

Abstract
Objective
Recently, we have shown that shear stress regulates the angiogenic potential of endothelial cells in vitro by an Angiopoietin-2 (Ang2)– dependent mechanism; however its pathophysiological significance in vivo was not clear. We hypothesized that Ang2 plays an important role in blood flow recovery after arterial occlusion in vivo by regulating angiogenesis and arteriogenesis.
Methods and Results
C57Bl/6J mice underwent femoral artery ligation and were injected with a specific Ang2 inhibitor, L1-10, or vehicle for 10 days. Ang2 mRNA was upregulated at day 2, and Ang2 protein was upregulated at day 2, 5, and 7 in the ligated hindlimb. L1-10 treatment significantly blunted blood flow recovery. L1-10 decreased smooth muscle cell coverage of neovessels without affecting capillary density, suggesting a specific role for Ang2 in arteriogenesis. Mechanistically, L1-10 decreased expression of intercellular and vascular cell adhesion molecules as well as infiltrating monocytes/macrophages in the ischemic tissue. Although L1-10 had no effect on the number of CD11b+ cells (monocytes/macrophages) mobilized in the bone marrow, it maintained elevated numbers of circulating CD11b+ cells in the peripheral blood.
Conclusions
These results suggest that Ang2 induced in ischemic tissue plays a critical role in blood flow recovery by stimulating inflammation and arteriogenesis.
Keywords: angiopoietin-2, hindlimb ischemia, arteriogenesis, angiogenesis, inflammation
Neovascularization, or blood vessel formation, plays an important role in normal physiology as well as in pathophysiology. In particular, neovascularization plays a role in diseases involving ischemia, including tumors, atherosclerotic plaques, and ischemic heart disease.1 Therefore, understanding the mechanisms of neovascularization and the ability to control neovascularization could be important in developing therapies for these diseases. The two main forms of neovascularization in the adult are angiogenesis and arteriogenesis.1 Angiogenesis is the formation of new blood vessels from preexisting blood vessels, whereas arteriogenesis is the enlarging of existing blood vessels to form collaterals and the recruitment of vascular smooth muscle cells (VSMCs).1 The exact molecular mechanisms that drive neovascularization remain unclear.
Numerous factors are thought to be involved in neovascularization, including ischemia, inflammation, and the secretion of growth factors.1 We have recently identified Angiopoietin-2 (Ang2) as a mechano-sensitive protein involved in shear stress–mediated tubule formation and migration of endothelial cells in vitro.2 However, the role of Ang2 in neovascularization in vivo remains controversial and needs to be clarified.3,4
In human diseases, Ang2 has been found to be upregulated within neovessels of advanced atherosclerotic lesions as compared to early lesions.5 In addition, Ang2 was upregulated in an animal model of myocardial ischemia.6 In both advanced atherosclerosis and myocardial ischemia, hypoxia and inflammation are present.7,8 In particular, monocytes/ macrophages have been shown to play an integral role in collateral formation during ischemia.9,10 Therefore, we hypothesized that Ang2 plays an important role in blood flow recovery during ischemia through the modulation of angiogenesis, arteriogenesis, and inflammation. To test this hypothesis, we used a mouse model of hindlimb ischemia to examine the role of Ang2 in blood flow recovery as well as angiogenesis and arteriogenesis in response to ischemia. From this, we demonstrate that Ang2 plays an important role in blood flow recovery during ischemia through the formation of collaterals and the recruitment of inflammatory cells. These findings implicate Ang2 as an important factor in regulating ischemic diseases, and Ang2 may provide a possible link between blood vessel formation and inflammation.
Methods
For expanded methods and results please see the supplement available at http://atvb.ahajournals.org.
Hindlimb Ischemia Model
Male C57Bl/6J mice (Jackson Laboratory) were used at 6 to 8 weeks of age. All protocols were approved by the Institutional Animal Care and Use Committee and done in accordance with federal guidelines on the principles for the care and use of animals in research. Hindlimb ischemia surgery was performed as previously described by us.11 L1-10 (4 mg/kg), an FC-fusion protein Ang2 specific inhibitor (Amgen), vehicle (PBS), or IgG1-FC control (R&D systems) was injected subcutaneously every other day starting with 1 day before surgery.
Real-Time Quantitative Polymerase Chain Reaction, Shear Studies, Matrigel Tubule Formation Assay, Preparation of Tissue/Cell Lysates, and Immunoblotting
Experiments were performed as previously described.2
Whole Mount Immunohistochemistry
On day 2, 5, 7, or 10 after surgery, the animal was euthanized by CO2 inhalation and pressure fixed with 10% formalin. The adductor muscle was removed and incubated overnight at 4°C with primary antibody, then incubated with fluorescently tagged secondary antibody for 4 hours. The whole tissue was imaged using a LSM 510 Meta confocal microscope (Zeiss). Antibodies used were: PECAM-1 (Chemicon), Ang2 (Santa Cruz), smooth muscle α-actin-fluorescein isothiocyanate (Sigma), and CD11b (BD Biosciences).
