

Abstract
A novel thermodynamic approach to the reversible unfolding of proteins in aqueous urea solutions has been developed based on the premise that urea ligands are bound cooperatively to the macromolecule. When successive stoichiometric binding constants have values larger than expected from statistical effects, an equation for moles of bound urea can be derived that contains imaginary terms. For a very steep unfolding curve, one can then show that the fraction of protein unfolded, B̄, depends on the square of the urea concentration, U, and is given by
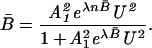 |
A12 is the binding constant as B̄→ 0, and λ is a parameter that reflects the augmentation in affinities of protein for urea as the moles bound increases to the saturation number, n. This equation provides an analytic expression that reproduces the unfolding curve with good precision, suggests a simple linear graphical procedure for evaluating A12 and λ, and leads to the appropriate standard free energy changes. The calculated ΔG° values reflect the coupling of urea binding with unfolding of the protein. Some possible implications of this analysis to protein folding in vivo are described.
Keywords: multiple binding, virtual equilibrium constants, denaturation, cooperativity
The effects of high concentrations of urea in aqueous solution on the properties of proteins have intrigued investigators since early in this century (1–3). In the same era, the reversibility of denaturation by various reagents, including urea, was discovered (4, 5). Some decades later when spectroscopic probes responsive to the conformational states of proteins had been developed, the quantitative dependence of denaturation on urea concentration could be ascertained. Such data provided a foundation for a thermodynamic approach to the unfolding process.
A representative unfolding curve for a protein [bacterial histidine-containing protein, HPr (6)] in aqueous solutions of increasing urea concentration is illustrated in Fig. 1. Since the transition is reversible, it has become customary to view each point as an equilibrium position of the interconvertible native, N, and denatured forms, D, respectively, and to define an equilibrium constant KN-D = (D)/(N). This constant can then be used to compute a standard free energy of unfolding, ΔGD-N° (7–12).
Figure 1.
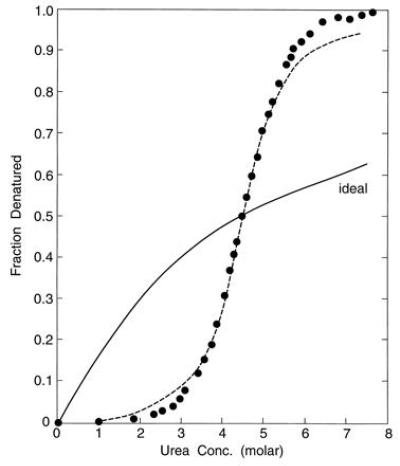
Unfolding curve for HPr, bacterial histidine-containing protein, in aqueous urea solutions. Experimental data from ref. 6. The broken line through the points presents the curve calculated from Eq. 27. The “ideal” curve is that for a transformation in which successive steps of ligand binding are energetically intrinsically identical.
As is apparent from Fig. 1, the KN-D is extremely sensitive to the concentration of urea in the aqueous solvent. An increase in urea concentration of less than 2-fold (from 3.5 to 6 M) changes KN-D by more than a 100-fold. This is surprising and perplexing, for urea is a very innocuous solute, very compatible with water. At the gross or macroscopic level, aqueous urea solutions show no exceptional properties. True, the dielectric increment of urea, +2.7 (13), implies that in a 3–6 M solution it raises the dielectric constant of the water from near 80 to the range of 88–96. This contrasts with the effects of most other common organic solutes in water, which reduce the dielectric constant slightly. On the other hand, even 1 M glycine in water raises the dielectric constant to 100, and 5 M β-alanine in water increases the dielectric constant to 250 (13). Yet, protein–ligand binding studies (14) show that the latter two solutes, in contrast to urea, stabilize the native conformation of the protein rather than facilitate denaturation.
The very steep rise in Fig. 1 is reminiscent of ligand–receptor behavior when there is very strong cooperativity between successively bound molecules. The strong cooperativity in the urea/HPr system is particularly evident if one compares the experimentally observed curve with that for an “ideal” transformation in which successive steps of ligand binding are energetically intrinsically identical and which manifests a halfway point at the same concentration of urea as does HPr unfolding. Below 2 M urea, the free energy of unfolding of the HPr is so unfavorable that the transformation is essentially undetectable, even though the ideal system attains a conversion near 30%. Above 2 M urea, a steep and increasing upward rise appears so that the conversion for unfolding quickly overtakes and surpasses that for the ideal system. The observed unfolding of the protein approaches completion at a urea concentration (≈6.5 M) that achieves only 60% conversion for the ideal transformation.
