deferentia (c) and prostate (d). In panel c, high- and low-affinity sites for prazosin at [3H]-silodosin-binding sites were represented as α1A and α1L, respectively. *Significantly different from WT mice (P<0.01). Mean±s.e.mean of 3–6 experiments.
In competition experiments, the specific binding of [3H]-silodosin to WT mouse vas deferens was a biphasic competition with prazosin (pKi=9.9 and 8.1) (Figure 3b) and RS-17053 (pKi=8.9 and 6.8), suggesting the presence of the two distinct α1A and α1L subtypes, as in the mouse cortex. From the binding capacities of 500 pM [3H]-silodosin in the absence or presence of 1 nM prazosin, the proportion of α1A and α1L subtypes was evaluated. Figure 3c shows that both subtypes occur at a similar ratio in tissue segments of WT, BKO and DKO mouse vas deferentia, but are absent in AKO mice.
[3H]-silodosin also bound to the prostate segments. Because only four to five segments were prepared from each mouse, only the binding capacities at 500 pM [3H]-silodosin were examined. As shown in Figure 3d, the densities were estimated at 60 fmol per mg total tissue protein in prostrate of WT, BKO and DKO mice, but binding was negligible in AKO mice. In WT mouse prostate, the specific binding of 500 pM [3H]-silodosin was reduced to 39±4% in the presence of 1 nM prazosin (n=4).
Contractile responses to noradrenaline in mouse vas deferens and prostate
In the vas deferentia isolated from WT mice, non-cumulative application of noradrenaline (1–100 μM) produced a transient contraction. This contraction was relatively insensitive to prazosin, and thus not inhibited by prazosin at concentrations less than 10 nM (pKB=7.7±0.2, n=5). However, the contraction was strongly attenuated by low concentrations of silodosin (pKB=9.7±0.2, n=5, Figure 4). The contractile responses to noradrenaline also showed low sensitivity to RS-17053, 5-methylurapidil and BMY 7378 (Figure 4b, Table 2). These results suggested that the contractile responses in the WT mouse vas deferens were mediated through prazosin low-affinity α1-adrenoceptors (α1L subtype). In AKO mice, the response of the vas deferens to noradrenaline was abolished, whereas the same amplitude of contraction was produced by 100 μM noradrenaline in BKO and DKO mouse vas deferentia (Figure 5). The contractile responses to 10 μM noradrenaline in BKO and DKO mouse vas deferentia were not affected by treatment with 10 nM prazosin or 100 nM BMY 7378, but were attenuated by 3 nM silodosin to less than 20% (n=3 in BKO and DKO mice, respectively). α,β-Methylene ATP (1 μM, a P2X purinoceptor agonist) also produced a transient contraction, which was not affected by gene targeting of the three α1-adrenoceptors (Figure 5b).
Figure 4.

Contractile responses to noradrenaline in mouse vas deferens (epididymal portion). (a) Effects of prazosin and silodosin on the contractile responses to 10 μM noradrenaline (NA) in the vas deferens isolated from WT mice. In contrast to silodosin (3 nM), prazosin (10 nM) failed to inhibit the contractile response to noradrenaline. (b) Effects of prazosin, silodosin, 5-methylurapidil (5-MU) and BMY 7378 on the concentration–response curves for noradrenaline in the WT mouse vas deferens. Mean±s.e.mean of 4–6 experiments. WT, wild type.
Table 2.
Pharmacological characteristics of noradrenaline-induced contractions in WT mouse vas deferens and prostate
| Antagonist |
pKB |
|
|---|---|---|
| Vas deferens | Prostate | |
| Prazosin | 7.7±0.2 | 8.2±0.3 |
| Silodosin | 9.7±0.2a | 9.6±0.3a |
| RS-17053 | NI | 6.9±0.2a |
| 5-Methylurapidil | 7.6±0.2 | 7.4±0.3 |
| BMY 7378 | NI | NI |
Abbreviations: RS-17053, N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-α, α-dimethyl-1H-indole-3-ethamine hydrochloride; WT, wild type.
Mean±s.e.mean of 4–6 experiments.
NI indicates no inhibition at 0.1 μM BMY 7378 and 1 μM RS-17053.
pKB values were calculated by the Schild analysis.
Estimated at 1 nM silodosin or 1 μM RS-17053 by the concentration ratio method (Furchgott, 1972).
