

Abstract
Numerous proteins bend DNA upon binding, a phenomenon of potential significance for regulation of gene expression and chromatin. DNA bending is commonly predicted from the presence of electrophoretic mobility anomalies in protein–DNA complexes. However, as compared with electrophoretic methods, several DNA binding oncoprotein families do not display comparable evidence of DNA bends in x-ray structural studies. Herein, circularization kinetics and affinity measurements with prebent DNA templates were employed to assess bending and DNA structural preferences for Max and other basic helix–loop–helix/leucine zipper proteins. In this way, proteins in the Myc/Max basic helix–loop–helix/leucine zipper family were found not to bend DNA in solution but to actually stabilize DNA in an unbent configuration that resists circularization. The mobility anomaly was found to be induced by the leucine zipper protein motif, rather than structural distortions of DNA. Thus rigid protein domain structures may induce anomalous electrophoretic mobility. Moreover, the energetic preference of non-DNA bending proteins for unbent templates suggests mechanisms whereby chromatin structure may regulate transcription.
Keywords: Myc, transcription, chromatin
A number of DNA binding proteins appear to preferentially stabilize DNA in a “bent” configuration. The electrophoretic mobility anomaly (1) is widely used to detect DNA bends and, in conjunction with phased intrinsically bent DNA sequences, may predict DNA bend orientation (2–5). DNA binding studies using minicircular “prebent” DNA substrates have shown large (200- to 300-fold) affinity changes dependent on preexisting bends (6, 7). In this way, proteins that bend DNA display enhanced affinity for prebent DNA. Within cells, prebent DNA could exist in chromatin as a consequence of nucleosome structure, supercoiling, and other features. Affinity constraints that are dictated by DNA structure, rather than sequence, may thus provide a link between chromatin structure and gene expression.
Evidence of protein-induced DNA bending has been obtained by crystallographic, electrophoretic, and ligation kinetic methodologies. In some cases, solution-phase demonstrations of DNA bending agree with crystal-based structure determinations. In several interesting cases, however, discrepancies have been noted with electrophoretic evidence of DNA bending (mobility anomalies), but no significant DNA bending in the corresponding x-ray structure (8–13). For example, the Myc oncoprotein, Max, and several other basic helix–loop–helix/leucine zipper (b-HLH-ZIP) proteins produce electrophoretic DNA–protein mobility anomalies (8, 9), while a DNA–Max cocrystal structure reveals no significant DNA bend (12). Similarly, hetero- and homodimeric protein–DNA complexes of Jun and Fos oncoproteins produced mobility anomalies in circular permutation electrophoresis assays (10, 11, 14), whereas x-ray structure determinations of Jun–Fos–DNA cocrystals reveal minimal DNA distortion from linearity (13). Recently, Sitlani and Crothers (15) demonstrated that Jun and Fos do not bend DNA, based on circularization kinetics analyses. In electrophoretic mobility studies, both b-HLH-ZIP and b-ZIP family proteins migrated so as to suggest the presence of oriented DNA bends of opposite direction depending on dimerization partner, based on phasing studies with intrinsically bent dA tracts (8, 11). These observations suggest that solution-phase DNA binding could differ from crystal-phase complexes, perhaps due to crystal packing forces (16) or alternatively that one of these assays may not always accurately predict DNA bending, a possibility further supported by circularization kinetics analyses (15). The broad application of the electrophoretic mobility anomaly (circular permutation) assay to the analysis of DNA bending thus motivates the search for explanations of this anomalous behavior.
The apparently discrepant predictions between electrophoretic and crystal structure determinations of DNA bending has been particularly noted in two protein families whose DNA binding motif contains a leucine zipper, the b-ZIP family and the b-HLH-ZIP family. The presence of this common domain is consistent with the possibility that the leucine zipper may play a role in mediating the unique structural behaviors of these protein–DNA complexes.
In this study, b-HLH-ZIP proteins were examined for evidence of DNA bending by two independent assays: circularization (ligation) kinetics and prebent DNA binding preferences. Proteins in this family produced sequence-specific inhibition of circularization, suggesting that b-HLH-ZIP proteins actually stabilize DNA in an unbent configuration. In addition, no preferential affinity for prebent DNA was observed, but rather, enhanced affinity existed for linear unbent DNA templates. Finally, an explanation for the anomalous mobility induced by this family was obtained with the observation that the mobility anomaly was dependent on the presence (and the length) of the leucine zipper.