Solutions of 4–8 M urea contain 25–50%, by weight of solute; the molar concentration of urea reaches 106-fold that of the protein. It behooves us, therefore, to examine the behavior of the protein if urea molecules are bound with high cooperativity. This can be approached from a purely phenomenological, classical, thermodynamic perspective, not dependent on a two-state conformational model for the protein.
Binding Equations for Strongly Coupled Successive Steps
The increasing effectiveness of urea (U) in the unfolding of proteins (P) can be formulated quantitatively in classical thermodynamic terms with a stepwise, stoichiometric representation of ligand binding to the protein:
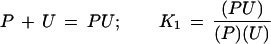 |
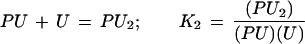 |
 |
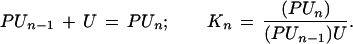 |
1 |
The subscript n denotes the moles of bound urea at saturation of the receptor. The moles B of ligand, urea, bound per mole of protein can be related to the concentration of free ligand, (U), by the equation (15, 16):
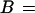 |
2 |
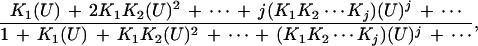 |
where Kj (which is always real and positive) represents the stoichiometric equilibrium constant for the successive stoichiometric step, j,
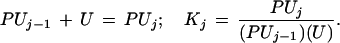 |
3 |
An alternative algebraic expression for B, following from the fundamental theorem of algebra (16) applied to Eq. 2, is
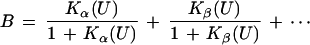 |
4 |
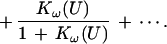 |
The coefficients Kα ⋯ Kω have the dimensions of equilibrium constants and can be combined to yield functions of the stoichiometric equilibrium constants K1, K2 ⋯ Kj. However, they do not correspond to any particular step in the successive equilibria of Eq. 1 nor to the binding of ligand by any specific site on the receptor, so they may be viewed as virtual equilibrium constants.
When the uptake of one mole of ligand enhances the affinity of the receptor for the succeeding mole—i.e., when the ratio of successive stoichiometric binding constants is greater than statistical (in which case the binding is frequently called “cooperative”), the constants Kα, Kβ ⋯ Kω are complex numbers appearing in conjugate pairs. For a bivalent receptor, for example, which becomes saturated when two moles of ligand are bound,
 |
5 |
where i = 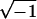 and the coefficients
a and b are real numbers. Alternatively, in
exponential notation,
and the coefficients
a and b are real numbers. Alternatively, in
exponential notation,
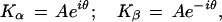 |
6 |
where A = 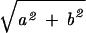 and θ = arctan (b/a). Eq. 4
for such a bivalent system becomes
and θ = arctan (b/a). Eq. 4
for such a bivalent system becomes
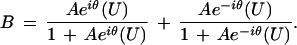 |
7 |
Recalling the relation
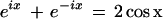 |
8 |
we can convert Eq. 7 into
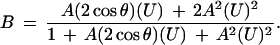 |
9 |
It should be noted in passing that one can show readily (15) that
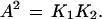 |
10 |
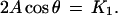 |
11 |
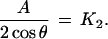 |
12 |
In the general case, a multivalent system where saturation of receptor is reached when n moles of ligand are bound, and the accentuation of successive stoichiometric equilibrium constants continuously exceeds statistical values, then (15, 16),
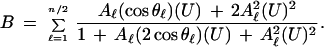 |
13 |
Originally there were n terms in this series, but they were condensed to n/2 pairs by steps corresponding to that used in the transformation of Eq. 7 to Eq. 9.
Application to Unfolding by Urea
As Fig. 1 illustrates, curves for protein unfolding in urea solutions are remarkably steep. The accentuations in successive stoichiometric equilibrium constants must be very large. For a bivalent system, for example, K2 must be much larger than K1. From Eqs. 11 and 12 we see that in the limit K2/K1 approaches infinity for θ = π/2. Under these circumstances, Eq. 9 becomes
 |
14 |
This corresponds to uptake of ligand in coupled pairs.