Figure 5.

Effects of α1-adrenoceptor gene knockout on the contractile responses in mouse vas deferens (epididymal portion). (a) Representative responses to 100 μM noradrenaline (NA) in WT, AKO, BKO and DKO mice. (b) Contractile responses induced by 100 μM noradrenaline and 1 μM α, β-methylene ATP (α,β-Me ATP). Mean±s.e.mean of 3–6 experiments. *Significantly different from other columns (P<0.01). AKO, α1A-adrenoceptor gene knockout; BKO, α1B-adrenoceptor gene knockout; DKO, α1D-adrenoceptor gene knockout; WT, wild type.
In WT mouse prostate, the cumulative application of noradrenaline produced concentration-dependent contraction (maximum amplitude of contraction=53±3 mg, pEC50=6.5±0.2, n=15). The contraction was relatively insensitive to prazosin, RS-17053 and 5-methylurapidil; low affinities were estimated (Figure 6, Table 2). However, the responses to noradrenaline were inhibited by low concentrations of silodosin (pKB=9.6±0.3, n=5) (Figure 6b). Furthermore, the response of the prostate was markedly attenuated in AKO mice, but not in BKO or DKO mice (Figures 7a and c). The residual contractile response to noradrenaline in AKO mice was not inhibited by 3 nM silodosin or 10 nM BMY 7378, but was insurmountably inhibited by 1 nM prazosin (Figure 7b). Amplitudes of contraction similar to those of WT mouse prostate were produced in prostate of BKO and DKO mice (Figure 7c), and the pEC50 values for noradrenaline were 6.5±0.1 (n=4) in BKO mice and 6.4±0.2 (n=3) in DKO mice. The concentration–response curves for noradrenaline in BKO and DKO mice were not attenuated by 1 nM prazosin and 10 nM BMY 7378, but were shifted to the right and downwards by 3 nM silodosin (pKB value=9.4±0.2 in BKO mice and 9.5±0.2 in DKO mice, n=3). The amplitudes of contractions induced by α,β-methylene ATP (10 μM) were not significantly different among WT, AKO, BKO and DKO mice (Figure 7c).
Figure 6.

Effects of prazosin (a), silodosin (b), 5-methylurapidil (c; 5-MU) and BMY 7378 (d) on the concentration–response curves for noradrenaline in WT mouse prostate. Mean±s.e.mean of 4–6 experiments. WT, wild type.
Figure 7.

Contractile responses to noradrenaline in mouse prostate. (a) Concentration–response curves for noradrenaline in WT and AKO mouse prostate. Mean±s.e.mean of 4–6 experiments. (b) Effects of silodosin, BMY 7378 and prazosin on the contractile response to noradrenaline in the AKO mouse prostate. This is a representative result obtained from three AKO mice. The response induced by 100 μM noradrenaline in the control was equivalent to 18.5 mg, which was taken as 100%. Prazosin (1 nM) produced insurmountable inhibition. (c) Contractile responses induced by 100 μM noradrenaline and 10 μM α,β-methylene ATP (α,β-Me ATP). Mean±s.e.mean of 3–6 experiments. *Significantly different from other columns (P<0.01). AKO, α1A-adrenoceptor gene knockout; WT, wild type.
Discussion
[3H]-Silodosin has very high apparent affinities for both α1A-adrenoceptor and α1L-adrenoceptor subtypes (Murata et al., 1999; Morishima et al., 2008). In recent radioligand binding studies with [3H]-silodosin, it has been demonstrated that α1L-adrenoceptors are pharmacologically distinct from the three classical α1-adrenoceptors (α1A, α1B and α1D) in the intact segments of the rat cerebral cortex (Morishima et al., 2008), rabbit ear artery (Hiraizumi-Hiraoka et al., 2004) and human and rabbit prostate (Morishima et al., 2007a; Su et al., 2008). In the present binding study with [3H]-silodosin, the α1L-adrenoceptor was recognized as a distinct site showing low affinity for prazosin, RS-17053 and BMY 7378, but high affinity for silodosin and tamsulosin in the intact tissue segments of the cerebral cortex, vas deferens and prostate of WT mouse, along with the α1A-adrenoceptor, which showed high affinity for prazosin, RS-17053, silodosin and tamsulosin. The same results were also obtained in BKO and DKO mice, whereas the [3H]-silodosin binding sites disappeared in AKO mice. Furthermore, the functional studies with mouse vas deferens and prostate revealed that the α1-adrenoceptors mediating contractile responses to noradrenaline showed an α1L-adrenoceptor profile and that the contractions were specifically abolished in AKO mice, but not in BKO and DKO mice. Adrenergic contractions in vas deferens and prostate have been reported through α1L-adrenoceptors in many mammalian species including mice (Ohmura et al., 1992; Hiraoka et al., 1999; Ford et al., 1996; Van der Graaf et al., 1997; Amobi et al., 2002; Gray and Ventura, 2006; Morishima et al., 2007a; Su et al., 2008). Thus, the present results strongly suggest that α1L-adrenoceptors occur and function independently from other α1-adrenoceptor subtypes, but that the expression of the α1L-adrenoceptor is closely related to that of the α1A-adrenoceptor gene, and that the α1B-adrenoceptor and the α1D-adrenoceptor are not involved in manifestation of the α1L-adrenoceptor phenotype.