MATERIALS AND METHODS
Protein Constructs and DNA Template Construction.
Recombinant Max protein was expressed as a His-fusion construct (provided by Chi Dang, Johns Hopkins University) and was purified after bacterial overexpression by nickel chelate chromatography (Qiagen, Chatsworth, CA) as described (9). Other b-HLH-ZIP proteins were also synthesized as described (9) with the addition of microphthalmia (Mi) that was expressed from a cDNA template using T3 RNA polymerase followed by in vitro translation using rabbit reticulocyte lysate (Promega) as described (17). DNA templates for circularization kinetic studies or affinity measurements were made as outlined in Fig. 2A and as described for circles containing the TATA sequence element (7). For these templates, a plasmid containing six dA tracts spaced either 31 or 37 bp between the center of the last dA tract and the center of the adenovirus major late promoter CACGTG (and several flanking residues) element was subject to PCR in the presence of [32P]dCTP. The PCR product was gel-purified, digested with XbaI to generate fragments exactly 156 bp long. Monomeric minicircles were generated by ligation of these fragments under dilute conditions (300 μl, total volume) as described (7), and circular species were sequentially purified through 4% and 6% native acrylamide gels. Probes to assess twist were engineered using PCR primers that lengthen or shorten the linear template length by 2 or 4 bp at a location adjacent to the XbaI restriction site. Circular products were verified by BAL-31 exonuclease resistance.
Figure 2.
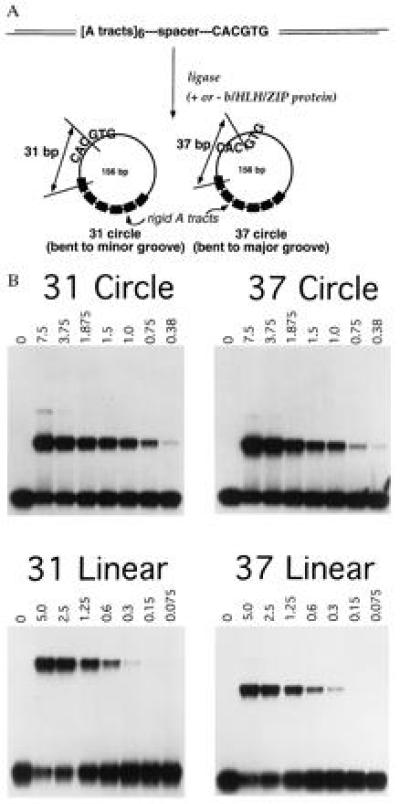
(A) Schema for construction of constrained minicircular DNAs. Linear templates for minicircle production contain the CACGTG sequence phased either 31 or 37 bp from the center of the last intrinsically bent dA tract. Minicircles were produced and labeled as described (7). (B) Lack of affinity differences for oriented prebent DNAs but modestly enhanced affinity for linear DNA templates. Minicircular or linearized probes containing the identical b-HLH-ZIP target sequence (derived from adenovirus major late promoter) were subject to electrophoretic mobility-shift analysis as described (9) using purified Max at concentrations indicated as pmol/10 μl of DNA binding reaction mixture. The upper bands represent protein–DNA complex and the lower bands represent unbound free probe. Evidence of preferential affinity for linear probes can be seen from the enhanced ability of Max to deplete unbound linear probes (lower bands at high Max concentrations).
Circularization Kinetics.
Ligation kinetics reactions were carried out in 200 μl in 50 mM Tris·HCl, pH 7.8/10 mM MgCl2/10 mM DTT/1 mM ATP/BSA (25 μg/ml)/3 μM Max/650 pg of template DNA (either containing or lacking CACGTG binding site). After a 30-min preincubation at 21°C, 400 units of T4 DNA ligase was added and aliquots were removed at the indicated time points. Reactions were stopped by addition of proteinase K (to 0.67 mg/ml), followed by native gel electrophoresis using Tris/glycine/EDTA. Position of circular monomers was verified by BAL-31 resistance.
Gel Electrophoretic Affinity Studies.