Laser Doppler Perfusion Imaging
Laser doppler perfusion imaging (LDPI) was performed as previously described.11
Flow Cytometric Cell Analysis
Total bone marrow (BM) and peripheral blood (PB) cells were obtained and subjected to red blood cell lysis buffer (Roche Diagnostics) according to the manufacturer’s instruction. Single cell suspensions of BM and PB cells were stained with PE-conjugated CD11b antibody (BD Biosciences) and analyzed using BD LSRII system. FACS data were analyzed with Flowjo (Tree Star Inc) using controls with isotype-matched IgG.
Statistical Analysis
Data are reported as average±SEM obtained from at least 3 independent studies. Statistical significance (P<0.05) was assessed by Student t test as well as 1-way ANOVA followed by Bonferroni test for comparisons over a time-course within the same group.
Results
Ang2 Is Upregulated in the Ischemic Hindlimb Whereas Ang1 Is Not
To investigate the role of angiopoietins in neovascularization, we examined the expression of Ang1 and Ang2 in the mouse hindlimb after femoral artery ligation. The adductor muscle was collected for mRNA 2, 5, or 7 days after hindlimb ischemia surgery. Real-time quantitative PCR revealed that Ang2 mRNA was upregulated 7-fold in the ischemic adductor muscle compared to the contralateral nonischemic control adductor on day 2, but returned to equivalent values on day 5 and day 7 (Figure 1A), suggesting an early role for Ang2 in hindlimb ischemia. In contrast, Ang1 mRNA levels were not significantly different between the ischemic and nonischemic adductor muscle (Figure 1B). C-fms mRNA, a macrophage marker, was upregulated in the ischemic adductor on day 2, 5, and 7, with day 7 starting to decline back to normal levels (Figure 1C), suggesting a role for macrophages in hindlimb ischemia recovery.
Figure 1.
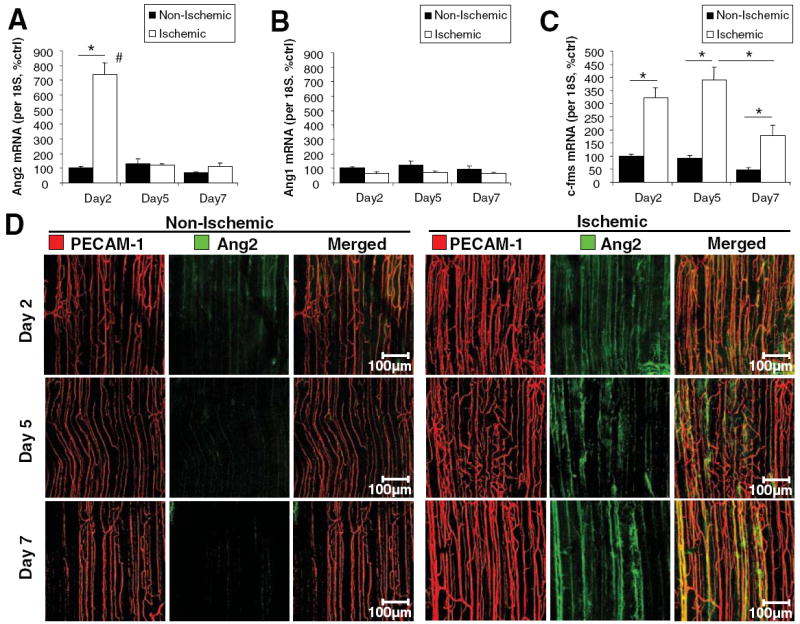
Ang2 and c-fms are upregulated in the ischemic hindlimb whereas Ang1 is not. C57Bl/6J mice underwent femoral artery ligation. RNA was collected after 2, 5, or 7 days from the ischemic and nonischemic adductor muscle. Real-time PCR was performed for Ang2 (A), Ang1 (B), and c-fms (C). D, Whole mount immunohistochemistry was performed after 2, 5, or 7 days on the nonischemic and ischemic adductor muscle for Ang2 (green) and PECAM-1 (red). Shown are confocal images of the proximal region of the adductor muscle. (mean±SEM, n=4 to 6; *P<0.05; #P<0.05 compared to day 5 and day 7).
Whole mount immunostaining of the mouse adductor muscle was performed for PECAM-1 and Ang2 protein expression (Figure 1D). PECAM-1 staining for endothelial cells revealed blood vessels in the nonischemic control that align parallel to the muscle fibers (Figure 1D). The ischemic adductor muscle had increased PECAM-1 staining intensity and a slight increase in blood vessel density compared to the nonischemic control (Figure 1D). By day 7, blood vessels in the ischemic adductor muscle are larger in diameter (Figure 1D). Ang2 protein expression was upregulated in the ischemic adductor muscle on day 2, 5, and 7, suggesting that Ang2 protein expression lags behind mRNA expression (Figure 1D).
