

Abstract
During a viral infection, reprogramming of the host cell gene expression pattern is required to establish an adequate antiviral response. The transcriptional coactivators p300 and CREB binding protein (CBP) play a central role in this regulation by promoting the assembly of transcription enhancer complexes to specific promoters of immune and proinflammatory genes. Here we show that the protein A238L encoded by African swine fever virus counteracts the host cell inflammatory response through the control of p300 transactivation during the viral infection. We demonstrate that A238L inhibits the expression of the inflammatory regulators cyclooxygenase-2 (COX-2) and tumor necrosis factor alpha (TNF-α) by preventing the recruitment of p300 to the enhanceosomes formed on their promoters. Furthermore, we report that A238L inhibits p300 activity during the viral infection and that its amino-terminal transactivation domain is essential in the A238L-mediated inhibition of the inflammatory response. Importantly, we found that the residue serine 384 of p300 is required for the viral protein to accomplish its inhibitory function and that ectopically expressed PKC-θ completely reverts this inhibition, thus indicating that this signaling pathway is disrupted by A238L during the viral infection. Furthermore, we show here that A238L does not affect PKC-θ enzymatic activity, but the molecular mechanism of this viral inhibition relies on the lack of interaction between PKC-θ and p300. These findings shed new light on how viruses alter the host cell antiviral gene expression pattern through the blockade of the p300 activity, which represents a new and sophisticated viral mechanism to evade the inflammatory and immune defense responses.
Positive and negative control of gene transcription plays a pivotal role in the functional differentiation of cells and in their ability to respond to extracellular signals and environmental stress (34, 53). During a viral infection, reprogramming of the host cell gene expression pattern occurs in order to establish an adequate antiviral response, but viruses have evolved strategies to subvert the host cell antiviral defense mechanisms, preventing both the inflammatory and immune responses (22).
CBP and p300 proteins play a central role in the regulation of gene transcription. They are transcriptional coactivators able to integrate multiple signal-induced pathways and coordinate gene expression, acting as crucial scaffolds for the formation of transcriptional initiation complexes (11).
CBP/p300 proteins do not specifically interact with promoter elements of target genes, but they are recruited to promoters by interaction with DNA-bound transcription factors, where they directly interact with the RNA Pol II complex (11, 36, 45). In this respect, it has been demonstrated that CBP/p300 interact with multiple transcription factors, including p53 (32), E2F (42), CREB (3), NFAT (24), NF-κB (25, 47), c-Jun (5), and c-Fos (4), coordinating the transcription of their target genes. The p300 coactivator carries out this function through different functional domains integrated in its amino-terminal (CH1 and KIX domains) and carboxyl-terminal (CH2 and CH3 domains) regions, as shown in the diagram in Fig. 1. CBP and p300 have been shown to be involved in several cellular events (reviewed in reference 27), such as the establishment of an adequate signal-induced immune and inflammatory response, by promoting the assembly of different transcription enhancer complexes (enhanceosomes) to specific promoters of immune and proinflammatory genes (8). Therefore, it is not surprising that several proteins encoded by different classes of DNA and RNA viruses have targeted both CBP and p300 as a mechanism to exploit or subvert cellular programs.
FIG. 1.

p300 protein structure and functional domains. Diagram of the p300 coactivator protein showing its functional domains clustered in two different regulatory regions: the amino-terminal region, containing the CH1 and KIX functional domains and also a bromo domain, and the carboxyl-terminal region, containing the CH2 and CH3 domains, which are part of the HAT catalytic domain. Both regulatory regions can act independently and interact simultaneously with the transcriptional machinery and/or with different transcription factors to build the transcriptional activity mediated by these coactivators. The amino acid (aa) position of each functional domain is also indicated in the scheme.
Viral proteins from at least four distinct viruses associate with CBP/p300: adenovirus E1A, SV40 large T antigen, E6 and E7 proteins from HPV, and HTLV-1 Tax protein (2, 10, 15, 33, 38). Interestingly, all of these viral proteins interacting with p300 and/or CBP modulate their acetyltransferase activity and their transactivation ability, thus affecting cell cycle regulatory proteins and promoting the subsequent cell malignant transformation (reviewed in reference 27).
ASFV, the sole member of the Asfarviridae family (17), encodes a protein, A238L, which inhibits NF-κB and NFAT activity when expressed in different cell types or during ASFV infection (44, 48). In previous reports, we have also shown that A238L is thus able to downregulate the transcriptional activation of the immunomodulatory genes for COX-2, TNF-α, and inducible nitric oxide synthase by a mechanism involving the transactivation mediated by NFAT, NF-κB, and c-Jun (28, 29, 31). We have also demonstrated very recently that A238L downregulates p300 transactivation through a mechanism that involves PKC-θ in T cells (28). However, the molecular mechanism underlying the A238L-mediated control of the expression of proinflammatory genes during the infection remains largely unclear. Since p300 is essential to promote the expression of COX-2 (16) and TNF-α (54) during the initiation of the inflammatory response mediated by macrophages and T lymphocytes, we decided to further investigate the molecular mechanism by which A238L exerts its inhibitory activity during the infection and to analyze the role of p300 in the A238L-mediated inhibition of the expression of these proinflammatory factors, using the Vero cell line and porcine macrophages as models of ASFV infection (19).
We demonstrate here that A238L inhibits the expression of the proinflammatory molecules COX-2 and TNF-α in ASFV-infected Vero cells, by blocking the recruitment of the coactivator p300 to their promoters, destabilizing the enhanceosome formed on such regulatory sequences. Furthermore, we report not only that A238L inhibits the activity mediated by the amino-terminal TAD of p300 during the viral infection but also that the amino-terminal CH1 regulatory region of p300 is essential in the A238L-mediated inhibition on COX-2 and TNF-α promoters. Furthermore, the expression of the viral protein during the infection of porcine alveolar macrophages with the ASFV virulent strain E70 efficiently interfered with the association between PKC-θ and the amino-terminal TAD of p300, thus impairing the phosphorylation and activation of the coactivator. Finally, we demonstrate that the viral protein requires the residue serine 384 to carry out its inhibitory function through the ASFV infection and that the expression of the constitutively active mutant S384D fully reverts the inhibition mediated by A238L in ASFV-infected Vero cells.
These findings shed new light on how viruses can interact with and modulate the activity of the transcriptional coactivator p300, thus altering the host cell immune and proinflammatory gene expression patterns. Hence, these results represent a new and unique viral mechanism of evasion, since to our knowledge, it has never been shown that the direct inhibition of the amino-terminal TAD of p300 by a viral product results in the control of the host protective response against the viral infection.
MATERIALS AND METHODS
Abbreviations.
AP-1, activating protein 1; ASFV, African swine fever virus; ATCC, American Type Culture Collection; CBP, CREB binding protein; COX-2, cyclooxygenase-2; CREB, cyclic AMP-response element binding protein; DMEM, Dulbecco's modified Eagle medium; HAT, histone acetyltransferase; hpi, hours postinfection; HPV, human papillomavirus; HTLV-1, human T-cell leukemia virus type 1; IgG, immunoglobulin G; Ion, calcium ionophore; MBP, myelin binding protein; MOI, multiplicity of infection; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-kappa B; PFU, plate-forming units; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; RLU, relative luciferase units; RNA Pol II, RNA polymerase II; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SV40, simian virus 40; TAD, transactivation domain; TFIIB, transcription factor II B; TNF-α, tumor necrosis factor alpha; wt, wild type.
Cell culture, viruses, and reagents.
Vero (African green monkey kidney) cells were obtained from the ATCC and grown in DMEM (Gibco) supplemented with 5% fetal bovine serum. Porcine alveolar macrophages were obtained by bronchoalveolar lavage of pigs and cultured in DMEM supplemented with 10% homologous swine serum, 2 mM l-glutamine, 100 U of gentamicin per ml, and nonessential amino acids (9). The Jurkat human leukemia T-cell line was obtained from the ATCC and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. Both media were supplemented with 2 mM l-glutamine, 100 U of gentamicin per ml, and nonessential amino acids. Cells were grown at 37°C in 7% CO2 in air saturated with water vapor. The generation of Jurkat cells (Jurkat-A238L) and cells from its control cell line (Jurkat-pcDNA) that stably expressed A238L was described previously (29). Jurkat cells were stimulated by PMA (Sigma) at 15 ng/ml and A23187 Ion (Sigma) at 1 μM. The Vero-adapted ASFV strain Ba71V was propagated and titrated by plaque assay on Vero cells as described previously (19). The A238L-defective mutant ΔA238L viruses cloned in both the Vero-adapted Ba71V strain and the E70 virulent strain were generated by insertion of the Escherichia coli β-glucuronidase gene into the A238L open reading frame, as previously described (23, 28). Both Ba71Vwt and Ba71VΔA238L viruses were used to infect Vero cells at a MOI of 5 PFU per cell. The virulent ASFV strain E70 was propagated and titrated by plaque assay on swine alveolar macrophages as described previously (9).
Western blot analysis.