For a multivalent system, the corresponding binding equation is
 |
15 |
This corresponds to progressive uptakes of coupled pairs of ligand molecules.
If one has a cluster of binding sites that in the absence of ligand are essentially identical, one can partition the free energy of binding into three terms (17):
 |
16 |
The first term on the right-hand side expresses the affinity for a ligand at any site isolated from any influence of other sites on the receptor. The second term takes account of the relative number of open and of occupied sites, which can affect the extent of binding of ligand even when there is no interaction between sites. The third term specifies the impact of interactions between occupied sites on the resultant affinity for ligand. A more explicit expression is
 |
17 |
which leads to
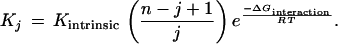 |
18 |
For a facilitative interaction, which leads to increases in affinity constants Kj, the free energy of interaction must be negative, so the exponent in Eq. 18 becomes a positive number. If the facilitative interaction continues to add to the affinity progressively with increasing moles bound ligand, B, then the exponent will increase with B.
It has been shown (18, 19) that when n, the number of sites on the receptor is large, equations for moles bound, B, can be reduced to simpler algebraic form. For the binding of pairs of urea ligands at the n/2 receptor sites of a protein, one can replace Eq. 15 by the expressions
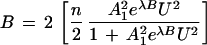 |
19 |
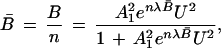 |
20 |
where A12 is the binding constant for the uptake of the first pair of urea ligands and λ is a factor that reflects the enhancement in affinities as B (or B̄) increases.
For convenience in evaluating the parameters A12 and λ for a specific unfolding curve, one can make the following algebraic rearrangements.
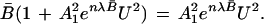 |
21 |
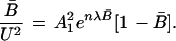 |
22 |
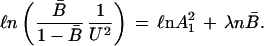 |
23 |
The ratio [B̄/(1 − B̄) × (1/U2)] and may be viewed as the operative equilibrium constant, Koper, during unfolding of protein in the presence of urea. Eq. 23 suggests that ℓn Koper be plotted as a function of fractional conversion B̄. In the limit of decreasing values of B̄,
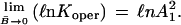 |
24 |
If the interaction parameter, λ, remains constant with increase in B̄, then nλ will be fixed and the data will follow a straight line, the slope of which is nλ. Also, as B̄ approaches 1,
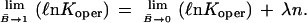 |
25 |
Free energy changes corresponding to Koper can be computed from the equation
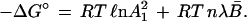 |
26 |
Comparison with Experiment
Fig. 2 presents a transform of the unfolding curve of Fig. 1 into the format specified by Eq. 23. The experimental points shown, encompassed within the range 0.15 < B̄ < 0.85, fall along a straight line, in conformance with Eq. 23 when λ is a constant.
Figure 2.
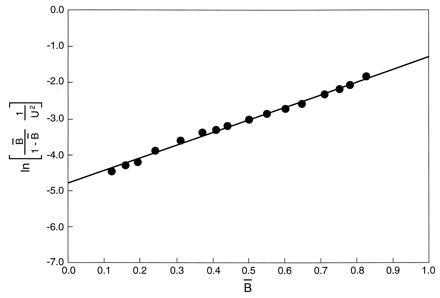
Unfolding data for HPr protein in urea solutions presented in terms of the logarithm of the operative binding constant, Koper, as a function of the fraction unfolded, B̄.
Experimental points for B̄ below 0.15 or above 0.85 are not displayed in Fig. 2. Since the value of B̄ depends on a difference between two experimental readings of a physical quantity (circular dichroic θ, fluorescence intensity, or absorbance), the error therein is larger as B̄ approaches the asymptotes where B̄ or 1 − B̄ tends to zero. If the points for B̄ < 0.15 are added to Fig. 2, they fall progressively below the line as B̄ → 0. Such a trend suggests a systematic error in the choice of an appropriate value for the physical quantity assigned to the fully folded protein, which is used in every computation of B̄. Similarly it has been found that points for B > 0.85 trace out a curve rising progressively above the straight line shown. Such a trend is likely due to a systematic error, in this region, in the choice of the value for the physical quantity assigned to the fully unfolded protein.