More recently, we reported in a study with the rat cerebral cortex that the α1L-adrenoceptor could be detected in the intact segments, but that the pharmacological profile disappeared after homogenization (Morishima et al., 2008). The same phenomenon was observed in this study with WT mouse cortex. Interestingly, in both species, the total binding density of [3H]-silodosin in the tissue segments of cerebral cortex was equal to the density of α1A-adrenoceptors detected in the membrane preparations (Figure 1b). Because contamination with α1B-adrenoceptors and α1D-adrenoceptors is negligible in the present [3H]-silodosin binding sites (Murata et al., 1999; Morishima et al., 2008), it seemed that the pharmacological profile of the α1L-adrenoceptors converted to that of the α1A-adrenoceptors by homogenization without a loss of yield. Moreover, the α1A-adrenoceptors in the membrane preparations were abolished in AKO mice. These results show that both α1A-adrenoceptor and α1L-adrenoceptor are derived from the same α1A-adrenoceptor gene, and further suggest that α1L-adrenoceptor is a pharmacological phenotype of α1A-adrenoceptor (Su et al., 2008).
[3H]-prazosin also bound to the intact segments of WT mouse cortex with a high affinity and the binding sites were divided into two components by silodosin but not by BMY 17053. A similar population of silodosin high- and low-affinity sites and the same densities were observed in DKO mouse cortex (Figure 1c). However, sites for silodosin were selectively abolished in AKO mice (no high-affinity sites) or BKO mice (no low-affinity sites), resulting in a complete loss of the corresponding subtypes. These results show that [3H]-prazosin at the concentrations used in this experiment can selectively recognize α1A-adrenoceptors and α1B-adrenoceptors in the intact segments of WT and DKO mouse cortices, as reported in the rat cerebral cortex (Morrow and Creese, 1986; Morishima et al., 2008).
The α1A-, α1B- and α1L-adrenoceptor subtypes were distinctly identified in the [3H]-silodosin and [3H]-prazosin binding sites in WT mouse, whereas in AKO mice, the α1A and α1L subtypes were specifically abolished and the α1B subtype selectively disappeared in BKO mice. Thus, we conclude that no quantitative/qualitative compensation by other subtypes of the same α1-adrenoceptor group (for example, upregulation or supersensitivity) is caused in knockout mice, which supports previous in vivo and in vitro studies with AKO, BKO and DKO mice (Cavalli et al., 1997; Rokosh and Simpson, 2002; Tanoue et al., 2002; Simpson, 2006). This conclusion also accounts for a complete loss of α1L-adrenoceptor-mediated responses in the vas deferens and prostate of AKO mice and no significant shift of pEC50 values for noradrenaline between WT and BKO or DKO mouse prostate. It can be noted that the expression of each α1-adrenoceptor subtype is differentially regulated in cardiac myocytes (Rokosh et al., 1996).
In contrast to AKO mouse vas deferens, a small but significant contraction was elicited by noradrenaline in AKO mouse prostate (Figure 7a). This residual contraction was insensitive to silodosin (1 nM, a specific concentration for α1A and α1L-adrenoceptors) and BMY 17053 (10 nM, a specific concentration for α1D-adrenoceptor) but potently inhibited by prazosin (1 nM, a specific concentration for α1A-, α1B-, or α1D-adrenoceptors) (Figure 7b). Therefore, a part of the contractile response to noradrenaline in mouse prostate may be mediated through α1B-adrenoceptors, although the major component is still α1L-adrenoceptor-mediated.