L1-10 Inhibits Both the Antagonist and Agonist Roles of Ang2
To investigate the specific role of Ang2 in neovascularization, we used an FC-fusion peptide called L1-10, which is a specific inhibitor of Ang2 binding to its receptor, Tie2. L1-10 is a related compound to L1-7, which was used in previous studies and shown to be a specific inhibitor of Ang2.12 When measuring the neutralization of the angiopoietin:Tie2 interaction, L1-10 showed >1000-fold selectivity for Ang2 over Ang1 (personal communication with Amgen, 2008). To further confirm the inhibitory effects of L1-10 on Ang2, we examined the phosphorylation of Tie2 by Ang2 binding both in vitro and in vivo. Ang2 is known to phosphorylate Tie2 when used at high concentrations, in vitro.13 In vivo, however, Tie2 has been shown to block Tie2 phosphorylation.3 Therefore, we examined the ability of L1-10 to block the Ang2:Tie2 interaction both in vitro and in vivo. HUVECs were treated with control, Ang1, Ang2, or L1-10, and the phosphorylation of Tie2 was examined. Ang2 stimulated Tie2 phosphorylation at concentrations of 800 ng/mL which was blocked by L1-10 (Figure 2A). However, L1-10 did not block Ang1-induced phosphorylation of Tie2 (Figure 2A). Total Tie2 levels were unaffected. To examine the functional role of L1-10, we examined its effect on shear-mediated tubule formation. Previously, we showed that oscillatory shear stress (OS) mediated tubule formation through a mechanism dependent on Ang2, but not Ang1, in endothelial cells.2 We found that L1-10 inhibited OS-mediated tubule formation of HUVECs (Figure 2B), suggesting that L1-10 inhibits the functional role of Ang2 in vitro.
Figure 2.

L1-10 inhibits both the agonist and antagonist roles of Ang2. A, HUVECs were treated with 800 ng/mL of Ang1 or Ang2±10 μg/mL L1-10 for 15 minutes. Cell lysates were used for Western analysis. B, Conditioned media collected from HUVECs that were exposed to laminar shear (LS, 15 dyn/cm2), oscillatory shear (OS, ±5 dyn/cm2), or static control (ST) for 24 hours were added to HUVECs in a Matrigel tubule formation assay. Shown are representative images, and tubule length was quantified and normalized to percent ST. C, Mice were injected via tail vein with vehicle or 20 μg Ang2±L1-10. After 30 minutes, the adductor muscle lysates were prepared for Western analysis with antibodies to phospho-Tie2, Tie2, and β-actin (see Supplemental Figure IC for expanded blot). (mean±SEM, n=3 to 6; *P<0.05).
To examine the effect of L1-10 on the Ang2:Tie2 interaction in vivo, we performed tail vein injection of 20 μg Ang2 with or without L1-10 and examined Tie2 phosphorylation in the adductor muscle. In basal state, Tie2 in mouse vasculature is known to be constitutively phosphorylated.14 We found similar findings in the adductor muscle when mice were injected with vehicle control (Figure 2C). Tail vein injection of Ang2 caused a significant decrease in Tie2 phosphorylation, suggesting that in vivo Ang2 acts as an antagonist of Tie2 signaling (Figure 2C). When Ang2 was injected with L1-10, Tie2 phosphorylation was significantly rescued, thereby blocking the Ang2 antagonism of Tie2 phosphorylation (Figure 2C). We found that L1-10 had no effect on VEGFR-2 signaling in endothelial cells or PDGFR-β signaling in aortic smooth muscle cells (supplemental Figure I), confirming its specificity for Ang2.
Ang2 Inhibition Impairs Blood Flow Recovery After Femoral Artery Ligation
To investigate the in vivo role of Ang2 in neovascularization after ischemia, we investigated the effect of inhibiting Ang2 on blood flow recovery and vascular remodeling after femoral artery ligation. We found that inhibiting Ang2 by treatment with L1-10 significantly impaired blood flow recovery in the ischemic hindlimb on days 5 and 7 postsurgery when compared to vehicle and FC control as shown by LDPI measurements (Figure 3). FC control had no effect compared to vehicle control. Therefore, vehicle was used as the control for all subsequent experiments and FC was only examined at the 10-day time point.
Figure 3.