Using the data for the interval 0.15 < B̄ < 0.85, the range of highest reliability, one can extrapolate the line observed to B̄ = 0 and thereby evaluate A12. For bacterial HPr, this mean value of the operative binding constant is 0.83 × 10−2. Using Eq. 25, one finds λn is 3.5.
Thus one can now return to the explicit expression for B̄ as a function of U2, Eq. 20:
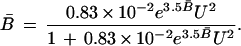 |
27 |
This provides an analytic representation of the graphical information. The curve corresponding to Eq. 27 is displayed in Fig. 1 as a broken line (- - -). It is apparent that in the range 0.15 < B̄ < 0.85, the agreement with the published experimental observations (6) is excellent, as would be expected from the close fit in Fig. 2. At the peripheries, B̄ < 0.15 and B̄ > 0.85, there are systematic deviations. As mentioned earlier, these may be manifestations of defects in choices of limiting values in the asymptotic approaches to fully folded and completely unfolded protein. Alternatively, the theoretical analysis may need further refinement.
Corresponding to the operative equilibrium constant,
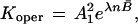 |
28 |
a standard free energy change ΔG° has been defined as:
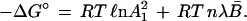 |
29 |
It is apparent that ΔG° varies with the extent of unfolding. In the limit of zero urea concentration, where B̄ = 0, the limiting value of ΔG° is 2840 cal·mol−1. This corresponds to −RT ℓn A12, the standard free change associated with the uptake of the first pair of urea ligands by the empty sites of the protein macromolecule in solution in pure water.
Implications
In the usual treatment of unfolding data in terms of a single N = D equilibrium, the ΔGN-D° obtained (6, 12) for bacterial HPr by extrapolation to zero urea concentration is 4210 cal·mol−1, substantially different from the 2840 cal·mol−1 computed in this paper. The discrepancy is only an apparent one, for one must recognize that the two ΔG° values refer to different processes. That for the N = D equilibrium is the free energy change for the conversion of native protein in its standard state (“hypothetical” 1 molar) to fully unfolded protein in its standard state (“hypothetical” 1 molar):
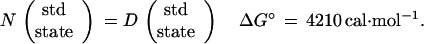 |
30 |
On the other hand, that calculated by the procedure in this paper is for the transformation
 |
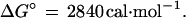 |
31 |
These are different chemical reactions so it is not surprising that they are associated with different values of ΔG°.
Nevertheless, it is still appropriate to compare ΔG° values for wild-type and mutant proteins.
For example, for the HPr protein, a mutant, S31A, in which the Ser-31 of the wild type was replaced by an Ala, shows an unfolding curve similar to that illustrated in Fig. 1 but displaced slightly toward lower urea concentrations (6). At zero urea concentration, the ΔG° value for S31A is 2640 cal·mol−1. Thus Δ(ΔG°) is −200 cal·mol−1 (2640 − 2840). The Δ(ΔG°) evaluated from the midpoint of unfolding curves (11, 12) is essentially the same, for the interaction parameter, the slope of the line in Fig. 2, is the same for S31A as for HPr. If the Δ(ΔG°) is computed on the basis of the two-state assumption, Eq. 30, a value of −460 cal·mol−1 is obtained (6). From either result one can see that the mutant protein is slightly more disposed toward unfolding.
The change in ΔG°, 200 cal·mol−1, cannot be ascribed to a weakening of certain bonds in the protein (20), for it may be instead a reflection of increased affinity of the receptor for urea molecules. Binding of solute molecules and opening of bonds in the protein are coupled during unfolding by urea, and from the experiments at hand it is not possible to deconvolute the net result into the independent contributions of different forms of interactions. To achieve that insight one needs electromagnetic sensors in addition.
The analysis in this paper has focused on the unfolding of the protein. Since the curve in Fig. 1 refers to a reversible conformational change, the folding process must also be a cooperative one, along the reverse pathway. Viewed from that perspective, it is apparent that the folding increases from 10% to 90% for a drop in concentration of urea from about 5.5 to 3.5 molar. This is a very sharp transition for a relatively small fractional drop in ligand concentration, to about 0.65 the starting value. If the binding of ligand were ideal, its concentration would have to change from about 40 molar to 0.4 molar; that is, the drop would have to be to about 0.01 of the starting value, to change the folding of the protein from 10% to 90%. Thus, with cooperative ligand binding the conformation of the macromolecule can be manipulated over a wide range by a small perturbation in ligand concentration.