This study clearly revealed that two different phenotypes (α1A and α1L-adrenoceptors) originate from a single α1A-adrenoceptor gene, yet no evidence explains why multiple phenotypes are produced from a single gene. However, it is interesting, in this context, to note the history of the β4-adrenoceptor. Like α1L-adrenoceptors, β4-adrenoceptors were originally identified as functional receptors resistant to several β-adrenoceptor antagonists, such as propranolol (Kaumann and Molenaar, 1996; Sarsero et al., 1998). However, the β4-adrenoceptor was absent after β1-adrenoceptor gene knockout (Kaumann et al., 2001). At present, the β1-adrenoceptor has been considered to have two distinct binding sites within the same receptor, orthosteric and allosteric sites with high and low affinity for catecholamines or propranolol (Konkar et al., 2000; Baker and Hill, 2007). The β4-adrenoceptor reflects the pharmacological profiles of an allosteric site of β1-adrenoceptors with low affinity for propranolol. Thus, it may be possible that the α1L phenotype represents the presence of an allosteric site in the α1A-adrenoceptor.
Another interesting finding in this study was that, even though α1L-adrenoceptors occurred as a separate entity, independent of α1A-adrenoceptors, in the intact segments, the pharmacological profile completely changed to that of classical α1A-adrenoceptors upon homogenization. Such a change in profile upon homogenization was not seen in β4-adrenoceptors (Kaumann et al., 2001; Joseph et al., 2003). Thus, if the α1L-adrenoceptor is an allosteric site variant of the α1A-adrenoceptor, it is likely that a more drastic conformational change is caused by homogenization in α1A-adrenoceptors than in β1-adrenoceptors, resulting in a greater change in profile. Alternatively, the α1L-adrenoceptor may be a phenotype constructed with some associated proteins and the construct may be disrupted by homogenization. This has been demonstrated in the receptor for calcitonin gene-related peptide, in which different phenotypes are produced by association with different receptor-modifying proteins (McLatchie et al., 1998; Poyner et al., 2002). Moreover, the α1L-adrenoceptor might be constructed under a particular environment with specific parameters, such as microdomains, relation to the cytoskeleton and extracellular ion environment, without involving an associated protein. Recently, it has been suggested that pharmacological properties may not necessarily remain constant between tissues expressing the same receptors or between different assay conditions (Kenakin et al., 1995; Muramatsu et al., 2005; Nelson and Challiss, 2007). Thus, the molecular mechanisms underlying the expression of the α1L-adrenoceptor phenotype should be explored in future studies.
In summary, this study using knockout mice demonstrated that α1L-adrenoceptors can be identified as binding and functional entities in mice, and strongly suggested that two distinct phenotypes (α1A-adrenoceptor and α1L-adrenoceptor) are derived from the same α1A-adrenoceptor gene.
Acknowledgments
We are grateful to Kissei Pharmaceutical Co., Ltd. for providing the silodosin and [3H]-silodosin. We also thank Ms Hiromi Degura and Ms Reiko Inaki for their secretarial assistance. This work was in part supported by a Grant-in-Aid for Scientific Research (IM and SM) and the 21st COE Research Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (IM), as well as by a grant from the Smoking Research Foundation of Japan (IM), by the US Department of Veterans Affairs Research Service, and by the US National Institutes of Health (HL31113) (PCS).
Abbreviations
- AKO
α1A-adrenoceptor gene knockout
- Bmax
maximum binding capacity
- BKO
α1B-adrenoceptor gene knockout
- BMY 7378
(8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4,5]decane-7,9-dione dihydrochloride
- DKO
α1D-adrenoceptor gene knockout
- RS-17053
N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-α,α-dimethyl-1H-indole-3-ethamine hydrochloride
Conflict of interest
The authors state no conflict of interest.