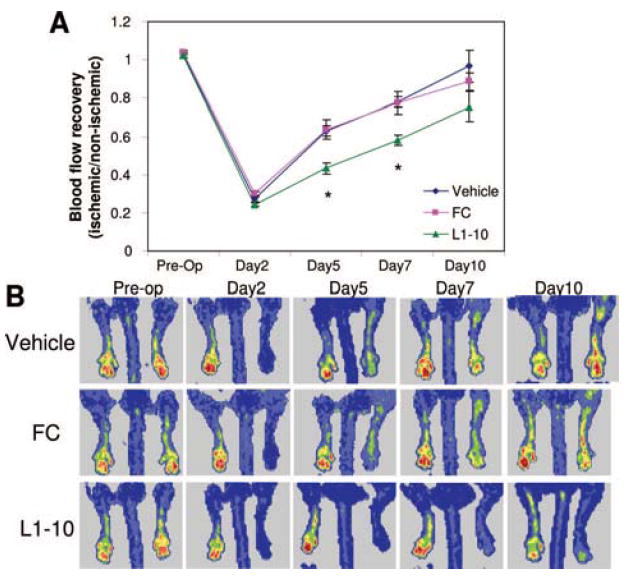
Ang2 inhibition impairs blood flow recovery during hindlimb ischemia. Mice underwent femoral artery ligation and were treated with vehicle, L1-10, or FC control. LDPI was measured before surgery (preop), and 2, 5, 7, and 10 days after surgery. A, Mean perfusion was expressed as a ratio of the ischemic paw to the contralateral nonischemic paw. B, Shown are representative LDPI images at the indicated time-points. (mean±SEM, n=6 to 12; *P<0.05 compared to vehicle and FC control).
Ang2 Inhibition Blocks Arteriogenesis
To examine how Ang2 inhibition impaired blood flow recovery, we examined blood vessel formation in the ischemic adductor muscle. The adductor muscle was isolated and whole mount immunostained for PECAM-1, an endothelial cell specific marker, and smooth muscle α-actin (SMA), a VSMC marker. Because arteriogenesis is the formation of collaterals requiring the participation of VSMCs, we denoted PECAM-1–positive vessels covered with VSMCs as a marker of arteriogenesis. We found that on day 7 and day 10, there was no significant difference in the VSMC-free blood vessels (capillaries), between the Ang2 inhibitor–treated and vehicle-treated ischemic adductor muscles (Figure 4). Although the morphology looked slightly different, with L1-10–treated vessels being less tortuous, the overall capillary density did not change. However, on days 7 and 10, L1-10 treatment significantly blunted arteriogenesis as determined by the decrease in the density of VSMC-covered blood vessels in the ischemic adductor muscle as compared to vehicle control (Figure 4). On day 10, L1-10 treatment significantly decreased arteriogenesis in the ischemic adductor muscle as compared to FC control, similar to vehicle control results (Figure 4B). These results suggest that Ang2 improves blood flow recovery via arteriogenesis or collateral formation.
Figure 4.
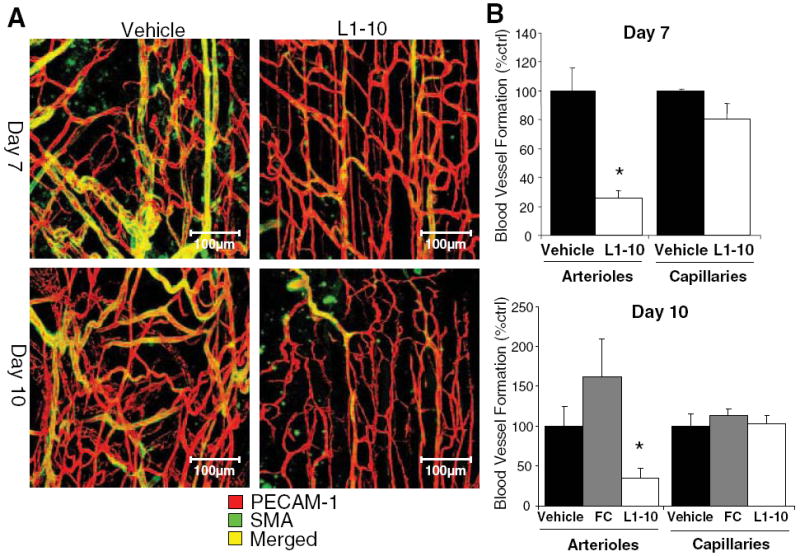
Ang2 inhibition blocks arteriogenesis and the formation of collaterals. Mice underwent femoral artery ligation and were treated with vehicle or L1-10. A, Shown are representative whole mount confocal images from the distal region of the ischemic adductor muscle (red for PECAM-1 and green for SMA). B, Yellow representing arterioles and red representing capillaries were quantified and expressed as a percent of vehicle. (mean±SEM, n=4 to 5; *P<0.05 compared to vehicle and FC control).
Ang2 Inhibition Reduces Inflammatory Cell Infiltration
Because monocytes/macrophages are known to play an important role in hindlimb ischemia and we found them upregulated in our model (Figure 1C), we investigated the effects of Ang2 inhibition on monocyte/macrophage infiltration during hindlimb ischemia. The ischemic adductor muscle was isolated and whole mount immunostained for PECAM-1 and CD11b, a common marker of macrophages and sometimes neutrophils. On day 7 and day 10 after hindlimb ischemia surgery, Ang2 inhibition significantly decreased the number of CD11b+ cells in the ischemic adductor muscle as compared to vehicle control (Figure 5), suggesting that Ang2 inhibition reduces inflammatory cell infiltration. FC control-treated group was not significantly different from that of vehicle (Figure 5B).