As has been known for decades, solutes other than urea can drive protein unfolding, most of them at far lower concentrations. Thus it has been shown that reversible denaturation of hemoglobin by salicylate (5) has a midpoint at 0.3 M concentration, about 1/15 that of the usual midpoint in a urea-driven unfolding curve. At, and below, 0.3 M in aqueous solution, salicylate produces no unusual effects on the gross, macroscopic character of the solvent. It seems likely that it effects unfolding by being bound to the protein.
This perspective suggests some heuristic speculations about what may control protein folding in vivo. If an appropriate ligand in the cytoplasm were bound to the polypeptide chain as it emerged from the ribosome, the chain could be maintained in an unfolded conformation. When the binding is cooperative, then a relatively small drop in ligand concentration could swing the protein conformation to a folded conformation. Such a change in ligand concentration could be effected by auxiliary factors (such as chaperones) capable of binding ligand competitively or of releasing ligand by forming an alternative complex with the protein. These coupled interactions could serve to regulate conformational changes in the macromolecule and the transition from unfolded to native form in vivo.
Footnotes
Abbreviation: HPr, histidine-containing protein.
References
- 1.Ramsden W, Chavasse N G. Trans Faraday Soc. 1913;9:93–96. [Google Scholar]
- 2.Anson M L, Mirsky A E. J Gen Physiol. 1929;13:121–132. doi: 10.1085/jgp.13.2.121. , 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins F G. Nature (London) 1930;126:328–330. , 383–384. [Google Scholar]
- 4.Northrop J H. J Gen Physiol. 1932;16:323–337. doi: 10.1085/jgp.16.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anson M L, Mirsky A E. J Gen Physiol. 1934;17:393–398. doi: 10.1085/jgp.17.3.393. , 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammen P K, Scholtz J M, Anderson J W, Waygood E B, Klevit R E. Protein Sci. 1995;4:936–944. doi: 10.1002/pro.5560040513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellman J A. C R Trav Lab Carlsberg Ser Chim. 1955;29:223–229. [PubMed] [Google Scholar]
- 8.Schellman J A. Biopolymers. 1987;26:549–559. doi: 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- 9.Aune K, Tanford C. Biochemistry. 1969;8:4586–4590. doi: 10.1021/bi00839a053. [DOI] [PubMed] [Google Scholar]
- 10.Pace C N. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 11.Matouschek A, Mathews J M, Johnson C M, Fersht A R. Protein Eng. 1994;7:1089–1095. doi: 10.1093/protein/7.9.1089. [DOI] [PubMed] [Google Scholar]
- 12.Scholtz J M. Protein Sci. 1995;4:35–43. doi: 10.1002/pro.5560040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohn E J, Edsall J T. Proteins, Amino Acids and Peptides. New York: Reinhold; 1943. pp. 144–146. [Google Scholar]
- 14.Klotz I M, Luborsky S W. J Am Chem Soc. 1959;81:5119–5124. [Google Scholar]
- 15.Klotz I M. Proc Natl Acad Sci USA. 1993;90:7191–7194. doi: 10.1073/pnas.90.15.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klotz I M. Ligand-Receptor Energetics: A Guide for the Perplexed. New York: Wiley; 1997. [Google Scholar]
- 17.Klotz I M, Walker F M, Pivan R B. J Am Chem Soc. 1946;68:1486–1490. doi: 10.1021/ja01212a030. [DOI] [PubMed] [Google Scholar]
- 18.Scatchard G. Ann NY Acad Sci. 1949;51:660–672. [Google Scholar]
- 19.Thompson C J, Klotz I M. Arch Biochem Biophys. 1971;147:178–185. doi: 10.1016/0003-9861(71)90325-0. [DOI] [PubMed] [Google Scholar]
- 20.Mirsky A E, Pauling L. Proc Natl Acad Sci USA. 1936;22:439–447. doi: 10.1073/pnas.22.7.439. [DOI] [PMC free article] [PubMed] [Google Scholar]