Nuclear extracts from Jurkat-pcDNA and Jurkat-A238L cells, unstimulated or stimulated with PMA/Ion, were prepared as previously described (29). To prepare whole-cell extracts of mock-infected or ASFV-infected porcine macrophages, the cells were washed twice with phosphate-buffered saline and lysed in radio-immunolabeling protein assay buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, and 0.25% Na-deoxycholate and supplemented with protease inhibitor mixture tablets (Roche). In each case, the protein concentration was determined by the bicinchoninic acid spectrophotometric method (Pierce). Cell lysates were fractionated by SDS-PAGE and electrophoretically transferred to an Immobilon extra membrane (Amersham), and the separated proteins reacted with specific primary antibodies. The antibodies used were the following: GAL4 (sc-577; Santa Cruz Biotechnology), p300 (sc-584; Santa Cruz Biotechnology), PKC-θ (sc-212; Santa Cruz Biotechnology), RNA Pol II (sc-9001; Santa Cruz Biotechnology), and TFIIB (sc-225; Santa Cruz Biotechnology). Membranes were exposed to horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences), followed by chemiluminescence (ECL; Amersham Biosciences) detection by autoradiography. TINA 2.0 software was used to perform densitometric analysis.
In vitro DNA-protein binding assay.
Binding of proteins to the specific probes of the TNF-α and COX-2 promoters was analyzed by a DNA-protein binding assay, by using streptavidin-coated beads to bind biotinylated DNA probe, which was incubated with nuclear extract proteins. Biotine-labeled, double-stranded oligonucleotide probes corresponding to the different binding sites were synthesized by Isogen. The probes used were the following: κ1 (5′-biotine-CCCCGGGGCTGTCCCAGGC-3′), κ2 (5′-biotine-GTGTGAGGGGTATCCTTGATG-3′), (−157)NFAT (5′-biotine-CGATGGAGAAGAAACCGAG-3′), and CRE/κ3 (5′-biotine-AGATGAGCTCATGGGTTTCTCCACC-3′) for the human TNF-α promoter; and distal NF-κB (5′-biotine-GGAGAGGGGATTCCCTGCGC-3′), proximal NF-κB (5′-biotine-GAGTGGGGACTACCCCCTCTG-3′), distal NFAT (5′-biotine-GAGGAGGGAAAAATTTGTG-3′), and proximal NFAT (5′-biotine-AGGCGGAAAGAAACAGTCATTT-3′) for the human COX-2 promoter. The control probe was an irrelevant DNA sequence (5′-biotine-TTACCAACTGAGCCATCTCC-3′). The binding assay was performed as described previously (31). Briefly, 500 μg of nuclear extract proteins from stimulated or unstimulated Jurkat-pcDNA and Jurkat-A238L cells was mixed with 5 μg of biotinylated probe and 50 μl of 4% streptavidin beaded agarose (Sigma) with 70% slurry. The mixture was incubated at room temperature for 1 h with shaking. Beads were then pelleted and washed three times with ice-cold phosphate-buffered saline. The bound proteins were eluted in loading buffer and separated by 4 to 15% PAGE, followed by Western blot analysis with the indicated antibodies.
Plasmid constructs.
The pcDNA-A238L expression plasmid was generated as described previously (29). The COX-2 promoter construct containing the full-length promoter sequence fused to a firefly luciferase reporter gene, named p2-1900(−1796, +104), was generated as described previously (35). The human TNF-α promoter construct containing the full-length promoter sequence fused to a firefly luciferase reporter gene, named pTNF(−1311)luc, was generated as described previously (49). The GAL4-p300 full-length construct and the mutants GAL4-p300(192-703), GAL4-p300(1-1301), and GAL4-p300(1239-2414) were kindly provided by Neil D. Perkins and generated as described previously (50). The GAL4-luciferase construct (pGAL4-Luc) contains five GAL4 DNA consensus binding sites derived from the yeast GAL4 gene fused to a luciferase reporter gene (43). The p300wt expression plasmid pCI-p300 and its HAT deletion mutant, pCI-p300ΔHAT, were generous gifts from Joan Boyes and were generated as described previously (7). The expression plasmids pCMV-E2-p300ΔCH1(Δ61-1032) and pCMV-E2-p300ΔCH2(Δ1139-1394) were kindly provided by Richard G. Pestell and were generated as described previously (2). The full-length and amino-terminal TAD p300 serine 384-to-alanine (S384A) and serine 384-to-aspartic acid (S384D) mutant expression plasmids, GAL4-p300(S384A) and GAL4-p300(S384D), respectively, were generated by using a QuickChange site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. Oligonucleotides used for mutagenesis were the following: p300(S384A), 5′-CCACATGACACACTGCCAGGCAGGCAAGTCTGCCAAGTGGC-3′ (forward) and 5′-GCCACTTGGCAGACTTGCCTGCCTGGCAGTGTGTCATGAGG-3′ (reverse); and p300(S384D), 5′-CCACATGACACACTGCCAGGACGGCAAGTCTGCCAAGTGGC-3′ (forward) and 5′-GCCACTTGGCAGACTTGCCGTCCTGGCAGTGTGTCATGAGG-3′ (reverse). Serine-to-alanine or -aspartic acid substitutions are underlined. The Renilla luciferase plasmid pRL-tk-luc (Promega) was used to evaluate transfection efficiency.
Transfection and luciferase assays.
Vero cells, Jurkat cells, and porcine alveolar macrophages were transfected with 250 ng of specific reporter plasmids and/or 1 μg of expression plasmids per 106 cells using the Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions and mixing in Opti-MEM (Invitrogen) in a six-well plate. The cells were incubated at 37°C during 4 h, washed, and incubated in serum-free medium. As a transfection control for luciferase assays, the Renilla luciferase control plasmid pRL-TK (Promega) was cotransfected in all of the experiments. Sixteen hours after transfection, Jurkat-pcDNA and Jurkat-A238L cells were stimulated with 15 ng/ml of PMA plus 1 μM Ion over 4 h (PMA/Ion), Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell, and macrophages were infected with E70wt or E70ΔA238L at a MOI of 5 PFU/cell. At the indicated times, cells were lysed with 200 μl of cell culture lysis reagent (Promega) and microcentrifuged at full speed for 5 min at 4°C, and 20 μl of each supernatant was used to determine firefly and Renilla luciferase activity in a Monolight 2010 luminometer (Analytical Luminescence Laboratory) using a dual luciferase assay system (Promega). Transfections were normalized to Renilla luciferase activity, and results were expressed as the RLU after normalization of protein concentration determined by the bicinchoninic acid method, as indicated in the figure legends. Transfection experiments were performed in triplicate, and the data are presented as the means of the RLU (means ± standard deviations).
mRNA analysis.
Total RNA was prepared from ASFV-infected Vero cells by the Trizol reagent RNA protocol (Invitrogen). Total RNA (1 μg) was reverse transcribed into cDNA with a RevertAid first-strand cDNA synthesis kit (MBI Fermentas) and used for PCR amplification with the addition of Taq DNA polymerase (Roche) following the manufacturer's instructions. Specific primers used in PCRs were the following: for human COX-2, 5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ (forward) and 5′-AGATCATCTCTGCCTGAGTATCTT-3′ (reverse); for human TNF-α, 5′-TCAGATCATCTTCTCGCACCC-3′ (forward) and 5′-GACTCGGCAAAGTCGAGATAG (reverse); and for human β-actin, 5-′GAGAAGATGACCCAGATCATG-3′ (forward) and 5-′TCAGGAGGAGCAATGATCTTG-3′ (reverse). The PCRs were performed by 30 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1 min. Amplified cDNAs were separated by agarose gel electrophoresis, and bands were visualized by ethidium bromide staining.
Coimmunoprecipitation.
Whole-cell extracts were prepared from porcine macrophages infected with E70wt or E70ΔA238L at a MOI of 5 (PFU/cell), and their protein concentrations were determined as described above. The extracts were incubated with a specific antibody against PKC-θ (sc-212; Santa Cruz Biotechnology), or a rabbit or mouse preimmune normal IgG as a negative control, at a final concentration of 4 μg/ml. The samples were incubated at 4°C overnight. Protein A/G-Sepharose beads (Sigma-Aldrich) were added, incubated for 3 h at 4°C, and centrifuged. The beads were washed three times with wash buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40). The immunoprecipitates were mixed with SDS loading buffer and analyzed by 4 to 15% SDS-PAGE, followed by Western blotting or in vitro kinase assay.
Solid-phase in vitro phosphorylation kinase assay.
We used 2 μg of MBP (sc-4113; Santa Cruz Biotechnology) or 200 ng of purified recombinant p300 (Active Motif) as the substrate for in vitro phosphorylation, in which immunoprecipitated PKC-θ from ASFV-infected macrophages was assayed. The cells were lysed in radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1% Nonidet P-40 and supplemented with phosphatase inhibitors (1 mM NaVO3, 10 mM NaF, and 10 mM Na2MoO4) and protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 1 μg of pepstatin, 2 μg of leupeptin, and 2 μg of aprotinin per ml). Cleared extracts were incubated overnight with 4 μg of specific antibody against PKC-θ (sc-212; Santa Cruz Biotechnology) to immunoprecipitate it. Precipitates were finally resuspended in kinase buffer containing 20 mM HEPES (pH 7.6), 20 mM MgCl2, 20 mM β-glycerophosphate, 20 μM ATP, and 1 μCi of [γ-32P]ATP (specific activity, 3,000 Ci/mol) supplemented with phosphatase inhibitors and mixed with the corresponding substrate. After 30 min at 30°C, the kinase reaction was terminated by washing with TNT buffer containing 20 mM Trizma base (pH 7.5), 200 mM NaCl, and 1% Triton X-100 and supplemented with protease inhibitor mixture tablets (Roche). Phosphorylated proteins were separated in SDS-12% PAGE, dried, and developed by autoradiography.