Editor's note. Readers may note that the content of this paper is very similar to that of a paper by Gray et al. (Gray KT, Short JL, Ventura S (2008). The α1A-adrenoceptor gene is required for the α1L-adrenoceptor-mediated response in isolated preparations of the mouse prostate. Br J Pharmacol 155: 103–109; doi:10.1038/bjp.2008.245) in this journal, and reaches similar conclusions. The first draft of this paper was received before acceptance of the paper by Gray et al., and the two manuscripts were handled independently by different editors and referees.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153 Suppl 2:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amobi N, Guillebaud J, Kaisary A, Lloyd-Davies RW, Turner E, Smith IC. Contractile actions of imidazoline alpha-adrenoceptor agonists and effects of noncompetitive alpha1-adrenoceptor antagonists in human vas deferens. Eur J Pharmacol. 2002;462:169–177. doi: 10.1016/s0014-2999(03)01346-3. [DOI] [PubMed] [Google Scholar]
- Argyle SA, McGrath JC. An alpha(1A)/alpha(1L)-adrenoceptor mediates contraction of canine subcutaneous resistance arteries. J Pharmacol Exp Ther. 2000;295:627–633. [PubMed] [Google Scholar]
- Baker JG, Hill SJ. Multiple GPCR conformations and signaling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF, et al. Decreased blood pressure response in mice deficient of the α1b-adrenergic receptor. Proc Natl Acad Sci USA. 1997;94:11589–11594. doi: 10.1073/pnas.94.21.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DV, Gever JR, Jasper JR, Kava MS, Lesnick JD, Meloy TD, et al. Human cloned alpha1A-adrenoceptor isoforms display alpha1L-adrenoceptor pharmacology in functional studies. Eur J Pharmacol. 1999;370:337–343. doi: 10.1016/s0014-2999(99)00154-5. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. Alpha-1 and alpha-2 adrenoceptor: response coupling in canine saphenous and femoral veins. Trends Pharmacol Sci. 1986;7:347–349. [PubMed] [Google Scholar]
- Ford AP, Arredondo NF, Blue DR, Jr, Bonhaus DW, Jasper J, Kava MS, et al. RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-alpha, alpha-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective alpha 1A-adrenoceptor antagonist, displays low affinity for functional alpha 1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol Pharmacol. 1996;49:209–215. [PubMed] [Google Scholar]
- Ford AP, Daniels DV, Chang DJ, Gever JR, Jasper JR, Lesnick JD, et al. Pharmacological pleiotropism of the human recombinant alpha1A-adrenoceptor: implications for alpha1-adrenoceptor classification. Br J Pharmacol. 1997;121:1127–1135. doi: 10.1038/sj.bjp.0701207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF.The classification on adrenoceptors (adrenergic receptors): an evaluation from the standpoint of receptor theory Handbuch der Experimentellen Pharmacology 1972Springer: New York; 283–335.In: Blaschko H, Muscholl E (eds)vol. 3 [Google Scholar]
- Gray KT, Ventura S. Alpha 1L-adrenoceptors mediate contractions of the isolated mouse prostate. Eur J Pharmacol. 2006;540:155–161. doi: 10.1016/j.ejphar.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, et al. International Union of Pharmacology. X. Recommendation for nomenclature of alpha 1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hiraizumi-Hiraoka Y, Tanaka T, Yamamoto H, Suzuki F, Muramatsu I. Identification of alpha-1L adrenoceptor in rabbit ear artery. J Pharmacol Exp Ther. 2004;310:995–1002. doi: 10.1124/jpet.104.066985. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Ohmura T, Oshita M, Watanabe Y, Morikawa K, Nagata O, et al. Binding and functional characterization of alpha 1-adrenoceptor subtypes in the rat prostate. Eur J Pharmacol. 1999;366:119–126. doi: 10.1016/s0014-2999(98)00895-4. [DOI] [PubMed] [Google Scholar]