Protein–DNA binding reactions (verified to be at equilibrium) were carried out as described (9) with purified recombinant proteins, incubated for 30 min at 30°C, resolved by native PAGE, and quantitated by PhosphorImager analysis. Kd values with standard deviation were determined from a minimum of three experiments plotting the slope of 1/r vs. 1/[A], where r is [bound DNA]/([bound DNA] + [unbound DNA]), and [A] is protein concentration (μM). Protein concentration was determined by titrating the recombinant protein species against DNA of known specific activity to protein–DNA saturation. To minimize nonspecific interactions that otherwise complicated these measurements, DNA binding was carried out in the presence of BSA (5 μg/10 μl) and poly[d(I-C)] (0.5 μg/10 μl) so that measured Kd values reflect the presence of these competitors. Thus Kd values are condition-specific but meaningful for comparisons with the different DNA templates.
Circular Permutation Analyses.
Circular permutation assays were performed as described (9) using four probes, all 154 bp, obtained by cleavage of pBend-MLP with BglII, NheI, SpeI, and BamHI followed by labeling with Klenow subunit of DNA polymerase I and [α-32P]dATP. Proteins for these assays were synthesized using programmed rabbit reticulocyte lysate and fragments of the microphthalmia transcription factor that span the b-HLH-ZIP region or end shortly into the leucine zipper (wild-type or Mice mutant) as well as E47s as described (17).
RESULTS
Prior studies demonstrated anomalous electrophoretic mobility of protein–DNA complexes with the b-HLH-ZIP proteins Myc/Max, Max–Max, TFEB, TFE3, and USF using either circular permutation assays or phasing studies with intrinsically bent DNA probes (8, 9, 18). To assess protein–DNA energetic preferences relative to DNA structure, circular-ligation kinetic studies were undertaken. In this assay (19), intramolecular ligation of linear DNA fragments was studied in the presence or absence of the b-HLH-ZIP protein using DNA fragments containing or lacking a target binding sequence (CACGTG, derived from adenovirus major late promoter, MLP). Phasing studies previously suggested that Max bends DNA toward the minor groove (8, 9). Therefore, circularization templates were employed that phase the binding site relative to intrinsically bent dA tracts, potentially discriminating between bends that would resemble the letter C vs. the letter S. For the b-HLH-ZIP protein Max, rather than accelerating ligation, sequence-specific inhibition of circularization was observed, regardless of phase orientation (Fig. 1). Max did not inhibit all ligation, however, since higher-order ligated species were preferentially formed (Fig. 1A), and Max did not impede circularization of comparable fragments lacking its binding sequence (Fig. 1B). DNA fragments underwent accelerated circularization in the presence of a different bending protein, TATA binding protein, when its binding site was present (data not shown). For Max, both the predicted (31 bp) and oppositely (37 bp) phased fragments displayed comparable sequence-dependent inhibition of ligation (Fig. 1 C and D, curves 31,+Max and 37,+Max vs. all other DNA/protein combinations). It is possible that Max produces bends that are intermediate in phase, thereby skewing the template in a configuration unfavorable for ligation; however, electrophoretic phasing studies (8, 9) demonstrated simple in-phase maximum and half-turn out-of-phase minimum mobility anomalies. In addition, a series of linear DNAs were constructed that vary the DNA template length by +2, +4, −2, or −4 bp relative to the 31-bp spaced template. In each case, monomeric circularization was impeded by the presence of protein (Fig. 1E). Moreover, in the presence of Max, the fastest ligating templates were the 156-bp, supporting the x-ray structural prediction (12) that Max does not substantially change the twist of DNA. These constructs help control for the possibility that differences in twist (with bending) might have impeded circularization of templates containing an integral number of helical turns (Fig. 1 A–D). In addition, these protein–DNA interactions appear to be sequence-specific and lack evidence of nonspecific aggregation based on electrophoretic and competition studies (data not shown and ref. 17). Therefore, these ligation kinetic studies support the x-ray structural prediction (12) that Max does not substantially bend DNA, despite its ability to produce anomalous electrophoretic mobility. Moreover, the phase-independent inhibition of ligation is consistent with the possibility that b-HLH-ZIP proteins preferentially stabilize DNA in an unbent (linear) configuration that resists circularization, favoring inter- rather than intramolecular ligation.
Figure 1.