RESULTS
p300 overexpression reverts the A238L-mediated inhibition of inducible inflammatory mediators.
A238L efficiently modulates the expression of NFAT/NF-κB target genes in different cell types (28, 29, 31). Regarding this aspect, we have recently described that A238L inhibits p300 intrinsic activity mediated by its amino-terminal TAD when ectopically expressed in Jurkat cells (30). However, the mechanism used by the viral protein to block the inflammatory response during the viral infection is not fully described. To further study this molecular mechanism, Vero cells were cotransfected with wt p300 (pCI-p300wt) or p300 HAT-defective mutant (pCI-p300ΔHAT) expression plasmids, together with different luciferase constructs under the control of COX-2 and TNF-α human promoter sequences. Sixteen hours after transfection, cells were infected either with the parental Ba71Vwt virus or with the Ba71VΔA238L knockout mutant virus (MOI of 5 PFU/cell), as described in Materials and Methods, and at 18 h after infection, luciferase activity was measured in whole-cell extracts. As shown in Fig. 2A, Ba71Vwt virus, which contains the A238L gene into its genome, was able to control the induction of COX-2 (left panel) and TNF-α (right panel) promoters simultaneously. However, this was not the case after Ba71VΔA238L virus infection, in which the lack of the A238L gene resulted in activation of the analyzed promoters. Interestingly, overexpression of wt p300, but not overexpression of the p300ΔHAT-defective mutant, restores the level of transcription of those promoters to the levels found in Ba71VΔA238L mutant virus-infected cells, indicating that the inhibitory effect of A238L during the viral infection involves the transcriptional activation mediated by the coactivator p300.
FIG. 2.
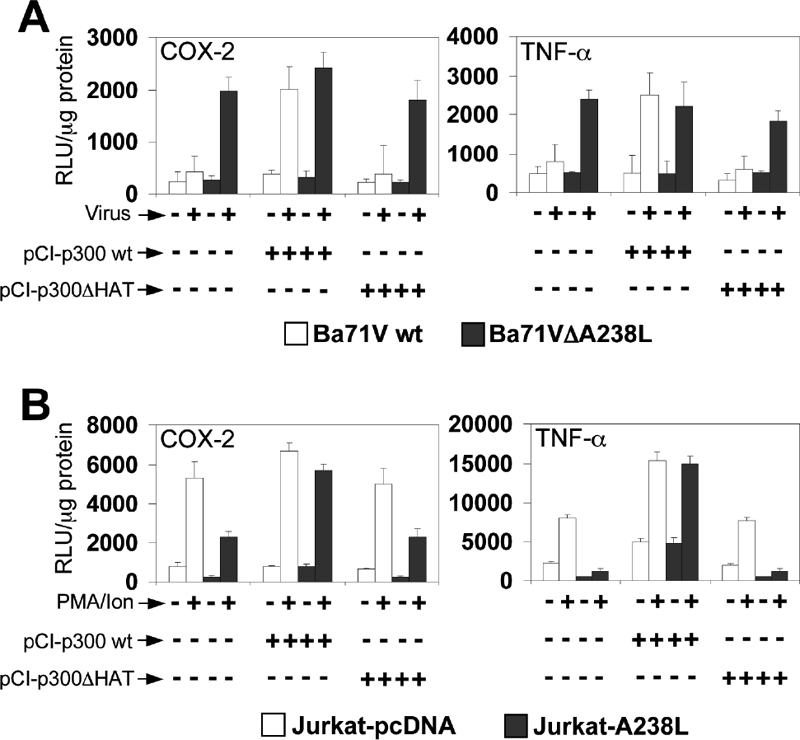
Effect of p300 overexpression in the transcriptional activation of COX-2 and TNF-α promoters. (A) Vero cells were transfected with COX-2 (left panel) or TNF-α (right panel) promoter reporter plasmids (250 ng/106 cells), together with pCI-p300wt or p300 HAT deletion mutant (pCI-p300ΔHAT) expression plasmids (1 μg/106 cells). Sixteen hours after transfection, the cells were mock infected (−) or infected (+) with the Vero-adapted isolate Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. Whole-cell extracts were prepared at 24 hpi and assayed for luciferase activity. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown. (B) Jurkat-pcDNA or Jurkat-A238L cells were transiently transfected with COX-2 (left panel) or TNF-α (right panel) promoter reporter plasmids (250 ng/106 cells), together with pCI-p300wt or p300 HAT deletion mutant (pCI-p300ΔHAT) expression plasmids (1 μg/106 cells). Sixteen hours after transfection, the cells were cultured in the absence (−) or presence (+) of 15 ng/ml of PMA plus 1 μM Ion (PMA/Ion) during 4 h and assayed for luciferase activity. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. Results from triplicate assays are shown in RLU per μg of protein (means ± standard deviations).
To corroborate the results obtained during the infection, we used Jurkat T cells stably expressing the viral protein (Jurkat-A238L) or control cells (Jurkat-pcDNA), which were cotransfected with the COX-2 and TNF-α promoter-driven luciferase constructs, together with the expression plasmids pCI-p300wt or pCI-p300ΔHAT. As expected, after PMA/Ion stimulation, the results were similar to those obtained in ASFV-infected Vero cells. As shown in Fig. 2B, A238L was able to inhibit the COX-2 (left panel) and TNF-α (right panel) reporter constructs in resting and stimulated Jurkat-A238L cells, compared to the expression levels obtained in the Jurkat-pcDNA control cell line. Moreover, the inhibition induced by the expression of A238L was reversed by enforced expression of p300, in which the HAT domain of p300 was required for full reversion of this inhibition, thus corroborating the results obtained during ASFV infection.
Taken together, these data indicate that the viral protein modulates p300-mediated inducible transcription of inflammatory genes both during the viral infection and when ectopically expressed in a different cell line.
A238L interferes with the recruitment of the transcriptional initiation complex on COX-2 and TNF-α.
To further investigate the mechanism by which A238L accomplishes the p300-dependent inhibition of COX-2 and TNF-α promoters, we analyzed the role of the viral protein in the recruitment of the coactivator p300, the inducible RNA Pol II, and the TFIIB basal transcription factor in the formation of an inducible transcriptional complex, also known as the enhanceosome (51), necessary for the complete transcriptional activation of the proinflammatory genes for COX-2 (16) and TNF-α (20). To achieve this, we performed DNA-protein binding experiments using biotinylated double-strand DNA probes corresponding to all the NFAT-, NF-κB-, or AP-1-responsive elements present in COX-2 and TNF-α human promoter sequences activated in competent cells, such as Jurkat human T cells. Probes were incubated with nuclear extracts from Jurkat-pcDNA or Jurkat-A238L cells treated with PMA/Ion or untreated, and the complex was pulled down with streptavidin-agarose beads, as described in Materials and Methods. Finally, the proteins in the complex were analyzed by Western blotting using specific antibodies against p300, RNA Pol II, and TFIIB. As shown in Fig. 3A, A238L strongly displaced the binding of p300 to the distal and proximal NFAT and NF-κB probes, especially from the distal NFAT probe, one of the most important enhancer elements in the COX-2 promoter regulation in T cells (35). In addition, RNA Pol II was recruited to both the distal and the proximal NFAT enhancer elements and, to a lesser degree, to the distal and proximal NF-κB sites, and it was displaced by the presence of the viral protein. As expected, TFIIB maintained the binding baseline levels in all the probes assayed and all experimental conditions, being unaffected by the presence of A238L. Concerning the TNF-α promoter, p300 binding to the NF-κB (κ1), the (−157)NFAT, and the composite CRE/κ3 response elements was higher after stimulation of Jurkat cells, and more importantly, the presence of A238L destabilized the formation of the enhanceosome, diminishing dramatically the recruitment of the coactivator from these three probes (Fig. 3B). Moreover, inducible RNA Pol II was found preferentially bound to the (−157)NFAT and CRE/κ3 regulatory elements, essential enhancers in the modulation of the TNF-α promoter during a viral infection (20, 21), and it was strongly displaced from these probes in the presence of the A238L protein. In the case of the NF-κB (κ1) probe, RNA Pol II was correctly recruited, probably due to the activity of other coactivators, such as CBP, which is able to perform its function regardless of p300 (13, 20). In parallel to the results obtained for the COX-2 promoter, basal transcription factor TFIIB was not displaced by A238L from the analyzed probes.
FIG. 3.

A238L inhibits the transcriptional initiation complex recruitment to the enhancer elements located in COX-2 and TNF-α. Nuclear extracts from Jurkat-pcDNA and Jurkat-A238L cells untreated (−) or treated (+) with 15 ng/ml of PMA plus 1 μM Ion (PMA/Ion) for 4 h were incubated with the indicated biotinylated probes. (A) Distal and proximal NFAT binding sites (d-NFAT and p-NFAT) and distal and proximal NF-κB binding sites (d-NFκB and p-NFκB) from the COX-2 promoter. (B) κ1 and κ2 NF-κB binding sites, the (−157)NFAT binding site, and the composite element CRE/κ3 from the TNF-α promoter. The complexes were pulled down with streptavidin-agarose beads, as described in Materials and Methods. After extensive washing, proteins in the complexes were analyzed by Western blotting using antibodies against p300, RNA Pol II, or TFIIB. Inputs were also included to show the presence of the analyzed proteins in the nuclear protein extracts. A control probe was included to rule out unspecific binding of the analyzed proteins (data not shown).
