

Abstract
UVA radiation is the major component of the UV solar spectrum that reaches the earth, and the therapeutic application of UVA radiation is increasing in medicine. Analysis of the cellular effects of UVA radiation has revealed that exposure of human cells to UVA radiation at physiological doses leads to increased gene expression and that this UVA response is primarily mediated through the generation of singlet oxygen. In this study, the mechanisms by which UVA radiation induces transcriptional activation of the human intercellular adhesion molecule 1 (ICAM-1) were examined. UVA radiation was capable of inducing activation of the human ICAM-1 promoter and increasing ICAM-1 mRNA and protein expression. These UVA radiation effects were inhibited by singlet oxygen quenchers, augmented by enhancement of singlet oxygen life-time, and mimicked in unirradiated cells by a singlet oxygen-generating system. UVA radiation as well as singlet oxygen-induced ICAM-1 promoter activation required activation of the transcription factor AP-2. Accordingly, both stimuli activated AP-2, and deletion of the putative AP-2-binding site abrogated ICAM-1 promoter activation in this system. This study identified the AP-2 site as the UVA radiation- and singlet oxygen-responsive element of the human ICAM-1 gene. The capacity of UVA radiation and/or singlet oxygen to induce human gene expression through activation of AP-2 indicates a previously unrecognized role of this transcription factor in the mammalian stress response.
Keywords: gene regulation, reactive oxygen species, UV response
Gene expression in mammalian cells is frequently regulated through sequence-specific transcription factors (1). One of the first transcription activator proteins to be discovered was the transcription factor AP-2 (2). AP-2 is involved in vertebrate morphogenesis and, in particular, in the differentiation and development of neural crest cells and epidermal keratinocytes (3, 4). In the latter cell type, AP-2 was found to be present in abundance and to be involved in gene expression by serving as an accessory transcription factor in regulation of the keratin 14 gene (4). AP-2 activity is inducible by retinoic acid, cAMP, and phorbol esters (5, 6), but, in comparison with other transcription factors such as AP1 or NF-κB (1, 7), little is known about environmental factors capable of triggering AP-2-mediated gene regulation.
Increased gene expression as a consequence of environmental stress is typically observed in mammalian cells upon exposure to UVC (<280 nm), UVB (280–320 nm), or UVA (320–400 nm) radiation (1, 8, 9). In human keratinocytes, this so-called UV response leads to an increased transcriptional expression of various genes, including the adhesion molecule intercellular adhesion molecule 1 (ICAM-1) (10, 11). The cis- and trans-acting genetic elements responsible for gene induction by UVC or UVB radiation have been well characterized (1). In contrast, the mechanisms by which UVA radiation induces transcriptional activation of human genes are poorly understood and most likely differ from those induced by short wavelength UV radiation because they involve different chromophores leading to different photobiological effects (12). Recent studies indicate that singlet oxygen is a primary effector in UVA radiation-induced gene expression (13–15), but the capacity of singlet oxygen to activate transcription factors and to induce promoter activation has not yet been demonstrated. The study of UVA radiation- and/or singlet oxygen-induced gene regulation, however, is important because the amount of UVA radiation reaching the earth’s surface is approximately 20 times greater than that of UVB radiation (16). In addition to solar UVA radiation, human skin is exposed to increasing doses of UVA radiation from high intensity UVA sources for cosmetic and therapeutic reasons (17–19).
In the present study, we analyzed UVA radiation-induced promoter activation by using promoter constructs based on the human ICAM-1 promoter, which contains numerous putative binding sites for transcription factors, including AP-2 (20, 21).
MATERIALS AND METHODS
Cell Culture.
Long-term cultured, normal human keratinocytes prepared from neonatal foreskin and human, transformed, primary, embryonal kidney cells (293 cells) were cultured as described (9, 22).
UV Irradiation.
For UV radiation, medium was replaced by PBS, and cells were exposed to UVA radiation (30 J/cm2) or UVB radiation (100 J/m2) as described (11, 23).
Chemical Treatments and Singlet Oxygen Generation.
All chemicals were purchased from Sigma except for sodium azide (Merck). Vitamin E (α-tocopheryl succinate) was dissolved in ethanol and added to cell cultures 24 h before irradiation in a concentration of 25 μM. Sodium azide (50 mM in PBS), d-mannitol (0–100 mM in PBS), catalase (0–1600 units/ml in PBS), and superoxide dismutase (0–750 units/ml in PBS) were present only during irradiation of cells. For irradiation in the presence of heavy water, deuterium oxide (99.9 atom % 2H) was used in a final concentration of 95% in PBS (13, 15).
