

Introduction
Resistance is the major factor limiting endocrine therapy. This short communication defines the different types of resistance, the mechanisms by which resistance may occur and how they may be identified.
Types
Endocrine resistance may be subdivided into the following types: primary or de novo, and secondary or acquired (occurring after an initial response to treatment). The term 'acquired' infers that, under the pressures of treatment, inductive molecular changes or clonal selection occurs, resulting in a resistant cellular phenotype with an independent growth advantage. Adaptive changes in key molecules of signalling pathways have been observed with endocrine treatment, such as loss of oestrogen receptors (ERs) and over-expression of c-erbB2 at relapse on tamoxifen [1]. Under these circumstances, the underlying mechanisms of resistance might be expected to be common to primary and secondary types.
Endocrine resistance is most often thought of in clinical terms, but resistance may also occur at pathological, proliferative and molecular levels. Although these parameters are positively correlated, they are not equivalent, and disconnects between them occur relatively frequently. For example, cell cycle response and clinical response to tamoxifen and letrozole in the P024 trial were discordant in over one-third of cases despite there being a highly significant correlation between the two types of response (P = 0.00037) [2]. Similarly, the degree of proliferative response was not significantly different in cases responding or not responding to endocrine treatment in the IMPACT (Immediate Preoperative 'Arimidex' [anastrozole], Tamoxifen, or Arimidex Combined with Tamoxifen) trial [3]. Furthermore, as illustrated below, clinically resistant tumours frequently exhibit molecular responses.
Mechanisms of resistance
Theoretically, there are multiple mechanisms whereby breast cancers appear unresponsive to endocrine therapy. These include inherent tumour insensitivity to hormone therapy, activation of hormone signalling pathways by nonendocrine pathways, ineffective or compromised therapy, and domination of cell survival [4]. To determine whether these mechanisms are apparent in primary breast cancers, the neoadjuvant setting in which treatment is given with tumour still within the breast has been particularly informative. Because of relatively easy access to tumour, biopsies may be taken sequentially before and during treatment and analyzed to monitor the effects of treatment on molecular processes. Some of these studies are worthy of more detailed consideration using ER and the expression of oestrogen-regulated genes, including KIAA0101, SERPINA3, IRS1, TFF3 and TFF1 as markers of (anti)oestrogenic responses, and Ki67 and expression of cell cycle/DNA synthesis genes such as CDC, CKS2, Cyclin B1, TYMS and PCNA as markers of proliferation [5-7].
Inherent resistance
Hormone stimulation of tumour growth is generally associated with oestrogen, whose action is mediated primarily through the ER. It follows therefore that in ER-negative tumours oestrogen is disconnected from growth (which is driven by other growth factors). Therapies directed either at oestrogen or ER are therefore unlikely to be successful in ER-negative breast cancer. This is supported by a large volume of literature, and we have reported that in the neoadjuvant setting ER-negative tumours rarely respond to a variety of endocrine therapies [8]. Additionally, as shown in Figure 1, an ER-negative tumour treated electively with neoadjuvant letrozole did not exhibit a major change in expression of oestrogen-regulated and proliferation-related genes, illustrating molecular resistance to treatment. The clinical relevance of these observations is that patients with ER-negative tumours should not be regarded as candidates for endocrine treatment.
Figure 1.
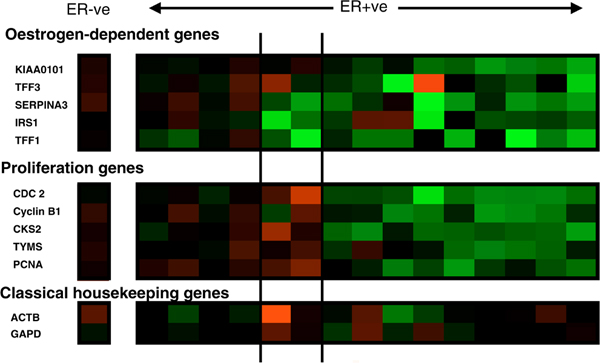
Changes in expression of oestrogen-regulated and proliferation-associated genes after 14 days of adjuvant treatment with letrozole. Green represents downregulation and red upregulation (these will show as relatively paler blocks if you are viewing this figure in black and white). Intensity of colour/shade represents degree of change. The single column to the left of the figure represents changes in an ER-negative case; the panels relate to 15 different clinically resistant ER-positive tumours: the left-hand panel illustrates a molecular resistant phenotype; the middle panel shows cases exhibiting decreases in the expression of oestrogen-regulated genes but not in cell cycle genes; and the right-hand panel shows molecular sensitivity in both oestrogen-regulated and cell cycle associated gene expression. ER, oestrogen receptor.