Taken together, these data demonstrate that A238L is able to destabilize the formation of the enhanceosome through blocking the binding of p300 to the NFAT, NFAT/AP-1, NF-κB, and/or composite elements, such as TNF-α CRE/κ3, and then preventing the recruitment of the RNA Pol II, which is essential for the complete transcriptional activation of COX-2 and TNF-α promoters.
The p300 amino-terminal TAD is inhibited during ASFV infection.
Since we have previously demonstrated that ectopically expressed A238L inhibits p300-mediated transactivation in T cells (30), we further planned to delineate the p300 region, through which A238L performs its inhibitory function in vivo, in order to more accurately analyze the biological role of the p300-mediated mechanism used by ASFV to evade the inflammatory response. To achieve this, we used different GAL4-p300 constructs containing the DNA binding domain of the GAL4 yeast transcriptional factor fused to the following p300 regions: the full-length sequence of p300 [named GAL4-p300(FL)]; the carboxyl-terminal half of p300 [named GAL4-p300(1239-2414)]; and the amino-terminal transactivation region, containing the CH1 and KIX regulatory domains [named GAL4-p300(192-703)]. First, Vero cells were cotransfected with the GAL4-luc reporter plasmid together with the indicated GAL4-p300 constructs. Sixteen hours after transfection, the cells were mock infected (0 h) or infected with the parental virus Ba71Vwt or with the deletion mutant Ba71VΔA238L, as described in Materials and Methods. We also added PMA as an inducer, to demonstrate that each of the constructs transfected are transcriptionally active during the infection and that the observed inhibition of the activity is due to the expression of A238L during the infection with the wt virus. At the indicated times after infection, whole-cell extracts were prepared and luciferase activity was measured. As shown in Fig. 4, the infection with the defective mutant Ba71VΔA238L increased the transcriptional activity of the GAL4-p300(FL) construct (Fig. 4A) and the activity mediated by the CH1/KIX region [GAL4-p300(192-703)] (Fig. 4B), without altering the transcriptional activity of the carboxyl-terminal half [GAL4-p300(1239-2414)] (Fig. 4C) of the coactivator. In contrast, the viral infection using the Ba71Vwt virus did not produce a significant change in the transactivation activity of all GAL4-p300 constructs assayed, which maintained their activity at baseline. This indicates that the A238L protein, expressed by the parental Ba71V virus, but not by the deletion mutant, is able to control the transcriptional activity of p300 during the viral infection, interfering specifically with the transcriptional activity of the CH1 and KIX domains located in the amino-terminal TAD of p300, since the activation of the constructions containing the CH1 and KIX regulatory domains [GAL4-p300(192-703)] is strongly upregulated during the infection with the Ba71VΔA238L mutant, where the viral protein is not expressed.
FIG. 4.
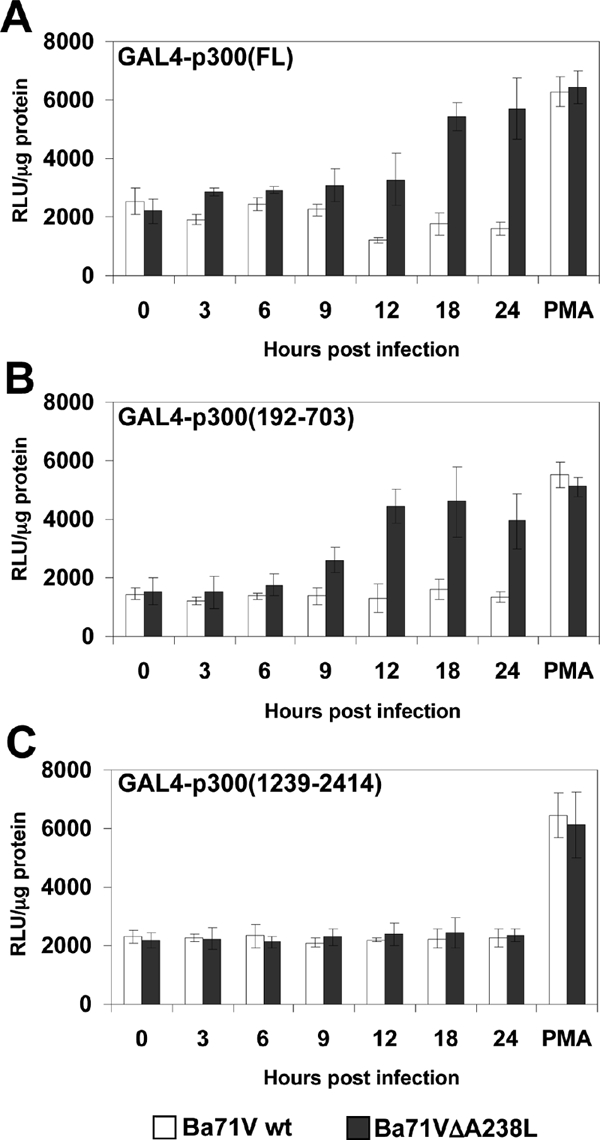
A238L inhibits p300 transactivation in ASFV-infected Vero cells. Vero cells were cotransfected with the reporter plasmid GAL4-luc (250 ng/106 cells) and with the following GAL4-fused mutant constructs of p300, at a final concentration of 250 ng/106 cells: panel A, full-length p300 [GAL4-p300(FL)]; panel B, mutant p300 construct encoding amino acids from 192 to 703 [GAL4-p300(192-703)]; and panel C, mutant p300 construct encoding amino acids from 1239 to 2414 [GAL4-p300(1239-2414)]. Sixteen hours after transfection, Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. A sample was infected and also activated with 15 ng/ml of PMA, to check the functionality of the constructs assayed. At the indicated poststimulation times, whole-cell extracts were prepared and luciferase activity was assayed. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown.
The CH1 and KIX regulatory domains of p300 are essential for A238L-mediated inhibition of proinflammatory gene promoters.
Once it was demonstrated that A238L blocks the transcriptional activity mediated by the amino-terminal TAD of p300, we further determined if this mechanism was responsible for the A238L-mediated inhibition of COX-2 and TNF-α promoters after ASFV infection. It has been previously shown that the CH1 and KIX domains, located in the amino-terminal region of p300, are essential in the enhancement of the transcriptional activity of NFAT, NF-κB, and c-Jun (56, 58). This suggests that the virus might exploit A238L to specifically inhibit these regulatory domains of p300, thus controlling COX-2 and TNF-α expression throughout the infection. To analyze this hypothesis, Vero cells were cotransfected with TNF-α and COX-2 reporter constructs, together with the expression plasmids pCMV (vector alone), pCMV-p300(ΔCH1), or pCMV-p300(ΔCH2), and afterwards the cells were mock infected or infected with Ba71Vwt or Ba71VΔA238L viruses at 5 PFU/cell. At the indicated postinfection times, luciferase activity was measured. When empty pCMV plasmid was cotransfected, infection with the defective Ba71VΔA238L virus induced COX-2 (Fig. 5A, left panel) and TNF-α (Fig. 5B, left panel) promoter activities after 6 hpi. However, this effect was not observed when Ba71Vwt was used, as expected since the A238L gene is expressed during the parental virus infection. Next, we found that overexpression of p300ΔCH1 was unable to reverse the inhibition induced by A238L throughout Ba71Vwt viral infection (Fig. 5A and B, middle panels). In contrast, the construction lacking the p300 carboxyl-terminal regulatory domain CH2 (p300ΔCH2), but expressing the CH1 domain, completely restored the expression of the luciferase reporter analyzed in Ba71Vwt-infected cells (Fig. 5A and B, right panels), thus counteracting the A238L-mediated inhibition on such COX-2 and TNF-α promoters, restoring the levels detected during the infection with Ba71VΔA238L. Therefore, these data indicate that the CH1 regulatory region, located in the amino-terminal TAD of p300, is essential for the control of the proinflammatory gene promoters mediated by the viral protein A238L during ASFV infection.
FIG. 5.

CH1 and KIX domains of p300 are required in the A238L-mediated inhibition of COX-2 and TNF-α gene expression. Vero cells were transiently cotransfected with COX-2 (A) or TNF-α (B) promoter reporter plasmids (250 ng/106 cells) as indicated in the figure, together with pCMV-p300(ΔCH1) or pCMV-p300(ΔCH2) deletion mutant expression plasmids (1 μg/106 cells). Sixteen hours after transfection, Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. At the indicated postinfection times, whole-cell extracts were prepared and luciferase activity was assayed. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown.
Serine 384 within the CH1 domain of p300 is essential in the A238L-mediated inhibition of the host inducible transcription.