Singlet oxygen was generated by thermal decomposition of the endoperoxide of the disodium salt of 3,3′-(1,4-naphthylidene) dipropionate (NDPO2), 1 mM in PBS, for 1 h in the dark at 37°C and yielded excited singlet molecular oxygen and 3,3′-(1,4-naphthylidene)dipropionate (NDP) (24). This singlet oxygen-generating system was shown to be well suited for use in cell cultures (14, 15) because it is water-soluble and nontoxic for up to 40 mM for 1 h of incubation. Infrared emission of singlet oxygen was measured with a liquid nitrogen-cooled germanium photodiode detector (model EO-817L, North Coast Scientific, Santa Rosa, CA) as described (24). The rate of singlet oxygen generation was monitored by the formation of NDP. Fifteen minutes after addition of 1 mM NDPO2, the rate of singlet oxygen generation was 3 μM per min. Control cells were stimulated with NDP, which had been generated by thermal decomposition from the same batch of NDPO2 used in these experiments.
Transfections and Reporter Gene Assay.
Transfection was mediated by Polybrene (Aldrich) (25). A commercially available luciferase reporter system (Serva) was used. Constructs containing various deletions of the ICAM-1 promoter were cloned as described (20, 26). For transfection of cells (triplicates of 2 × 105 cells), 5 μg of DNA was sufficient for 293 cells whereas 20 μg was required for keratinocytes. Transfection efficiency was monitored as described (26) by cotransfecting cells with the simian virus 40 promotor and enhance region–β-galactosidase control plasmid (Serva) using the β-galactosidase assay system (Serva). Routinely, 10 μg of DNA of this control plasmid was cotransfected in 293 cells and keratinocytes. Promoter activation was expressed as mean ± SD of relative specific luciferase activity, which was based on protein content (26). Sham-irradiated cells were used as controls and were set equal to 1.
Nuclear Extracts and Gel Electrophoretic Mobility-Shift Assays (GEMSAs).
Nuclear extracts were prepared as described by Dignam et al. (27). Protein concentration of the extracts was determined by the method of Bradford (28). GEMSAs were performed according to Goodwin (29). The AP-2 consensus oligonucleotide AP-2p (top strand, 5′-GACCCTCTCGGCCCGGGCACCCT-3′) was deduced from the ICAM-1 promoter (20). For competition experiments, synthetic, double-stranded oligonucleotides containing consensus sequences for AP-2 deduced from the simian virus 40 promoter (AP-2c; top strand, 5′-GATCGAACTGACCGCCCGCGGCCCGT-3′) and AP1 (AP1c; top strand, 5′-CGCTTGATGAGTCAGCCGGAA-3′) were obtained from Serva. Poly(dI·dC) × (dI·dC) (Pharmacia) was used as nonspecific competitor DNA.
RNA Extraction and Reverse Transcriptase–PCR.
Total RNA was isolated using RNeasy Total RNA Kits (Qiagen, Hilden, Germany). Expression of ICAM-1 mRNA was measured by differential reverse transcriptase–PCR as described (9, 23, 30) using a primer pair specific for ICAM-1 (5′-TGACCAGCCCAAGTTGTTGG-3′, 5′-ATCTCTCCTCACCAGCACCG-3′).
Immunofluorescence Flow Cytometry.
Keratinocyte ICAM-1 surface expression was assessed by immunofluorescence flow cytometry using anti-ICAM-1 mAb 84H10 (Dianova, Hamburg, Germany) as described (31).
RESULTS
Deletion of the AP-2 Site Abrogates UVA Radiation-Induced ICAM-1 Promoter Activation.
UVA and UVB radiation-induced activation of the human ICAM-1 promoter were compared in human, transformed, primary embryonal kidney cells (293 cells) using various ICAM-1 promoter-based luciferase reporter gene constructs in the range of 6000 to 277 bp upstream of the transcription start site. These constructs contained consensus-binding sequences for various transcription factors (Fig. 1A). Exposure of cells to UVA or UVB radiation induced ICAM-1 promoter activation 4- to 5-fold if cells were transfected with the pIC6000, pIC339, or pIC277 construct (Fig. 1B). Deletion of the putative AP-2-binding site in the pIC277 construct (pIC277ΔAP-2) resulted in loss of the UVA-induced, but not UVB-induced, ICAM-1 promoter-based luciferase activity. In contrast, deletion of the putative NF-κB site in the pIC277 construct (pIC277ΔNF-κB) affected neither UVA nor UVB radiation-induced ICAM-1 promoter activation. Essentially identical results were obtained when human keratinocytes were used instead of 293 cells (Table 1). In keratinocytes, UVA radiation-induced, but not UVB radiation-induced, ICAM-1 promoter activation was prevented completely if the putative AP-2-binding site was deleted from the pIC277 construct.
Figure 1.