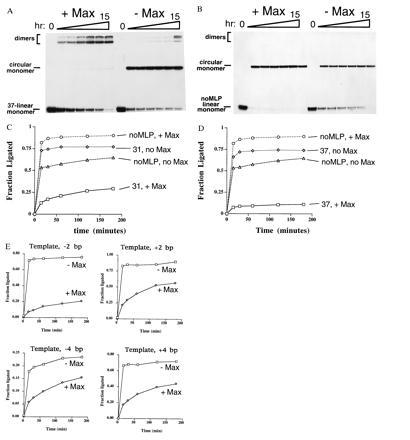
Inhibition of circularization by Max. Ligation kinetic studies were carried out using 156-bp linear DNAs containing the CACGTG (derived from adenovirus major late promoter, MLP) Max recognition sequence either in phase (31 bp) or out of phase (37 bp) relative to dA tracts within the same fragment. Fragments lacking the binding site are indicated as noMLP. Incubations were ended at 0, 15 min, 30 min, 1 hr, 2 hr, 3 hr, and 15 hr. (A) Electrophoretic profile of typical ligation reactions for fragments containing the Max binding site. Higher-order forms, including linear or circular dimers are indicated at the top of the gel. (B) Ligation reactions for fragments lacking the Max binding site (noMLP linear monomer). (C) Phase-independent inhibition of circularization. Ligation products were quantified by PhosphorImager and the molar fraction present as circular monomer was plotted as a function of time. Probes either contained the CACGTG binding site for Max at a spacing of 31 bp (in phase) from the center of the last dA tract (indicated as 31) or lacked the CACGTG binding site (indicated as noMLP), and ligations were run either in the presence or absence of purified Max protein as indicated. (D) Ligation conditions identical to C except that the CACGTG-containing template was spaced 37 bp (out of phase) from the center of the adjacent A tract (indicated as 37). DNA templates lacking the CACGTG binding site are indicated as noMLP. (E) Control for twist in circularization kinetics. Linear templates were produced exactly as above, but varying in length −2, −4, +2, or +4 bp relative to the 156-bp 31 linear template. Ligation kinetics were carried out as above, containing or lacking Max as indicated. In each case, the presence of Max impeded circularization, suggesting that twist does not explain the lack of circularization of the 156-bp templates.
If b-HLH-ZIP proteins did bend DNA, they would likely display differential affinities for DNA templates in varied prebent orientations, as was shown for CAP (6) and TATA binding protein (7). To test this, protein–DNA affinities were compared using minicircular or corresponding linear DNA probes. Prebent minicircles were produced analogously to studies of Escherichia coli CAP protein (6) and exactly as we described for TATA-containing minicircles (7) where significantly enhanced affinities were observed for “correctly” prebent templates. The center of the CACGTG binding site was positioned to be either in-phase or out of phase with dA tracts (Fig. 2A) so that the circles would constrain the binding site center to be prebent toward the major groove (37 circle) or toward the minor groove (31 circle). Minicircular DNA probes were subjected to electrophoretic mobility shift analysis in their circular and endonuclease-cleaved linear forms. As shown in Fig. 2B, no significant differences were observed in the binding of Max to minicircles with binding sites prebent in opposite orientations. In fact, binding was reproducibly ≈3-fold enhanced for the corresponding linearized probes derived from the identical minicircle DNA fragments. For example, Max’s affinity for circle 37 was 1.24 ± 0.08 μM, as compared with 0.371 ± 0.01 μM for linear 37, a Kd preference of 3.3-fold in favor of the linear template. It is noteworthy that the magnitude of the DNA prebend in these circular constructs is predicted to be small (360° per 156 bp or ≈2° per bp) because the dA tract segment makes up approximately one-third of the circle sequence and should contribute 18–22° of bend (20–25), a roughly proportional contribution of bend. Therefore, with more highly distorted DNA (as might occur in a supercoiled structure, for example), the preference for unbent DNA may be even more pronounced. Essentially identical affinity preferences were observed for TFEB and other b-HLH-ZIP proteins including USF, TFE3, a polyalanine chimeric mutant of Myc with enhanced basic domain α-helicity (26), and heterodimers of Myc/Max (data not shown) despite our prior observations that all of these proteins produce significant phase-specific mobility anomalies (8, 9).