- Joseph SS, Lynham JA, Molenaar P, Grace AA, Colledge WH, Kaumann AJ. Intrinsic sympathomimetic activity of (−)-pindolol mediated through a (−)-propranolol-resistant site of the beta1-adrenoceptor in human atrium and recombinant receptors. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:496–503. doi: 10.1007/s00210-003-0835-z. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Engelhardt S, Hein L, Molenaar P, Lohse M. Abolition of (−)-CGP 12177-evoked cardiostimulation in double beta1/beta2-adrenoceptor knockout mice. Obligatory role of beta1-adrenoceptors for putative beta4-adrenoceptor pharmacology. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:87–93. doi: 10.1007/s002100000336. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Molenaar P. Differences between the third cardiac beta-adrenoceptor and the colonic beta 3-adrenoceptor in the rat. Br J Pharmacol. 1996;118:2085–2098. doi: 10.1111/j.1476-5381.1996.tb15648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Morgan P, Lutz M. On the importance of the ‘antagonist assumption' to how receptors express themselves. Biochem Pharmacol. 1995;50:17–26. doi: 10.1016/0006-2952(95)00137-o. [DOI] [PubMed] [Google Scholar]
- Konkar AA, Zhai Y, Granneman JG. Beta1-adrenergic receptors mediate beta3-adrenergic-independent effects of CGP 12177 in brown adipose tissue. Mol Pharmacol. 2000;57:252–258. [PubMed] [Google Scholar]
- Lomasney JW, Cotecchia S, Lefkowitz RJ, Caron MG. Molecular biology of alpha-adrenergic receptors: implications for receptor classification and for structure–function relationships. Biochim Biophys Acta. 1991;1095:127–139. doi: 10.1016/0167-4889(91)90075-9. [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Michelotti GA, Price DT, Schwinn DA. Alpha-1 adrenergic receptor regulation: basic science and clinical implications. Pharmacol Ther. 2000;88:281–309. doi: 10.1016/s0163-7258(00)00092-9. [DOI] [PubMed] [Google Scholar]
- Morishima S, Suzuki F, Yoshiki H, Anisuzzaman AS, Sathi ZS, Tanaka T, et al. Identification of the alpha(1L)-adrenoceptor in rat cerebral cortex and possible relationship between alpha(1L)- and alpha(1A)-adrenoceptors. Br J Pharmacol. 2008;153:1485–1494. doi: 10.1038/sj.bjp.0707679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima S, Tanaka T, Yamamoto H, Suzuki F, Akino H, Yokoyama O, et al. Identification of alpha-1L and alpha-1A adrenoceptors in human prostate by tissue segment binding. J Urol. 2007a;177:377–381. doi: 10.1016/j.juro.2006.08.080. [DOI] [PubMed] [Google Scholar]
- Morishima S, Tanaka T, Yamamoto H, Suzuki F, Akino H, Yokoyama O, et al. α1-Adrenoceptor in lower urinary tract. J Pharmacol Sci. 2007b;103 Suppl I:40P. [Google Scholar]
- Morrow AL, Creese I. Characterization of alpha 1-adrenergic receptor subtypes in rat brain: a reevaluation of [3H]WB4101 and [3H]prazosin binding. Mol Pharmacol. 1986;29:321–330. [PubMed] [Google Scholar]
- Muramatsu I, Murata S, Isaka M, Piao HL, Zhu J, Suzuki F, et al. Alpha1-adrenoceptor subtypes and two receptor systems in vascular tissues. Life Sci. 1998;62:1461–1465. doi: 10.1016/s0024-3205(98)00090-3. [DOI] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Kigoshi S. Pharmacological profiles of a novel alpha 1-adrenoceptor agonist, PNO-49B, at alpha 1-adrenoceptor subtypes. Naunyn Schmiedebergs Arch Pharmacol. 1995;351:2–9. doi: 10.1007/BF00169057. [DOI] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Kigoshi S, Hashimoto S, Oshita M. Pharmacological subclassification of alpha 1-adrenoceptors in vascular smooth muscle. Br J Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu I, Tanaka T, Suzuki F, Li Z, Hiraizumi-Hiraoka Y, Anisuzzaman AS, et al. Quantifying receptor properties: the tissue segment binding method—a powerful tool for the pharmacome analysis of native receptors. J Pharmacol Sci. 2005;98:331–339. doi: 10.1254/jphs.cpj05001x. [DOI] [PubMed] [Google Scholar]