After demonstrating that the CH1 and KIX regulatory domains seem to be essential in the inhibition executed by Ba71wt virus on the transcriptional enhancement of COX-2 and TNF-α promoters, we further analyzed the molecular mechanism underlying A238L-mediated inhibition, dissecting the specific p300 regulatory domain affected by the viral protein. Since we have very recently shown that the phosphorylation of serine 384 is essential in the regulation of the amino-terminal TAD activity of p300 (30), we hypothesized that this residue might play an important role in the modulation carried out by the ASFV Ba71Vwt on the activity of the p300-induced promoters, COX-2 and TNF-α. To address this point, we separately cotransfected Vero cells with TNF-α and COX-2 promoter activity reporter constructs, together with the expression plasmids pCMV, pCMV-GAL4-p300wt, or pCMV-GAL4-p300(S384A). As shown above, wt virus Ba71V retains COX-2 (Fig. 6A, top panel) and TNF-α (Fig. 6B, top panel) promoter activities at baseline when control pCMV empty plasmid was cotransfected, whereas, as expected, infection with Ba71VΔA238L strongly induced the transcriptional activation of both of the promoters studied. Interestingly, overexpression of wt p300 reversed the inhibition induced by A238L during the viral infection (Fig. 6A and B, mid-top panels) but overexpression of the p300(S384A) mutant was not only unable to augment the activity of the analyzed promoters during parental Ba71V viral infection but also dramatically diminished the induction of the analyzed promoters during the infection with Ba71VΔA238L (Fig. 6A and B, mid-bottom panels), confirming that this residue is absolutely required for p300-mediated induction of COX-2 and TNF-α promoters. Since an accumulative effect in the inhibition mediated by the p300(S384A) mutant and A238L was not observed, it can be proposed that A238L might block the phosphorylation of serine 384, thus supporting the hypothesis that involves this serine in the A238L-mediated inhibition. To further confirm this hypothesis, we generated mutant constructs of p300 in which serine 384 has been substituted by aspartic acid, which is a phosphoserine mimicking substitution that can produce constitutively active mutants (37). The S384D mutant construct was then used in experiments similar to those described above, showing that in ASFV-infected Vero cells, the ectopic expression of the S384D mutant construct not only reversed the inhibition induced by the A238L protein (expressed during the Ba71Vwt virus infection) on the activity of the COX-2 (Fig. 6A, bottom panel) and TNF-α (Fig. 6B, bottom panel) promoters, but it also induced a stronger activation of these promoters. Taken together, these results not only indicate that the amino-terminal TAD of p300 is responsible for the activation of these proinflammatory promoters but also indicate the involvement in this activation of serine 384, located within this domain.
FIG. 6.

Serine 384 within the CH1 regulatory domain of p300 is essential in the A238L-mediated inhibition of the transcription of proinflammatory genes. Vero cells were transiently cotransfected with COX-2 (A) or TNF-α (B) promoter reporter plasmids (250 ng/106 cells) as indicated in the figure, together with pCMV-GAL4-p300wt or the mutant pCMV-GAL4-p300(S384A) or pCMV-GAL4-p300(S384D) expression plasmids (1 μg/106 cells). Sixteen hours after transfection, Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. At the indicated postinfection times, whole-cell extracts were prepared and luciferase activity was assayed. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown.
p300 amino-terminal serine 384 role on the endogenous COX-2 and TNF-α mRNA expression and p300 transactivation inhibited by A238L during the ASFV infection.
Considering the importance of the mechanism by which A238L regulates COX-2 and TNF-α promoters through the serine 384 of p300 during ASFV infection, we further explored the consequence of this regulatory mechanism on the inflammatory gene expression in vivo in ASFV-infected cells. To address this point, we transfected Vero cells with plasmids encoding the different point mutant full-length p300 expression vectors: p300wt, the p300(S384A) mutant, the p300(S384D) mutant, and empty pCMV as a control. Sixteen hours after transfection, the cells were infected with Ba71Vwt or Ba71VΔA238L viruses, and at the indicated postinfection times, total RNA was isolated and analyzed by reverse transcriptase PCR to measure COX-2 and TNF-α mRNA levels, as described in Materials and Methods. Figure 7 shows the expression of specific mRNA for endogenous COX-2 and TNF-α, demonstrating that in cells transfected with the control plasmid pCMV, the absence of A238L during the infection when the deletion mutant Ba71VΔA238L is used allowed the expression of both COX-2 and TNF-α at 6 and 18 hpi (Fig. 7, far left panel). In contrast, when wt Ba71V was used, the presence of A238L during the infection clearly downregulated the expression of both inflammatory molecules. Not only that, but in agreement with the data shown above, p300wt expression reverted the A238L-induced inhibition for both COX-2 and TNF-α (Fig. 7, mid-left panel). More interestingly, we found higher levels of COX-2 and TNF-α transcripts in infected cells expressing the construction p300(S384D), whereas in cells transfected with the p300(S384A) construct, COX-2 and TNF-α mRNA levels were clearly diminished after infection with either Ba71Vwt or Ba71VΔA238L viruses (Fig. 7, mid-right and far right panels, respectively). These results demonstrate that the molecular mechanism by which ASFV modulates the endogenous expression of the inflammatory response in the infected cell relies on the viral protein A238L through the control of serine 384 from p300, as might be expected from the data obtained above using reporter assays to analyze the activity of the promoters of the specific genes studied.
FIG. 7.

p300 amino-terminal serine 384 role on the endogenous COX-2 and TNF-α mRNA expression inhibited by A238L during the ASFV infection. Vero cells were transfected with the different full-length p300 expression vectors: mid-left panel, p300wt; mid-right panel, p300(S384A) mutant; far right panel, p300(S384D) mutant; and far left panel, empty pCMV as a control. Sixteen hours after the transfection, the cells were infected with Ba71Vwt or Ba71VΔA238L viruses, and at the indicated postinfection times, total RNA was isolated and analyzed by reverse transcriptase PCR to measure COX-2 and TNF-α mRNA levels. A control using oligonucleotides for β-actin was included to rule out differences in PCR amplification. A control of viral p72 (capsid protein) and A238L were also used to rule out differences in the infectivity (data not shown). Amplified DNA was separated on agarose gels. The densitometric analysis shows the ratio between amplified COX-2 or TNF-α mRNA and the β-actin loading control present in each sample, from three independent experiments (means ± standard deviations).
Next, and due to the important role observed for serine 384 in the control of p300 function and expression of COX-2 and TNF-α, and in an attempt to explore the contribution of this residue in the intrinsic transactivation ability of p300 modulated by ASFV, we analyzed the transcriptional activity during the viral infection of both the full-length [GAL4-p300(FL)] and amino-terminal [GAL4-p300(192-703)] constructs in their wt version and serine 384-to-alanine or -aspartic acid substitution mutant versions. Figure 8 shows that the S384A mutation abrogates the transcriptional activity induced during the infection with Ba71Vwt and Ba71VΔA238L viruses for both p300 full-length (Fig. 8A) and amino-terminal TAD (Fig. 8B) constructs (center panels), compared with the results obtained with the p300wt construct (Fig. 8A and B, left panels). Moreover, we could demonstrate that the transactivation of both the full-length and amino-terminal TAD constructs of p300, induced by ASFV infection in the presence of the p300(S384D) mutant construct, not only reached similar levels to those observed by the wt version of p300 after infection with Ba71Vwt and Ba71VΔA238L but also completely recovered the inhibitory effect induced by the presence of A238L upon infection with Ba71Vwt (Fig. 8A and B, right panels), indicating that the regulatory mechanism induced by ASFV through the expression of A238L is abolished when the constitutively active, artificial phosphorylated version of serine 384 (S384D) is present during the infection.
FIG. 8.

Mutation of p300 serine 384 to alanine results in a dominant negative, whereas mutation of serine 384 to aspartic acid results in a constitutively active version of the amino-terminal TAD of p300. Vero cells were cotransfected with the reporter plasmid GAL4-luc (250 ng/106 cells) and with the following GAL4-fused mutant constructs of p300, at a final concentration of 250 ng/106 cells: GAL4-p300wt and the S384A mutant or S384D mutant, in both the full-length (A) and amino-terminal [GAL4-p300(192-703)] (B) constructs, as indicated in the figure. Sixteen hours after transfection, Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. At the indicated postinfection times, whole-cell extracts were prepared and luciferase activity was assayed. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown.
Taken together, these data demonstrate that the phosphorylation of serine 384 is an essential step in the intrinsic activation of p300 during the ASFV infection. Therefore, the viral protein is directly interfering with the transcriptional activity of the CH1 and KIX domains included in the amino-terminal TAD of p300, by a mechanism that involves signaling through the phosphorylation of the serine 384, thus likely controlling the proinflammatory gene expression pattern in the host cell.
PKC-θ reverses the inhibitory effect of A238L on the transcriptional activation of COX-2 and TNF-α promoters.
Transcriptional activation and repression of p300 is regulated by phosphorylation by many different kinases, with PKC-θ being a fundamental member for p300 activity enhancement (30). Although the relevance of this kinase has not been addressed in Vero cells, we next explored whether PKC-θ might be involved in the molecular mechanism by which the viral protein controls simultaneously the activity of COX-2 and TNF-α promoters, by interfering with the phosphorylation of serine 384 located in the amino terminal of p300. For this, we performed experiments similar to those described above, using ASFV-infected Vero cells, in which we analyzed the activity of the COX-2 and TNF-α promoter-driven reporter constructs, when cotransfected together with either pEF vector alone (Fig. 9, left panels) or an expression plasmid for PKC-θ, either in the wt (Fig. 9, middle panels) or in a constitutively active version [PKC-θ(A/E)] (Fig. 9, right panels), which has been shown to be required for full function of this kinase when ectopically overexpressed in vitro (55). The expression of active PKC-θ(A/E) completely reverted the inhibitory effect of the viral protein on the transcriptional activity of the COX-2 (Fig. 9A) and TNF-α (Fig. 9B) promoters observed in Vero cells infected with the BA71Vwt virus, which is able to control the activity of those promoters due to the expression of A238L during the infection. However, the enforced expression of the activated kinase PKC-θ(A/E) (Fig. 9A and B, right panels) completely restored the transcription of the three analyzed promoters to the levels observed in the control Vero cells infected with Ba71VΔA238L, which is unable to control the transcriptional activation of these promoters, due to the lack of the A238L gene, when pEF vector alone (Fig. 9A and B, left panels) or wt PKC-θ (Fig. 9A and B, middle panels) were overexpressed.