Figure 5.
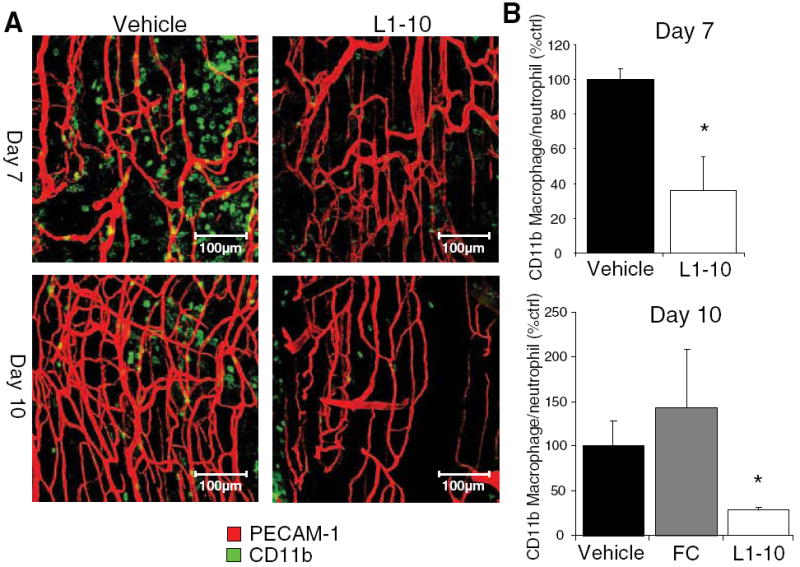
Ang2 inhibition reduces inflammatory cell infiltration. Mice underwent femoral artery ligation and were treated with vehicle or L1-10. A, Shown are representative whole mount confocal images of the distal region of the ischemic adductor muscle (Red for PECAM-1 and green for CD11b). B, CD11b+ cells were quantified and expressed as a percent of vehicle control. (mean±SEM, n=4 to 6; *P<0.05 compared to vehicle and FC control).
Ang2 Inhibition Reduces ICAM-1 and VCAM-1 Expression After Hindlimb Ischemia
To identify a mechanism for the decrease in infiltration of CD11b+ cells in the ischemic tissue of L1-10–treated mice, we first examined the expression of the adhesion molecules ICAM-1 and VCAM-1. Using real-time PCR, we found that on day 3 after ligation the L1-10 treated mice had significantly less ICAM-1 and VCAM-1 mRNA expression in the ischemic adductor muscle (Figure 6A and 6B). These findings suggest that L1-10 inhibits monocyte infiltration into the tissue by downregulating ICAM-1 and VCAM-1 expression.
Figure 6.
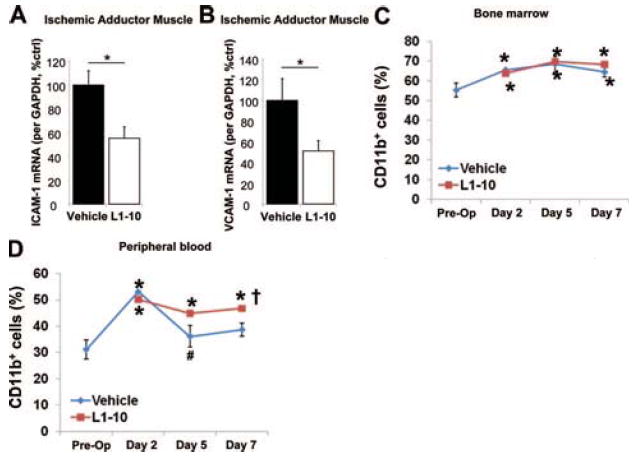
Effect of L1-10 on tissue ICAM-1 and VCAM-1 mRNA expression and CD11b+ cells in the bone marrow and peripheral blood after an ischemic event. Mice underwent femoral artery ligation and were treated with vehicle or L1-10. RNA was collected after 3 days from the ischemic adductor muscle. Real time PCR was performed for ICAM-1 (A) and VCAM-1 (B) (mean±SEM, n=4 to 6; *P<0.05). Cells from bone marrow (C) and peripheral blood (D) collected before (preop) and 2, 5, and 7 days after surgery were analyzed by FACS using CD11b antibody (mean±SEM, n=5; *P<0.05 vs Preop, #P<0.05 vs vehicle day 2, †P<0.05 vs vehicle day 7).