- Murata S, Taniguchi T, Muramatsu I. Pharmacological analysis of the novel, selective alpha1-adrenoceptor antagonist, KMD-3213, and its suitability as a tritiated radioligand. Br J Pharmacol. 1999;127:19–26. doi: 10.1038/sj.bjp.0702489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Taniguchi T, Suzuki F, Akagi Y, Muramatsu I. Evaluation of α1-adrenoceptors in the rabbit iris: pharmacological characterization and expression of mRNA. Br J Pharmacol. 1999;127:1367–1374. doi: 10.1038/sj.bjp.0702675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CP, Challiss RA. ‘Phenotypic' pharmacology: the influence of cellular environment on G protein-coupled receptor antagonist and inverse agonist pharmacology. Biochem Pharmacol. 2007;73:737–751. doi: 10.1016/j.bcp.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Ohmura T, Oshita M, Kigoshi S, Muramatsu I. Identification of alpha 1-adrenoceptor subtypes in the rat vas deferens: binding and functional studies. Br J Pharmacol. 1992;107:697–704. doi: 10.1111/j.1476-5381.1992.tb14509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshita M, Kigoshi S, Muramatsu I. Pharmacological characterization of two distinct alpha 1-adrenoceptor subtypes in rabbit thoracic aorta. Br J Pharmacol. 1993;108:1071–1076. doi: 10.1111/j.1476-5381.1993.tb13507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao H, Taniguchi T, Nakamura S, Zhu J, Suzuki F, Mikami D, et al. Cloning of rabbit alpha(1b)-adrenoceptor and pharmacological comparison of alpha(1a)-, alpha(1b)- and alpha(1d)-adrenoceptors in rabbit. Eur J Pharmacol. 2000;396:9–17. doi: 10.1016/s0014-2999(00)00171-0. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Ramsay D, Carr IC, Pediani J, Lopez-Gimenez JF, Thurlow R, Fidock M, et al. High-affinity interactions between human alpha1A-adrenoceptor C-terminal splice variants produce homo- and heterodimers but do not generate the alpha1L-adrenoceptor. Mol Pharmacol. 2004;66:228–239. doi: 10.1124/mol.66.2.228. [DOI] [PubMed] [Google Scholar]
- Rokosh DG, Simpson PC. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci USA. 2002;99:9474–9479. doi: 10.1073/pnas.132552699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokosh DG, Stewart AF, Chang KC, Bailey BA, Karliner JS, Camacho SA, et al. α1-Adrenergic receptor subtype mRNAs are differentially regulated by α1-adrenergic and other hypertrophic stimuli in cardiac myocytes in culture and in vivo: repression of α1B and α1D but induction of α1C. J Biol Chem. 1996;271:5839–5843. doi: 10.1074/jbc.271.10.5839. [DOI] [PubMed] [Google Scholar]
- Sadalge A, Coughlin L, Fu H, Wang B, Valladares O, Valentino R, et al. Alpha 1d Adrenoceptor signaling is required for stimulus induced locomotor activity. Mol Psychiatry. 2003;8:664–672. doi: 10.1038/sj.mp.4001351. [DOI] [PubMed] [Google Scholar]
- Sarsero D, Molenaar P, Kaumann AJ. Validity of (−)-[3H]-CGP 12177A as a radioligand for the ‘putative beta4-adrenoceptor' in rat atrium. Br J Pharmacol. 1998;123:371–380. doi: 10.1038/sj.bjp.0701609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PC.Lessons from knockouts: the alpha-1-Ars Adrenergic Receptors in the 21st Century 2006Humana Press: Totowa, NJ; 207–240.In: Perez DM (ed) [Google Scholar]
- Stam WB, Van der Graaf PH, Saxena PR. Analysis of alpha 1L-adrenoceptor pharmacology in rat small mesenteric artery. Br J Pharmacol. 1999;127:661–670. doi: 10.1038/sj.bjp.0702598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TH, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman ASM, Tanaka T, et al. Native profiles of α1A-adrenoceptor phenotypes in rabbit prostate Br J Pharmacol 2008 10.1038/bjp.2008.318e-pub ahead of print: advance online 25 July 2008; doi [DOI] [PMC free article] [PubMed]
- Suzuki F, Taniguchi T, Nakamura S, Akagi Y, Kubota C, Satoh M, et al. Distribution of alpha-1 adrenoceptor subtypes in RNA and protein in rabbit eyes. Br J Pharmacol. 2002;135:600–608. doi: 10.1038/sj.bjp.0704503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, et al. The α1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–775. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Graaf PH, Deplanne V, Duquenne C, Angel I. Analysis of alpha1-adrenoceptors in rabbit lower urinary tract and mesenteric artery. Eur J Pharmacol. 1997;327:25–32. doi: 10.1016/s0014-2999(97)89674-4. [DOI] [PubMed] [Google Scholar]
- Zhong H, Minneman KP. Alpha1-adrenoceptor subtypes. Eur J Pharmacol. 1999;375:261–276. doi: 10.1016/s0014-2999(99)00222-8. [DOI] [PubMed] [Google Scholar]