Since b-HLH-ZIP proteins impede circularization and display enhanced affinity for linear rather than circular DNA, from where might the electrophoretic mobility anomaly arise? Prior studies have indicated a similar discrepancy between x-ray structural data and electrophoretic behavior for the b-ZIP factors Jun and Fos, which produce phased electrophoretic mobility anomalies (10, 11), but minimal DNA bending in x-ray structural studies (13). Since b-HLH-ZIP and b-ZIP factors share the presence of a C-terminal leucine zipper motif, it appeared plausible that this domain might account for the electrophoretic behavior. To examine this possibility, the b-HLH-ZIP domain of microphthalmia (Mi) (17) was studied, because it was found to retain sequence specific DNA recognition even with much of its leucine zipper removed (Fig. 3). In addition, the b-HLH protein E47 lacks a leucine zipper altogether but is capable of binding the same DNA sequence as b-HLH-ZIP proteins such as Max and Mi (27). As shown in Fig. 3, circular permutation analysis with these three polypeptides and identical DNA probes revealed that the mobility anomaly correlated directly with the presence (complete or partial) of the leucine zipper at the C terminus of the polypeptide. Anomalous migration was significant in the presence of the intact b-HLH-ZIP peptide (lanes b-HLH-ZIP), did not occur in the complete absence of a leucine zipper (lanes b-HLH), and was intermediate with only a portion of the leucine zipper (lanes b-HLH-Z). A small amount of USF present in the reticulocyte lysate of the E47 electrophoretic mobility shift analysis reactions displays anomalous mobility with the identical probes, the magnitude of which is significant, relative to the distance from the origin. Comparative x-ray structural studies of the b-HLH-ZIP protein USF (28) have demonstrated that a zipperless (b-HLH) fragment binds DNA essentially indistinguishably from the full zipper-containing (b-HLH-ZIP) form, rendering it unlikely that loss of anomalous mobility is due to differing DNA structures. Moreover, x-ray cocrystal structures of the b-HLH proteins MyoD (29) and E47 (30) also reveal essentially unbent DNA.
Figure 3.
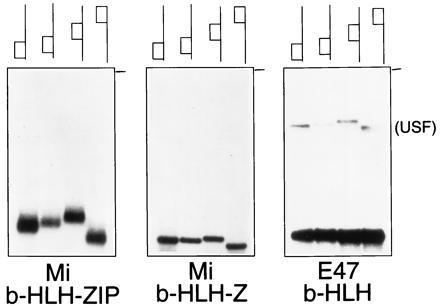
Leucine zipper produces a protein–DNA mobility anomaly. Circular permutation analysis was carried out using four probes (depicted schematically above each lane) with identical CACGTG binding sites from adenovirus major late promoter, located at different positions along identical length DNA fragments. The three tested proteins were the complete b-HLH-ZIP domain from microphthalmia, a microphthalmia truncation that deletes all but the most N-terminal 7 amino acids of the leucine zipper (lanes Mi b-HLH-Z), and the b-HLH protein E47. A small amount of USF can be seen in the E47 lanes (derived from reticulocyte lysate) and demonstrates a significant mobility anomaly with the same probes (anomaly is measured as ratio of differences from origin). The origin is indicated by a dash to the right of each panel.
DISCUSSION
The leucine zipper is a highly plausible candidate for producing an electrophoretic mobility anomaly, even in phasing assays. Protein-dependent mobility changes have also been suggested from studies of Crothers and colleagues using the yeast b-ZIP transcription factors GCN4 (31) and Jun/Fos (15), consistent with the possibility that the leucine zipper could mediate this electrophoretic behavior. For Jun–Fos–DNA complexes, Sitlani and Crothers (15) have recently demonstrated lack of DNA bending in solution through circularization kinetic analyses, also consistent with a role for the leucine zipper in having produced the electrophoretic mobility anomaly. A significant effect of spacing (between the protein binding site and the dA tracts) was shown to influence the electrophoretic behavior, thus potentially allowing for experimental designs that circumvent or minimize the potential contribution of motifs such as the leucine zipper in producing anomalous mobility. The leucine zipper dimerization motif positions the protein dimer at the center of dyad symmetry on the DNA template with its longitudinal axis perpendicular to the DNA backbone. Moreover, the perpendicular configuration is essential to recognition of both DNA half sites. Since the leucine zipper coiled coil motif is relatively nonglobular (as compared, for example, to the HLH or helix–turn–helix motifs), it is likely that it would behave as a “spoke” in electrophoretic mobility studies, retarding migration of the DNA complex in a fashion that is dependent on its position relative to the DNA ends. For example, with a leucine zipper protein bound in the center of a DNA probe, the complex might resemble the letter T, whereas when bound at the end it would resemble the letter L. The sieving properties of such a T-like complex are likely to be more complex than for the L-shaped complex, potentially explaining electrophoretic migration differences. Even in intrinsically bent DNA templates (as in phasing studies using dA tracts), such a protein spoke would be expected to produce phase-dependent mobility changes, a phenomenon that was observed with b-HLH-ZIP proteins (8, 9). It would also appear plausible that a leucine-zipper-mediated effect would vary as a function of distance from the intrinsically bent dA tracts, a finding compatible with the spacing-dependent mobility changes in Jun–Fos–DNA complexes recently reported (15). While the length of the leucine zipper appeared to correlate with the magnitude of the mobility anomaly in these studies (Fig. 3), it may be noteworthy that the mobility anomaly produced with bZIP proteins did not correlate with the size of the protein outside of the leucine zipper motif (32). These differences could relate to the presumably more globular structure of regions outside this coiled coil motif.