Ang2 Inhibition Maintains Elevated Number of CD11b+ Cells in Circulation After Femoral Artery Ligation
To examine whether Ang2 and L1-10 had any effect on the numbers of monocytes mobilized in bone marrow (BM) and circulating in peripheral blood (PB), the number of CD11b+ cells in BM and PB were determined by FACS. After the femoral artery ligation, the numbers of CD11b+ cells were significantly increased in both BM and PB at day 2 postsurgery (Figure 6C and 6D). In the BM, the increase of CD11b+ cells was sustained until day 7 in both vehicle and L1-10–treated mice (Figure 6C). In PB, whereas the increase of CD11b+ cells returned to normal at day 5 in vehicle-treated mice, the increase was sustained up to day 7 in L1-10–treated mice (Figure 6D).
Discussion
The novel findings of this study are: (1) Ang2 is upregulated at early time points in the mouse ischemic hindlimb both at the mRNA level and protein level, (2) the Ang2:Tie2 interaction is specifically inhibited by L1-10, both in vitro and in vivo, and (3) L1-10 inhibits shear-mediated tubule formation in vitro. In the mouse femoral artery ligation model, L1-10 treatment (4) impairs blood flow recovery in the ischemic hindlimb, (5) inhibits collateral formation as indicated by VSMC coverage of neovessels, (6) inhibits ICAM-1 and VCAM-1 expression as well as infiltration of CD11b+ cells in the ischemic adductor muscle, (7) does not affect the number of CD11b+ cells in the bone marrow, and (8) maintains elevated levels of CD11b+ cells in peripheral blood for 7 days after ischemia. Collectively, these findings suggest that the upregulation of Ang2 after mouse femoral artery ligation plays an important role in blood flow recovery by stimulating the expression of ICAM-1/VCAM-1 and the infiltration of monocytes/macrophages into the ischemic tissue as well as the coverage of capillary endothelial cells with VSMCs. However, it does not appear to play a significant role in the mobilization of monocytes from the bone marrow.
It has been found that Ang1 is involved in vessel stabilization, whereas Ang2 is involved in vascular remodeling and vessel destabilization.15 However, the complex role of Ang2 in vascular remodeling still remains unclear. We previously found that Ang2 was upregulated by oscillatory shear in endothelial cells in vitro,2 but it was not clear whether Ang2 expression would change in response to a sudden occlusion of blood flow in vivo and whether it plays a critical role in blood flow recovery in ischemic tissue. Here, we used femoral artery ligation in C57Bl/6J mice to test this hypothesis. Our results show that Ang2 is produced in ischemic vessels (most likely from endothelial cells) after femoral artery ligation, which is known to cause disturbed or reversed shear stress in collaterals.16 Ang2 has been previously found to be induced by hypoxia in endothelial cells both in vitro and in vivo,17 suggesting that Ang2 plays an important role in hypoxia- and ischemia-induced vascular remodeling. The presence of ischemia coupled with disturbed fluid shear stress could be possible mechanisms by which Ang2 is upregulated during hindlimb ischemia.
We found that inhibiting Ang2 impaired blood flow recovery in the ischemic hindlimb, suggesting that Ang2 is necessary for collateral formation after arterial occlusion. However, the role of Ang2 in hindlimb ischemia is somewhat conflicting. Recently, Reiss et al found that transgenic over-expression of Ang2 in endothelial cells impaired revascularization during hindlimb ischemia.3 However, the same authors previously reported that Ang2 promotes neovessel formation during cerebral ischemia.4 They concluded that the dose of Ang2 is a critical determinant of whether there is vessel growth or regression.3 We speculate that perhaps a physiological and transient upregulation of Ang2 can stimulate revascularization, as seen in our hindlimb ischemia model. In contrast, supraphysiological amounts or excessive duration of Ang2 exposure could cause chronic inflammation or continued vessel destabilization, promoting vessel regression and vascular leakage.15,18,19 Our finding that Ang2 mRNA expression is transiently upregulated further supports the initial role of Ang2 in neovascularization.
The reduced reperfusion in mice treated with L1-10 could be attributable to inhibited recruitment and proliferation of VSMCs. Mice deficient in Ang2 tend to be lethal within two weeks after birth because of postnatal defects in vascular growth.20 However, the few that survive show defects in pericyte and smooth muscle cell recruitment in lymphatic vessels,21 implicating Ang2 as an important regulator of smooth muscle cell recruitment. Whereas L1-10 had an inhibitory effect on smooth muscle cell coverage of endothelial cells, implicating it in collateral formation or arteriogenesis, L1-10 did not show a significant effect on capillary density in our model. This suggests that Ang2 mediates blood flow recovery mainly through arteriogenesis without affecting angiogenesis. It has been reported that the majority of blood flow recovery during hindlimb ischemia is achieved by arteriogenesis and not angiogenesis.22 Therefore, the decrease in blood flow recovery in mice treated with L1-10 is most likely mediated by decreased arteriogenesis.