FIG. 9.

PKC-θ overexpression recovers the normal expression levels of COX-2 and TNF-α inhibited by A238L. Vero cells were transiently cotransfected with COX-2 (A) or TNF-α (B) promoter reporter plasmids (250 ng/106 cells) as indicated in the figure, together with pEF-PKC-θ(wt) or the constitutively active mutant pEF-PKC-θ(A/E) expression plasmids (1 μg/106 cells). Sixteen hours after transfection, Vero cells were infected with Ba71Vwt or Ba71VΔA238L at a MOI of 5 PFU/cell. At the indicated postinfection times, whole-cell extracts were prepared and luciferase activity was assayed. Extracts were normalized to Renilla luciferase activity as described in Materials and Methods. RLU per μg of protein from triplicate transfections (means ± standard deviations) are shown.
These data indicate that PKC-θ plays a key role in the activation of COX-2 and TNF-α promoters during the ASFV infection and strongly suggest the involvement of this kinase in the specific pathway targeted by the viral protein A238L to counteract the host cell antiviral inflammatory response during the infection.
A238L interferes with the association between the ASFV-induced PKC-θ and the amino-terminal region of p300.
To further explore the role of PKC-θ in ASFV infection, we first investigated the expression and activity of PKC-θ during the ASFV infection of porcine alveolar primary macrophages, the natural target cell of the virus. Nuclear extracts from macrophages infected with either the virulent parental E70wt isolate or the deletion mutant lacking the A238L gene (E70ΔA238L) were prepared. PKC-θ was immunoprecipitated from those extracts and used in an in vitro kinase assay, using as substrates either purified p300 or purified MBP. The results (Fig. 10A) showed that ASFV infection induces PKC-θ translocation after 24 hpi, an event apparently not regulated by A238L, since the presence of the protein (infection with E70wt) does not correlate with any change of the nuclear location of the kinase compared to that observed with the A238L-defective mutant virus infection. In parallel, the presence or absence of the viral protein does not affect the level of kinase activity; similar levels were displayed in all the samples analyzed. To determine whether the molecular mechanism by which A238L performs its inhibitory function during ASFV infection involves a link between the amino-terminal domain of p300 and PKC-θ, we used two different GAL4-p300 constructs containing the GAL4 DNA binding domain fused to the full-length sequence of p300 [GAL4-p300(FL)] or fused to the amino-terminal transactivation region, containing the CH1 and KIX regulatory domains [GAL4-p300(192-703)]. After transfecting these constructs, cells were mock infected or infected during 24 h with either E70wt or E70ΔA238L viruses. Nuclear extracts were prepared, and PKC-θ was immunoprecipitated. The precipitates were analyzed by Western blotting using an anti-GAL4 antibody to detect the levels of PKC-θ associated with either the full-length p300 protein (Fig. 10B, left panel) or its amino-terminal TAD [GAL4-p300(192-703)] (Fig. 10B, right panel). The results clearly showed the absence of the band corresponding to GAL4-p300 in macrophages infected with the parental E70 virus, which efficiently express the viral protein (data not shown). However, the absence of A238L during the infection with the deletion mutant E70ΔA238L correlates with the presence of a band corresponding with both of the p300 constructions assayed, demonstrating that A238L mediates the impairment of the association between the coimmunoprecipitated PKC-θ and both GAL4-p300(FL) and GAL4-p300(192-703).
FIG. 10.

A238L interferes with the association between PKC-θ and the amino-terminal region of p300 in ASFV-infected macrophages. (A) Porcine alveolar primary macrophages were mock infected or infected for 24 h with E70wt or E70ΔA238L, at a MOI of 5 PFU/cell. Then, nuclear extracts were prepared and used for immunoprecipitation (IP) with 4 μg of rabbit polyclonal specific antibody against PKC-θ or rabbit preimmune normal IgG as a negative control of immunoprecipitation. Immunoprecipitated PKC-θ was used in an in vitro kinase assay, using as substrate either purified p300 or purified MBP. Proteins were separated by SDS-8% PAGE and developed by autoradiography. Densitometric analysis shows the ratio between phosphorylated [32P]p300 and immunoprecipitated PKC-θ, from three independent experiments (means ± standard deviations). (B) Porcine alveolar macrophages were transfected with either the GAL4-p300(FL) expression plasmid (left panels) or the GAL4-p300(192-703) mutant expression plasmid (right panels). Twenty-four hours later, the cells were mock infected or infected for 24 h with E70wt or E70ΔA238L, at a MOI of 5 PFU/cell. Then, nuclear extracts were prepared and immunoprecipitated with 4 μg of rabbit polyclonal specific antibody against PKC-θ or rabbit preimmune normal IgG as a negative control of immunoprecipitation. Immunoprecipitates were analyzed by Western blotting (WB) with the same antibody (αPKCθ) to determine the levels of the kinase in the precipitate and with an anti-GAL4 antibody (αGAL4) to detect the levels of PKC associated with p300(FL) (left panels) or p300(192-703) (right panels). The densitometric analysis shows the ratio between coimmunoprecipitated PKC and total GAL4-p300 present in the precipitate, from three independent experiments (means ± standard deviations).
DISCUSSION
Since the CBP/p300 family of coactivators plays a pivotal role in the regulation of the cell gene expression pattern (34, 53), it would be predicted that several viral proteins, such as adenoviral E1A (41), SV40 T large antigen (18), E6 and E7 proteins from HPV (6, 46), or Tax protein from HTLV-1 (38), might target the CBP/p300 activation pathway to block the host defensive response. Previously, we have reported that the ASFV protein A238L plays a critical role in mediating inhibition of functional NFAT, NF-κB, and AP-1 response elements in the promoters of several proinflammatory molecules (28, 29, 31). In addition, we have very recently shown the A238L-mediated inhibition of the activity of the amino-terminal TAD of p300 through interference of PKC-θ phosphorylation in Jurkat T cells (30). CBP/p300 are essential during the initiation of the inflammatory response mediated by T lymphocytes and macrophages, promoting the expression of several immunoregulators, such as TNF-α (54) and COX-2 (16). Our present results show the blockade of the transcriptional activation of COX-2 and TNF-α proinflammatory gene promoters during ASFV infection and show that the viral protein A238L in ASFV-infected Vero cells mediates this blockade. Interestingly, this inhibition was reverted by p300 overexpression, thus suggesting that A238L likely uses a unique p300-dependent mechanism to block the establishment of an effective host proinflammatory response. In fact, a HAT activity-defective mutant of p300 was unable to recover the activity of any of the promoters assayed which were inhibited by the viral protein, therefore indicating that the inhibitory effect displayed by A238L specifically affects the p300 coactivator function and not the acetyltransferase activity.
p300 also plays a crucial role in the formation of transcriptional initiation complexes on specific promoters (8), interacting with members of the basal transcriptional machinery, including the TATA binding protein and associated factors such as TFIIB (39), and with the RNA Pol II holoenzyme (11). Regarding this aspect, our data show that the presence of the viral protein destabilized the formation of enhanceosomes on the TNF-α and COX-2 promoters, by displacing the recruitment of p300 from transcription factors bound to DNA probes similar to the NFAT, NF-κB and AP-1 binding sites present in these promoters, thus inhibiting the recruitment of the inducible RNA polymerase (RNA Pol II), a critical step in the p300-mediated initiation of gene expression. The most important alterations in the transcriptional complex were observed in the binding of the transcriptional coactivator to the NFAT probes of the COX-2 promoter and also in the (−157)NFAT and composite CRE/κ3 probes in the TNF-α promoter, indicating that A238L exploits a common mechanism to destabilize the enhanceosome but also that the viral protein is altering each transcriptional complex in different ways, therefore preventing the binding of p300 to the main inducible enhancers in each case. These results prompted us to assume the possibility of A238L directly blocking the transcriptional activity of p300 during the viral infection. In connection to this, our data here showed not only that the viral protein holds at baseline the transactivation mediated by p300 during the viral infection but also that this inhibition is carried out through the CH1 and KIX regulatory regions, located within the amino-terminal TAD of the coactivator, whose activity was blocked by A238L without altering the activity of the carboxyl-terminal TAD. It has been previously demonstrated that other viral proteins, such as adenoviral E1A, SV40 T large antigen, E6 and E7 from HPV, and HTLV-1 Tax protein, inhibit CBP and/or p300 function by interacting with their carboxyl-terminal region, where these viral products preferentially prevent CBP/p300 binding to CH3-interacting transcription factors, such as p53 or E2F, thus promoting the malignant transformation of the infected cells (10, 18, 46). In contrast, A238L inhibits the functionality of the amino-terminal TAD without altering carboxyl-terminal activity, showing for the first time to our knowledge that a viral protein that controls the amino-terminal transactivation of p300 generates an immunosuppressive effect during the viral infection.