Certain b-HLH-ZIP and b-ZIP proteins have specifically been suggested to produce oriented DNA bends of opposite direction depending on dimerization partner, based on phasing studies with intrinsically bent dA tracts (8, 10). For example, Jun–Fos heterodimers produced DNA complexes with mobility anomalies suggesting bending toward the major groove, whereas Jun–Jun homodimers induced an oppositely oriented mobility anomaly, relative to intrinsically bend dA tract sequences (10). Similarly, Myc homodimers appeared to produce a backward oriented mobility anomaly relative to Myc–Max heterodimers (8). Interestingly, in both of these cases the homodimer species is substantially less stable than the heterodimer, due to specificities presumably dictated by coiled coil interactions within the leucine zipper (33). In the case of c-Myc, it is unclear whether a homodimeric species is present to a significant degree within cells. Therefore, the oppositely oriented mobility anomalies produced by these sets of less-stable homodimers could reflect significantly different dimerization structures dictated by repulsion at the typical homodimerization interface.
These studies demonstrate the potential capacity of certain rigid protein motifs to induce significant mobility anomalies. While the leucine zipper appears to be one such motif, it remains possible that others, or even the net configuration of an extended protein, might produce similar anomalous migration behaviors. Therefore, caution should be exercised in interpreting the presence of DNA bending from such studies. Assessing DNA bending by nonelectrophoretic means, such as circularization kinetics, may substantially improve the predictability of DNA bending behavior (15).
Finally, the observation that b-HLH-ZIP proteins do not bend DNA has allowed for the demonstration of a modest reproducibly enhanced affinity for unbent DNA. While the magnitude of the preference for undistorted DNA is likely to vary among different DNA binding motifs in proteins, the opportunity to bind nonlinear B form DNA sites is likely to arise commonly within cells. Therefore, the structural (as distinct from strictly sequence directed) specificity of DNA binding factors may play a role in transcriptional regulation that resides at the level of chromatin structure.
Acknowledgments
We acknowledge Drs. Philip Sharp, Stephen Harrison, Jeffrey Parvin, Kathryn Calame, Tom Ellenberger, and Myles Brown for useful discussions; Drs. Jason Kahn and Donald Crothers for helpful discussions and advice early in this project; and Dr. Chi Dang for the Max expression DNA construct. This work was supported by Grant AR43369 from the National Institutes of Health (to D.E.F.). D.E.F. is a Pew Foundation Scholar and a Scholar of the James S. McDonnall Foundation.
Footnotes
Abbreviation: b-HLH-ZIP, basic helix–loop–helix/leucine zipper.