We found that Ang2 inhibition reduced monocyte/macrophage infiltration into the ischemic hindlimb. To identify a mechanism for the reduced inflammatory cells in L1-10–treated mice, we examined the expression of the important adhesion molecules, ICAM-1 and VCAM-1. Tie2 activation leads to the recruitment of A20-binding inhibitor of nuclear factor-κB (ABIN-2), which inhibits the NF-κB pathway,23 and it has been found that Ang2 interferes with Tie2 phosphorylation resulting in activation of the NF-κB pathway and ICAM-1 and VCAM-1 expression.19 In our in vivo model, we found that Ang2 blocked Tie2 activation and that inhibiting Ang2 resulted in decreased ICAM-1 and VCAM-1 expression, most likely through the inhibition of the NF-κB pathway. Therefore, during ischemia the increased Ang2 may block Tie2-mediated inhibition of NF-κB, resulting in increased ICAM-1 and VCAM-1 expression and the subsequent monocyte adhesion. In addition, there is evidence that Ang2 can act as a chemoattractant for Tie-2+ monocytes,24 suggesting that Ang2 could also be recruiting monocytes in addition to promoting adhesion and infiltration.
To further investigate the link between Ang2 and monocytes, we examined the effect of Ang2 inhibition on bone marrow–derived and circulating CD11b+ cells. We found that compared to vehicle, L1-10 had no effect on the number of CD11b+ cells in the BM. However, both vehicle- and L1-10–treated mice had an increase in CD11b+ cells 2 days after ischemia in the peripheral blood. Whereas CD11b+ cells declined in vehicle-treated mice at day 5 and day 7, L1-10–treated mice had continually elevated CD11b+ cells within the PB. During ischemia, CD11b+ cells may home to the ischemic area and induce arteriogenesis. L1-10 may block the migration of circulating CD11b+ cells into the ischemic tissue through reduced ICAM-1 and VCAM-1 expression, which results in maintained elevated levels of CD11b+ cells in the PB and decreased CD11b+ cells in the tissue. Our interpretation is further supported by our findings that L1-10 decreases CD11b+ cells in the tissue and decreases ICAM-1 and VCAM-1 expression.
Heil et al found that mice deficient in CCR2, a receptor important for monocyte binding, had impaired blood flow recovery and collateral formation attributable to reduced monocyte migration into the tissue.10 Monocytes and macrophages seem to play a critical role in vascular wall remodeling by degrading the extracellular matrix, and the degradation products may promote the proliferation of VSMCs. Growth factors released by macrophages, such as FGF-2, have been shown to also promote the proliferation of VSMCs.9 Therefore, the inhibited arteriogenesis and collateral formation seen in L1-10–treated mice could be as a result of the decreased inflammation.
An interesting area of future study would be to examine the role of endothelial progenitor cells (EPCs) in Ang2-mediated neovascularization. Ang2 has been found to promote the angiogenicity and migration of EPCs and therefore could be playing a role in the mobilization of EPCs to the ischemic tissue, contributing to the neovascularization.25
In conclusion, we found that Ang2 plays an important role in blood flow recovery and reperfusion after an artery occlusion, and this was mediated through arteriogenesis and the recruitment of inflammatory cells to the ischemic area. Elucidating the role of Ang2 in neovascularization could be important in understanding ischemic diseases. In particular, the spatial and temporal controlled expression of Ang2 could have novel therapeutic applications for diseases involving impaired blood vessel formation.
Supplementary Material
Data Supplement (unedited) at: http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.108.175463/DC1
Acknowledgments
We thank Amgen for supplying us with L1-10. We thank Drs Craig Duvall, Hyuk Sang Kwon, Kathy Griendling, and Alejandra San Martin for their help in this study.
Sources of Funding This work was supported by funding from NIH grants HL87012, HL75209, and UO1HL80711 (to H.J.).
Footnotes
Disclosures None.
Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular Biology is online at http://atvb.ahajournals.org/subscriptions/
Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Fax: 410-528-8550. E-mail: journalpermissions@lww.com
Reprints: Information about reprints can be found online at http://www.lww.com/reprints
References
- 1.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 2.Tressel SL, Huang RP, Tomsen N, Jo H. Laminar shear inhibits tubule formation and migration of endothelial cells by an angiopoietin-2 dependent mechanism. Arterioscler Thromb Vasc Biol. 2007;27:2150–2156. doi: 10.1161/ATVBAHA.107.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiss Y, Droste J, Heil M, Tribulova S, Schmidt MH, Schaper W, Dumont DJ, Plate KH. Angiopoietin-2 impairs revascularization after limb ischemia. Circ Res. 2007;101:88–96. doi: 10.1161/CIRCRESAHA.106.143594. [DOI] [PubMed] [Google Scholar]
- 4.Beck H, Acker T, Wiessner C, Allegrini PR, Plate KH. Expression of angiopoietin-1, angiopoietin-2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol. 2000;157:1473–1483. doi: 10.1016/S0002-9440(10)64786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calvi C, Dentelli P, Pagano M, Rosso A, Pegoraro M, Giunti S, Garbarino G, Camussi G, Pegoraro L, Brizzi MF. Angiopoietin 2 induces cell cycle arrest in endothelial cells: a possible mechanism involved in advanced plaque neovascularization. Arterioscler Thromb Vasc Biol. 2004;24:511–518. doi: 10.1161/01.ATV.0000116864.86607.35. [DOI] [PubMed] [Google Scholar]
- 6.Matsunaga T, Warltier DC, Tessmer J, Weihrauch D, Simons M, Chilian WM. Expression of VEGF and angiopoietins-1 and -2 during ischemia-induced coronary angiogenesis. Am J Physiol Heart Circ Physiol. 2003;285:H352–H358. doi: 10.1152/ajpheart.00621.2002. [DOI] [PubMed] [Google Scholar]
- 7.Moreno PR, Purushothaman KR, Zias E, Sanz J, Fuster V. Neovascularization in human atherosclerosis. Curr Mol Med. 2006;6:457–477. doi: 10.2174/156652406778018635. [DOI] [PubMed] [Google Scholar]
- 8.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 9.Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heil M, Ziegelhoeffer T, Wagner S, Fernandez B, Helisch A, Martin S, Tribulova S, Kuziel WA, Bachmann G, Schaper W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ Res. 2004;94:671–677. doi: 10.1161/01.RES.0000122041.73808.B5. [DOI] [PubMed] [Google Scholar]
- 11.Duvall CL, Weiss D, Robinson ST, Alameddine FM, Guldberg RE, Taylor WR. The Role of Osteopontin in Recovery from Hind Limb Ischemia. Arterioscler Thromb Vasc Biol. 2008;28:290–295. doi: 10.1161/ATVBAHA.107.158485. [DOI] [PubMed] [Google Scholar]
- 12.Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, Graham K, Jacobsen F, Stevenson S, Ho J, Chen Q, Hartmann T, Michaels M, Kelley M, Li L, Sitney K, Martin F, Sun JR, Zhang N, Lu J, Estrada J, Kumar R, Coxon A, Kaufman S, Pretorius J, Scully S, Cattley R, Payton M, Coats S, Nguyen L, Desilva B, Ndifor A, Hayward I, Radinsky R, Boone T, Kendall R. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004;6:507–516. doi: 10.1016/j.ccr.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 13.Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene. 2000;19:4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- 14.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81:567–574. doi: 10.1161/01.res.81.4.567. [DOI] [PubMed] [Google Scholar]
- 15.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 16.Scholz D, Cai WJ, Schaper W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis. 2001;4:247–257. doi: 10.1023/a:1016094004084. [DOI] [PubMed] [Google Scholar]
- 17.Mandriota SJ, Pyke C, Di Sanza C, Quinodoz P, Pittet B, Pepper MS. Hypoxia-inducible angiopoietin-2 expression is mimicked by iodonium compounds and occurs in the rat brain and skin in response to systemic hypoxia and tissue ischemia. Am J Pathol. 2000;156:2077–2089. doi: 10.1016/S0002-9440(10)65079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roviezzo F, Tsigkos S, Kotanidou A, Bucci M, Brancaleone V, Cirino G. Papapetropoulos A. Angiopoietin-2 causes inflammation in vivo by promoting vascular leakage. J Pharmacol Exp Ther. 2005;314:738–744. doi: 10.1124/jpet.105.086553. [DOI] [PubMed] [Google Scholar]
- 19.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 20.Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 21.Shimoda H, Bernas MJ, Witte MH, Gale NW, Yancopoulos GD, Kato S. Abnormal recruitment of periendothelial cells to lymphatic capillaries in digestive organs of angiopoietin-2-deficient mice. Cell Tissue Res. 2007;328:329–337. doi: 10.1007/s00441-006-0360-8. [DOI] [PubMed] [Google Scholar]
- 22.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
- 23.Hughes DP, Marron MB, Brindle NP. The antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-kappaB inhibitor ABIN-2. Circ Res. 2003;92:630–636. doi: 10.1161/01.RES.0000063422.38690.DC. [DOI] [PubMed] [Google Scholar]
- 24.Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178:7405–7411. doi: 10.4049/jimmunol.178.11.7405. [DOI] [PubMed] [Google Scholar]
- 25.Kim KL, Shin IS, Kim JM, Choi JH, Byun J, Jeon ES, Suh W, Kim DK. Interaction between Tie receptors modulates angiogenic activity of angiopoietin2 in endothelial progenitor cells. Cardiovasc Res. 2006;72:394–402. doi: 10.1016/j.cardiores.2006.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement (unedited) at: http://atvb.ahajournals.org/cgi/content/full/ATVBAHA.108.175463/DC1