Since we have very recently demonstrated that serine 384 seems to be essential in the full activation of the amino terminal of p300 in Jurkat cells (30), and to further explore the molecular basis of this regulatory viral mechanism during the viral infection, we generated site-directed mutant constructs in which serine 384 was substituted to alanine or aspartic acid, in both the full-length and amino-terminal versions of p300, to investigate whether the signaling pathway involving this residue was being interfered with by A238L to downregulate the host inflammatory response. Aspartic acid and alanine are generally accepted as standard substitutions of serine to mimic the phosphorylated and nonphosphorylated states, respectively (37). By using these constructs, we could demonstrate that the serine in position 384, or a form that mimics a phosphorylated state of p300 in this residue, is needed to revert the A238L-mediated inhibition on the activity of the analyzed inflammatory gene promoters. This could represent a new signaling pathway through which p300 integrates different stimulus-associated signals and triggers a proinflammatory gene expression pattern. In connection to this, we demonstrated that the transcriptional activity of p300 was completely abrogated when serine 384 was substituted to alanine, whereas substitution by aspartic acid resulted in a dramatically increased p300 activity during the infection. Interestingly, and partially explaining the link between the viral protein function and serine 384, we showed that A238L was unable to inhibit the transcriptional activity of the aspartic mutant construct during the viral infection, thus connecting the importance of this residue in the establishment of an adequate p300-mediated proinflammatory response to the fact that the virus exploits this pathway to evade the host response during the viral infection. We present evidence here that this is indeed the case with data obtained during the infection, which demonstrate that COX-2 and TNF-α endogenous mRNAs are inhibited during ASFV infection by the presence of the viral protein and that this inhibition is further reverted by the overexpression of both p300 and p300(S384D) but not by p300(S384A).
The transactivation mediated by the coactivator p300 is accurately regulated by posttranslational modifications (14, 26, 52, 57), with phosphorylation being one of the most important regulation signals, since the activity of CBP/p300 is controlled by different kinases, such as mitogen-activated protein kinases, protein kinase A, Akt, IKK-α, and PKC-δ (1, 12, 40, 47, 59). Although the functional effect of the kinases involved in p300 transactivation is known, the precise phosphorylation sites and the molecular mechanisms underlying this regulation by phosphorylation remain unclear. Different external signals from the environment trigger specific signaling cascades that will shape the p300/CBP activation pattern. We state here the relevance of PKC-θ in the activation of the amino-terminal domain of p300 via phosphorylation of the residue serine 384. We think that this mechanism is part of a complex signaling network regulating p300 under pathological conditions, such as viral infection, and represents a very important step in the activation of an antiviral innate immune response, coordinated by p300 and, as we show here, blocked by the viral product A238L.
In relation to that, we have very recently demonstrated that PKC-θ upregulates the activity of p300 in stimulated T-cell lymphoma (30), and this prompted us to hypothesize that A238L may be blocking the host cell inflammatory response by interfering with the PKC-θ signaling on p300 during ASFV infection in susceptible cells. Here we described that the viral protein blocks the accessibility of PKC-θ to its target in p300, the serine 384, likely by binding to p300, in a region containing the serine 384, which probably remains hidden in the presence of the viral protein. In addition to this, we demonstrated that expression of a constitutively active form of PKC-θ completely recovers the activity of the COX-2 and TNF-α promoters, which was inhibited by A238L in ASFV-infected Vero cells. Furthermore, PKC-θ was immunoprecipitated from porcine primary macrophages, which constitute the natural target of the ASFV in vivo, after infections with both E70wt and E70ΔA238L and was used in in vitro kinase assays. Interestingly, PKC-θ activity is not affected by ASFV infection, maintaining its activity during the course of both infections, thus suggesting that A238L is not affecting the enzymatic activity of the kinase. Not only that, but we also present evidence to assess that the presence of the viral protein impairs the association of PKC-θ and the amino-terminal “(192-703)” region of p300, thus blocking the amino-terminal transactivation activity of p300 in porcine macrophages infected with a virulent strain of ASFV.
In conclusion, these data demonstrate the complex molecular mechanism accomplished by the viral protein A238L to counteract the host cell inflammatory response. Since we previously reported that A238L interacts with the amino-terminal TAD of p300, blocking the access of PKC-θ to this domain, and since we demonstrate here that the proinflammatory gene promoters of COX-2 and TNF-α need a PKC-θ-mediated activation of p300 to reach their optimum level of expression, which is interfered with by the viral protein, we can conclude that the interference carried out by A238L on this signaling pathway is responsible for the blockage on the host cell inflammatory antiviral response. Importantly, the analysis of the inhibitory mechanism executed by A238L revealed a new checkpoint involved in the regulation of the activity of the p300 amino-terminal TAD that involves serine 384, which in turn controls the activity of the main transcriptional factors responsible for the triggering of an adequate signal-induced antiviral response.
This can lead us to further analyze, in the near future, different signaling pathways involved in the transcription of inflammatory and immunoregulatory genes controlled by CBP and p300 coactivators, which could be important both in the antiviral host cell inflammatory response and in related pathologies.
Acknowledgments
This work was supported by grants from Laboratorios Esteve, Ministerio de Educación y Ciencia (BFU2004-00298/BMC), and by an institutional grant from the Fundación Ramón Areces. A.G.G. was funded by Centro de Investigación en Sanidad Animal (CISA).
We thank Neil D. Perkins for the generous gift of GAL4-p300 constructs. We thank Marisa Nogal for excellent and continuous technical assistance and Maria J. Bustos for purification of several viral strains used here. The helpful advice of Angel L. Carrascosa is also very much appreciated.
Footnotes
Published ahead of print on 12 November 2008.
REFERENCES
- 1.Ait-Si-Ali, S., D. Carlisi, S. Ramirez, L. C. Upegui-Gonzalez, A. Duquet, P. Robin, B. Rudkin, A. Harel-Bellan, and D. Trouche. 1999. Phosphorylation by p44 MAP kinase/ERK1 stimulates CBP histone acetyl transferase activity in vitro. Biochem. Biophys. Res. Commun. 262157-162. [DOI] [PubMed] [Google Scholar]
- 2.Arany, Z., D. Newsome, E. Oldread, D. M. Livingston, and R. Eckner. 1995. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 37481-84. [DOI] [PubMed] [Google Scholar]
- 3.Arany, Z., W. R. Sellers, D. M. Livingston, and R. Eckner. 1994. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77799-800. [DOI] [PubMed] [Google Scholar]
- 4.Bannister, A. J., and T. Kouzarides. 1995. CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J. 144758-4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannister, A. J., T. Oehler, D. Wilhelm, P. Angel, and T. Kouzarides. 1995. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene 112509-2514. [PubMed] [Google Scholar]
- 6.Bernat, A., N. Avvakumov, J. S. Mymryk, and L. Banks. 2003. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 227871-7881. [DOI] [PubMed] [Google Scholar]
- 7.Boyes, J., P. Byfield, Y. Nakatani, and V. Ogryzko. 1998. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396594-598. [DOI] [PubMed] [Google Scholar]
- 8.Carey, M. 1998. The enhanceosome and transcriptional synergy. Cell 925-8. [DOI] [PubMed] [Google Scholar]
- 9.Carrascosa, A. L., J. F. Santaren, and E. Vinuela. 1982. Production and titration of African swine fever virus in porcine alveolar macrophages. J. Virol. Methods 3303-310. [DOI] [PubMed] [Google Scholar]
- 10.Chakravarti, D., V. Ogryzko, H. Y. Kao, A. Nash, H. Chen, Y. Nakatani, and R. M. Evans. 1999. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 96393-403. [DOI] [PubMed] [Google Scholar]
- 11.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 1142363-2373. [DOI] [PubMed] [Google Scholar]
- 12.Chawla, S., G. E. Hardingham, D. R. Quinn, and H. Bading. 1998. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science 2811505-1509. [DOI] [PubMed] [Google Scholar]
- 13.Chen, J. J., W. C. Huang, and C. C. Chen. 2005. Transcriptional regulation of cyclooxygenase-2 in response to proteasome inhibitors involves reactive oxygen species-mediated signaling pathway and recruitment of CCAAT/enhancer-binding protein delta and CREB-binding protein. Mol. Biol. Cell 165579-5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevillard-Briet, M., D. Trouche, and L. Vandel. 2002. Control of CBP co-activating activity by arginine methylation. EMBO J. 215457-5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colgin, M. A., and J. K. Nyborg. 1998. The human T-cell leukemia virus type 1 oncoprotein Tax inhibits the transcriptional activity of c-Myb through competition for the CREB binding protein. J. Virol. 729396-9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng, W. G., Y. Zhu, and K. K. Wu. 2004. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood 1032135-2142. [DOI] [PubMed] [Google Scholar]