References
- 1.Wu H-M, Crothers D M. Nature (London) 1984;308:509–513. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 2.Zahn K, Blattner F R. Science. 1987;236:416–422. doi: 10.1126/science.2951850. [DOI] [PubMed] [Google Scholar]
- 3.Koo H-S, Crothers D M. Proc Natl Acad Sci USA. 1988;85:1763–1767. doi: 10.1073/pnas.85.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gartenberg M R, Ampe C, Steitz T A, Crothers D M. Proc Natl Acad Sci USA. 1990;87:6034–6038. doi: 10.1073/pnas.87.16.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson J F, Landy A. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1992;89:6343–6347. doi: 10.1073/pnas.89.14.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parvin J, McCormick R, Sharp P A, Fisher D E. Nature (London) 1995;373:724–727. doi: 10.1038/373724a0. [DOI] [PubMed] [Google Scholar]
- 8.Wechsler D S, Dang C V. Proc Natl Acad Sci USA. 1992;89:7635–7639. doi: 10.1073/pnas.89.16.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher D E, Parent L A, Sharp P A. Proc Natl Acad Sci USA. 1992;89:11779–11783. doi: 10.1073/pnas.89.24.11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerppola T K, Curran T. Science. 1991;254:1210–1214. doi: 10.1126/science.1957173. [DOI] [PubMed] [Google Scholar]
- 11.Kerppola T K, Curran T. Cell. 1991;66:317–326. doi: 10.1016/0092-8674(91)90621-5. [DOI] [PubMed] [Google Scholar]
- 12.Ferre-D’Amare A R, Prendergast C D, Ziff E B, Burley S K. Nature (London) 1993;363:38–45. doi: 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- 13.Glover J N M, Harrison S C. Nature (London) 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 14.Kerppola T K, Curran T. Mol Cell Biol. 1993;13:5479–5489. doi: 10.1128/mcb.13.9.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1996;93:3248–3252. doi: 10.1073/pnas.93.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerppola T K, Curran T. Nature (London) 1995;373:199–200. doi: 10.1038/373199a0. [DOI] [PubMed] [Google Scholar]
- 17.Hemesath T J, Steingrimmson E, McGill G, Vaught J, Hodgkinson C A, Arnheiter H, Copeland N G, Jenkins N A, Fisher D E. Genes Dev. 1994;8:2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 18.Roman C, Matera G, Cooper C, Artandi S, Blain S, Ward D C, Calame K. Mol Cell Biol. 1992;12:817–827. doi: 10.1128/mcb.12.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 20.Zinkel S S, Crothers D M. Nature (London) 1987;328:178–181. doi: 10.1038/328178a0. [DOI] [PubMed] [Google Scholar]
- 21.Salvo J J, Grindley N D F. Nucleic Acids Res. 1987;15:9771–9779. doi: 10.1093/nar/15.23.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levene S D, Wu H-M, Crothers D M. Biochemistry. 1986;25:3988–3995. doi: 10.1021/bi00362a003. [DOI] [PubMed] [Google Scholar]
- 23.Zahn K, Blattner F R. Science. 1987;236:416–422. doi: 10.1126/science.2951850. [DOI] [PubMed] [Google Scholar]
- 24.Nelson H C, Finch J T, Luisi B F, Klug A. Nature (London) 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 25.Koo H-S, Crothers D M. Proc Natl Acad Sci USA. 1988;85:1763–1767. doi: 10.1073/pnas.85.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher D E, Parent L, Sharp P A. Cell. 1993;72:467–476. doi: 10.1016/0092-8674(93)90122-7. [DOI] [PubMed] [Google Scholar]
- 27.Fisher D E, Carr C S, Parent L A, Sharp P A. Genes Dev. 1991;5:2342–2352. doi: 10.1101/gad.5.12a.2342. [DOI] [PubMed] [Google Scholar]
- 28.Ferre-D’Amare A D, Pognonec P, Roeder R G, Burley S K. EMBO J. 1994;13:180–189. doi: 10.1002/j.1460-2075.1994.tb06247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma P C, Rould M A, Weintraub H, Pabo C O. Cell. 1994;77:451–459. doi: 10.1016/0092-8674(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 30.Ellenberger T, Fass D, Arnaud M, Harrison S C. Genes Dev. 1994;8:970–980. doi: 10.1101/gad.8.8.970. [DOI] [PubMed] [Google Scholar]
- 31.Gartenberg M R, Ampe C, Steitz T A, Crothers D M. Proc Natl Acad Sci USA. 1990;87:6034–6038. doi: 10.1073/pnas.87.16.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerppola T K, Curran T. Mol Cell Biol. 1993;13:5479–5489. doi: 10.1128/mcb.13.9.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Shea E K, Rutkowski R, Kim P S. Cell. 1992;68:699–708. doi: 10.1016/0092-8674(92)90145-3. [DOI] [PubMed] [Google Scholar]