However, many endocrine-resistant tumours have ER-positive phenotypes, and the challenge is to discriminate between ER-positive responsive and ER-positive resistant tumours, and to clarify the underlying cause of resistance in the latter.
It is possible to envisage an aberrant receptor that may bind oestrogen or ER antibodies but not transduce a signal (RNAs encoding variant and mutant ER have been reported in breast cancer [9]). In this case, tumours would be inherently insensitive to hormone stimulation and refractory to endocrine deprivation therapy despite having ER-positive status.
Another scenario is that ER is functional and driving oestrogen-dependent processes, but that growth is not one of them, this being stimulated by other independent pathways. If this were the case, then oestrogen-deprivation therapies would reduce expression of classically oestrogen-regulated genes but not those associated with cellular proliferation. This phenotype was observed in two out of 15 ER-positive tumours treated with neoadjuvant letrozole (Figure 1).
Activation of hormone signaling pathways by nonendocrine pathways
In model systems, activation of other messenger systems may influence ER signalling such that it may occur in the presence of low oestrogen levels (hypersensitivity) or even in the absence of hormone. Thus, even in the absence of oestrogen, the HER (human epidermal growth factor receptor) signalling can result in ER phosphorylation (a critical step in ER activation) [10]. It is therefore interesting that endocrine therapy often fails to reduce proliferation in ER-positive breast cancers over-expressing c-erbB2 [11-13].
Ineffective or compromised therapy
Failure to respond to endocrine therapy may not necessarily be caused by inherent tumour resistance but because the treatment itself is inefficient or compromised. There could be several reasons for this, including a lack of drug potency, poor pharmocokinetics/pharmacogenetics [14] and compensatory endocrine loops (for example, the use of aromatase inhibitors in premenopausal women).
Inherent resistance, ligand-independent ER signalling and inefficient therapy could all be associated with a molecular phenotype in which the expression of oestrogen-regulated and proliferation-associated genes do not change markedly with treatment. This phenotype can be seen in Figure 1, in which four out of 15 tumours exhibit a phenotype of molecular resistance. It should be noted that lack of change with treatment may result from high expression not being reduced with therapy or a low basal level of expression being maintained, or a hybrid split according to oestrogen regulation and proliferation genes. By possessing knowledge not only of changes with treatment but also of the pretreatment and post-treatment levels of expression, it is possible to determine the specific type of resistance (see below).
Hypersensitivity and cell survival
However, the most common phenotype in letrozole-resistant tumours is for both oestrogen-regulated and proliferation-associated genes to decrease with treatment. This observation suggests that there are other mechanisms of resistance. These include hypersensitivity, in which tumours continue to grow in low oestrogen environments. The scenario would be that, despite therapy producing an overall reduction in proliferation, residual cell cycle progression plus effective cell survival is sufficient to maintain tumour growth.
It is clear that multiple pathways/processes may confer endocrine resistance. It therefore follows that their identification will be equally diverse.
Identification of resistance
Table 1 lists some resistance mechanisms and measurements that might help to identify them. Measurement of ER status should be routine because patients with ER-negative tumours rarely respond to endocrine therapy. Additionally, expression profiles of oestrogen-regulated genes, growth factors and their receptors can provide some indication of ER functionality and the involvement of other growth factor pathways.
Table 1.
Identification of resistance
| Mechanism | Phenotype | Assessment |
| Inherent resistance | ER negativity | ER measurement |
| Defective ER | ER read-outs | |
| Alternative growth signalling | GF/receptor assays | |
| ER modified | ER phosphorylation | Phosphorylation status |
| ER co-activators | ER read-outs | |
| ER co-repressors | ||
| Ineffective treatment | Inherent lack of potency | Target read-outs |
| Compensatory mechanisms | Target read-outs | |
| Poor pharmacokinetics | Pharmacogenetics |
ER, oestrogen receptor; GF, growth factor.
The status of ER signalling and its output may be assessed by measuring the degree/type of ER phosphorylation and levels of ER co-activators/co-repressors.
The efficiency of treatment may be monitored by measuring oestrogen levels (in the case of aromatase inhibitors), downstream signalling of ER (in the case of selective ER modulators and selective ER downregulators) and genetic polymorphisms (where there is evidence that pharmacogenetics influence drug levels or metabolism).
Based on the above considerations, it should be apparent that no single marker will identify resistance or its mechanism – multiple measurements and readouts will be necessary. Finally, it should be noted that single static measurements of biomarkers either before or during treatment are unlikely to be definitive, and (especially to determine efficiency of treatment) dynamic sequential assessments of tumour molecular biology or histopathology will be required. Tissue acquisition will be a logistical challenge if these analyses are to be undertaken on a routine basis.