- 17.Dixon, L. K., C. C. Abrams, G. Bowick, L. C. Goatley, P. C. Kay-Jackson, D. Chapman, E. Liverani, R. Nix, R. Silk, and F. Zhang. 2004. African swine fever virus proteins involved in evading host defence systems. Vet. Immunol. Immunopathol. 100117-134. [DOI] [PubMed] [Google Scholar]
- 18.Eckner, R., J. W. Ludlow, N. L. Lill, E. Oldread, Z. Arany, N. Modjtahedi, J. A. DeCaprio, D. M. Livingston, and J. A. Morgan. 1996. Association of p300 and CBP with simian virus 40 large T antigen. Mol. Cell. Biol. 163454-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enjuanes, L., A. L. Carrascosa, M. A. Moreno, and E. Vinuela. 1976. Titration of African swine fever (ASF) virus. J. Gen. Virol. 32471-477. [DOI] [PubMed] [Google Scholar]
- 20.Falvo, J. V., B. M. Brinkman, A. V. Tsytsykova, E. Y. Tsai, T. P. Yao, A. L. Kung, and A. E. Goldfeld. 2000. A stimulus-specific role for CREB-binding protein (CBP) in T cell receptor-activated tumor necrosis factor alpha gene expression. Proc. Natl. Acad. Sci. USA 973925-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falvo, J. V., A. M. Uglialoro, B. M. Brinkman, M. Merika, B. S. Parekh, E. Y. Tsai, H. C. King, A. D. Morielli, E. G. Peralta, T. Maniatis, D. Thanos, and A. E. Goldfeld. 2000. Stimulus-specific assembly of enhancer complexes on the tumor necrosis factor alpha gene promoter. Mol. Cell. Biol. 202239-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finlay, B. B., and G. McFadden. 2006. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124767-782. [DOI] [PubMed] [Google Scholar]
- 23.Garcia, R., F. Almazan, J. M. Rodriguez, M. Alonso, E. Vinuela, and J. F. Rodriguez. 1995. Vectors for the genetic manipulation of African swine fever virus. J. Biotechnol. 40121-131. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Rodriguez, C., and A. Rao. 1998. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med. 1872031-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerritsen, M. E., A. J. Williams, A. S. Neish, S. Moore, Y. Shi, and T. Collins. 1997. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 942927-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Girdwood, D., D. Bumpass, O. A. Vaughan, A. Thain, L. A. Anderson, A. W. Snowden, E. Garcia-Wilson, N. D. Perkins, and R. T. Hay. 2003. P300 transcriptional repression is mediated by SUMO modification. Mol. Cell 111043-1054. [DOI] [PubMed] [Google Scholar]
- 27.Goodman, R. H., and S. Smolik. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 141553-1577. [PubMed] [Google Scholar]
- 28.Granja, A. G., M. L. Nogal, C. Hurtado, C. Del Aguila, A. L. Carrascosa, M. L. Salas, M. Fresno, and Y. Revilla. 2006. The viral protein A238L inhibits TNF-alpha expression through a CBP/p300 transcriptional coactivators pathway. J. Immunol. 176451-462. [DOI] [PubMed] [Google Scholar]
- 29.Granja, A. G., M. L. Nogal, C. Hurtado, V. Vila, A. L. Carrascosa, M. L. Salas, M. Fresno, and Y. Revilla. 2004. The viral protein A238L inhibits cyclooxygenase-2 expression through a nuclear factor of activated T cell-dependent transactivation pathway. J. Biol. Chem. 27953736-53746. [DOI] [PubMed] [Google Scholar]
- 30.Granja, A. G., N. D. Perkins, and Y. Revilla. 2008. A238L inhibits NF-ATc2, NF-{kappa}B, and c-Jun activation through a novel mechanism involving protein kinase C-{theta}-mediated up-regulation of the amino-terminal transactivation domain of p300. J. Immunol. 1802429-2442. [DOI] [PubMed] [Google Scholar]
- 31.Granja, A. G., P. Sabina, M. L. Salas, M. Fresno, and Y. Revilla. 2006. Regulation of inducible nitric oxide synthase expression by viral A238L-mediated inhibition of p65/RelA acetylation and p300 transactivation. J. Virol. 8010487-10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu, W., and R. G. Roeder. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90595-606. [DOI] [PubMed] [Google Scholar]
- 33.Hamamori, Y., V. Sartorelli, V. Ogryzko, P. L. Puri, H. Y. Wu, J. Y. Wang, Y. Nakatani, and L. Kedes. 1999. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 96405-413. [DOI] [PubMed] [Google Scholar]
- 34.Hill, C. S., and R. Treisman. 1995. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell 80199-211. [DOI] [PubMed] [Google Scholar]
- 35.Iniguez, M. A., S. Martinez-Martinez, C. Punzon, J. M. Redondo, and M. Fresno. 2000. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J. Biol. Chem. 27523627-23635. [DOI] [PubMed] [Google Scholar]
- 36.Janknecht, R., and T. Hunter. 1996. Versatile molecular glue. Transcriptional control. Curr. Biol. 6951-954. [DOI] [PubMed] [Google Scholar]
- 37.Kock, J., M. Kann, G. Putz, H. E. Blum, and F. Von Weizsacker. 2003. Central role of a serine phosphorylation site within duck hepatitis B virus core protein for capsid trafficking and genome release. J. Biol. Chem. 27828123-28129. [DOI] [PubMed] [Google Scholar]
- 38.Kwok, R. P., M. E. Laurance, J. R. Lundblad, P. S. Goldman, H. Shih, L. M. Connor, S. J. Marriott, and R. H. Goodman. 1996. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature 380642-646. [DOI] [PubMed] [Google Scholar]
- 39.Kwok, R. P., J. R. Lundblad, J. C. Chrivia, J. P. Richards, H. P. Bachinger, R. G. Brennan, S. G. Roberts, M. R. Green, and R. H. Goodman. 1994. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 370223-226. [DOI] [PubMed] [Google Scholar]
- 40.Liu, Y., C. E. Denlinger, B. K. Rundall, P. W. Smith, and D. R. Jones. 2006. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 28131359-31368. [DOI] [PubMed] [Google Scholar]
- 41.Lundblad, J. R., R. P. Kwok, M. E. Laurance, M. L. Harter, and R. H. Goodman. 1995. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 37485-88. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Balbas, M. A., U. M. Bauer, S. J. Nielsen, A. Brehm, and T. Kouzarides. 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19662-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minden, A., A. Lin, F. X. Claret, A. Abo, and M. Karin. 1995. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 811147-1157. [DOI] [PubMed] [Google Scholar]
- 44.Miskin, J. E., C. C. Abrams, L. C. Goatley, and L. K. Dixon. 1998. A viral mechanism for inhibition of the cellular phosphatase calcineurin. Science 281562-565. [DOI] [PubMed] [Google Scholar]
- 45.Nakajima, T., C. Uchida, S. F. Anderson, C. G. Lee, J. Hurwitz, J. D. Parvin, and M. Montminy. 1997. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 901107-1112. [DOI] [PubMed] [Google Scholar]
- 46.Patel, D., S. M. Huang, L. A. Baglia, and D. J. McCance. 1999. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 185061-5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perkins, N. D., L. K. Felzien, J. C. Betts, K. Leung, D. H. Beach, and G. J. Nabel. 1997. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 275523-527. [DOI] [PubMed] [Google Scholar]
- 48.Revilla, Y., M. Callejo, J. M. Rodriguez, E. Culebras, M. L. Nogal, M. L. Salas, E. Vinuela, and M. Fresno. 1998. Inhibition of nuclear factor kappaB activation by a virus-encoded IkappaB-like protein. J. Biol. Chem. 2735405-5411. [DOI] [PubMed] [Google Scholar]
- 49.Rhoades, K. L., S. H. Golub, and J. S. Economou. 1992. The regulation of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. J. Biol. Chem. 26722102-22107. [PubMed] [Google Scholar]
- 50.Snowden, A. W., L. A. Anderson, G. A. Webster, and N. D. Perkins. 2000. A novel transcriptional repression domain mediates p21(WAF1/CIP1) induction of p300 transactivation. Mol. Cell. Biol. 202676-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thanos, D., and T. Maniatis. 1995. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 831091-1100. [DOI] [PubMed] [Google Scholar]
- 52.Thompson, P. R., D. Wang, L. Wang, M. Fulco, N. Pediconi, D. Zhang, W. An, Q. Ge, R. G. Roeder, J. Wong, M. Levrero, V. Sartorelli, R. J. Cotter, and P. A. Cole. 2004. Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 11308-315. [DOI] [PubMed] [Google Scholar]
- 53.Tjian, R., and T. Maniatis. 1994. Transcriptional activation: a complex puzzle with few easy pieces. Cell 775-8. [DOI] [PubMed] [Google Scholar]
- 54.Tsytsykova, A. V., and A. E. Goldfeld. 2002. Inducer-specific enhanceosome formation controls tumor necrosis factor alpha gene expression in T lymphocytes. Mol. Cell. Biol. 222620-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villalba, M., S. Kasibhatla, L. Genestier, A. Mahboubi, D. R. Green, and A. Altman. 1999. Protein kinase ctheta cooperates with calcineurin to induce Fas ligand expression during activation-induced T cell death. J. Immunol. 1635813-5819. [PubMed] [Google Scholar]
- 56.Vo, N., and R. H. Goodman. 2001. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 27613505-13508. [DOI] [PubMed] [Google Scholar]
- 57.Yaciuk, P., and E. Moran. 1991. Analysis with specific polyclonal antiserum indicates that the E1A-associated 300-kDa product is a stable nuclear phosphoprotein that undergoes cell cycle phase-specific modification. Mol. Cell. Biol. 115389-5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang, T., R. J. Davis, and C. W. Chow. 2001. Requirement of two NFATc4 transactivation domains for CBP potentiation. J. Biol. Chem. 27639569-39576. [DOI] [PubMed] [Google Scholar]
- 59.Yuan, L. W., and J. E. Gambee. 2000. Phosphorylation of p300 at serine 89 by protein kinase C. J. Biol. Chem. 27540946-40951. [DOI] [PubMed] [Google Scholar]