Past, present and future
The status of endocrine resistance can be put into perspective by assessing where it has come from, where it is now and where it is going. My personal view is that we have come from a past of having no understanding of the mechanisms of resistance and no rational method of identifying resistance. We now have a basic understanding of resistance mechanisms and their diversity, and we currently use ERs to help identify resistance. However, we can look forward to a future in which we will perform molecular phenotyping of resistant tumours on an individual basis and devise novel rational circumventing treatments using that knowledge.
Abbreviations
ER: oestrogen receptor.
Competing interests
The author declares that they have no competing interests.
Acknowledgments
Acknowledgements
This article has been published as part of Breast Cancer Research Volume 10 Supplement 4, 2008: Controversies in Breast Cancer 2008. The full contents of the supplement are available online at http://breast-cancer-research.com/supplements/10/S4
References
- Dowsett M, Johnston S, Martin LA, Salter J, Hills M, Detre S, Gutierrez MC, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Smith I. Growth factor signalling and response to endocrine therapy: the Royal Marsden Experience. Endocr Relat Cancer. 2005;12(suppl 1):S113–S117. doi: 10.1677/erc.1.01044. [DOI] [PubMed] [Google Scholar]
- Tao Y, Klause A, Vickers A, Bae K, Ellis M. Clinical and biomarker endpoint analysis in neoadjuvant endocrine therapy trials. J Steroid Biochem Mol Biol. 2005;95:91–95. doi: 10.1016/j.jsbmb.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, Griffith C, Boeddinghaus I, Salter J, Detre S, Hills M, Ashley S, Francis S, Walsh G, IMPACT Trialists Short-term changes in Ki-67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence-free survival. Clin Cancer Res. 2005;11:951s–958s. [PubMed] [Google Scholar]
- Miller WR. Biological rationale for endocrine therapy in breast cancer. Best Pract Res Clin Endocrinol Metab. 2004;18:1–32. doi: 10.1016/S1521-690X(03)00044-7. [DOI] [PubMed] [Google Scholar]
- Miller WR, Larionov AA, Renshaw L, Anderson TJ, White S, Murray J, Murray E, Hampton G, Walker JR, Ho S, Krause A, Evans DB, Dixon JM. Changes in breast cancer transcriptional profiles after treatment with the aromatase inhibitor, letrozole. Pharmacogenet Genomics. 2007;17:813–826. doi: 10.1097/FPC.0b013e32820b853a. [DOI] [PubMed] [Google Scholar]
- Miller WR, Larionov A, Renshaw L, Anderson TJ, White S, Hampton G, Walker JR, Ho S, Krause A, Evans DB, Dixon JM. Aromatase inhibitors – gene discovery. J Steroid Biochem Mol Biol. 2007;106:130–142. doi: 10.1016/j.jsbmb.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Miller WR, Larionov A, Anderson TJ, Walker JR, Krause A, Evans DB, Dixon JM. Predicting response and resistance to endocrine therapy: profiling patients on aromatase inhibitors. Cancer. 2008;112(suppl):689–694. doi: 10.1002/cncr.23187. [DOI] [PubMed] [Google Scholar]
- Miller WR, Anderson TJ, Hawkins RA, Keen J, Dixon JM. Neoadjuvant endocrine treatment: the Edinburgh experience. In: Dowsett M, Howell A, editor. Primary Medical Therapy for Breast Cancer: Clinical and Biological Aspects. Amsterdam: Elsevier; 1999. pp. 89–99. [Google Scholar]
- Fuqua SA, Cui Y. Estrogen and progesterone receptor isoforms: clinical significance in breast cancer. Breast Cancer Res Treat. 2004;87(suppl 1):S3–S10. doi: 10.1007/s10549-004-1577-4. [DOI] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Dowsett M, Harper-Wynne C, Boeddinghaus I, Salter J, Hills M, Dixon M, Ebbs S, Gui G, Sacks N, Smith I. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res. 2001;61:8452–8458. [PubMed] [Google Scholar]
- Ellis MJ, Tao Y, Young O, White S, Proia AD, Murray J, Renshaw L, Faratian D, Thomas J, Dowsett M, Krause A, Evans DB, Miller WR, Dixon JM. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol. 2006;24:3019–3025. doi: 10.1200/JCO.2005.04.3034. [DOI] [PubMed] [Google Scholar]
- Miller WR, Anderson TJ, White S, Evans D, Krause A, Dixon JM. Growth factor signalling in clinical breast cancer and its impact on response to conventional therapies: the Edinburgh experience. Endocr Relat Cancer. 2005;12(suppl 1):S119–S123. doi: 10.1677/erc.1.00995. [DOI] [PubMed] [Google Scholar]
- Ingle JN. Pharmacogenetics and pharmacogenomics of endocrine agents for breast cancer. Breast Cancer Res. 2008;10(suppl 4):S17. doi: 10.1186/bcr2177. [DOI] [PMC free article] [PubMed] [Google Scholar]