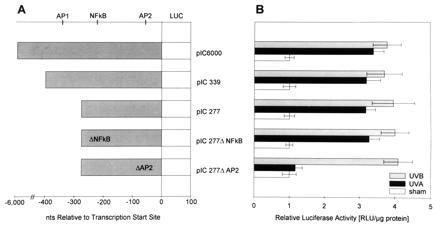
ICAM-1 promoter activation in 293 cells. (A) Localization of putative binding sites for transcription factors as detected by computer-aided analysis in the 5′-regulatory region of the ICAM-1 gene and in deletion constructs of the ICAM-1 promoter-based luciferase reporter gene constructs. The constructs were named after the number of base pairs relative to the transcription start site. Deletions of interior putative binding sites are indicated by Δ in the construct’s name and within the map. (B) The 293 cells were transiently transfected with the indicated ICAM-1-based luciferase reporter gene constructs and were exposed to 30 J/cm2 of UVA radiation (solid bars), 100 J/m2 of UVB radiation (gray bars), or sham-irradiation (open bars). ICAM-1 promoter activation was determined as described. Data are given as mean ± SD of relative, specific luciferase activity based on total protein contents and represent four experiments.
Table 1.
ICAM-1 promoter activation in normal human keratinocytes after exposure to UVA and UVB radiation
| Exposure | Relative, specific luciferase activity
per microgram of protein
|
|
|---|---|---|
| pIC277ΔNFkB | pIC277ΔAP2 | |
| Sham | 1.0 ± 0.12 | 1.0 ± 0.15 |
| UVA | 3.1 ± 0.58 | 1.3 ± 0.26 |
| UVB | 2.8 ± 0.28 | 3.4 ± 0.40 |
The recombinant plasmids pIC277ΔNFkB and pIC277ΔAP2 containing the named deletions of the ICAM-1 promoter and the luciferase reporter gene were transiently transfected into human keratinocytes as described. The transfected cells were sham-irradiated or irradiated with UVA (30 J/cm2) or UVB (100 J/m2). The cells were harvested 48 h after exposure and were analyzed for luciferase activity. Data are given as relative, specific luciferase activity (per microgram of protein) and represent the mean ± SD of four independent experiments.
UVA-Irradiated Keratinocytes Express a Protein That Binds to an AP-2 Consensus Sequence.
Nuclear extracts were prepared from human keratinocytes 2 h after exposure of cells to UVA radiation and were used for GEMSA. UVA-irradiated cells contained a protein capable of shifting a radiolabeled, synthetic, double-stranded oligonucleotide (AP-2p) containing an AP-2 consensus sequence deduced from the 5′-regulatory region of the human ICAM-1 gene (Fig. 2A). Similar results were obtained when the extracts were incubated in the presence of oligonucleotides that contained an AP-2-binding site derived from the simian virus 40 promoter (AP-2c; Fig. 2A). Binding was competed with specific competitor as well as unspecific competitor poly(dI·dC) × (dI·dC) in molar excess, which decreases the buildup of nonspecific high molecular weight aggregates and thereby affects migration of specific protein-DNA complexes. Binding was not competed with a consensus oligonucleotide containing an AP1 site.
Figure 2.
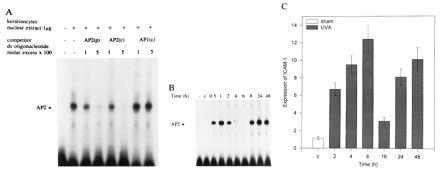
GEMSAs of nuclear extracts from UVA-irradiated (30 J/cm2) cultured human keratinocytes. (A) Binding of nuclear extracts to a double-stranded, radiolabeled oligonucleotide containing an AP-2 consensus sequence deduced from the sequence of the human ICAM-1 promoter (AP-2p) was studied in the presence or absence of unlabeled competitor, a commercially available oligonucleotide containing an AP-2 consensus sequence deduced from the simian virus 40 promoter (AP-2c), or an AP1 consensus oligonucleotide (AP1c) in 100 or 500 M excess. Nuclear extracts were prepared 2 h after irradiation as described. Data represent one of three essentially identical experiments. (B) Time course of the activation of transcription factor AP-2 in keratinocytes after UVA irradiation (30 J/cm2). Nuclear extracts were prepared at different time points as indicated and were analyzed by GEMSA using a radiolabeled oligonucleotide containing an AP-2-binding site. Free radiolabeled oligonucleotides (−) and the sham-irradiated control extract (lane c) were analyzed in lanes 1 and 2, respectively. Data represent one of five essentially identical experiments. (C) Time course of induction of ICAM-1 mRNA expression in cultured human keratinocytes after UVA irradiation (30 J/cm2). Sham-irradiated cells (lane c) served as control. Total RNA was extracted at the indicated times. Expression of ICAM-1 mRNA was analyzed by differential reverse transcriptase-PCR as described. Data are given as mean ± SD of relative ICAM-1 mRNA expression (normalized for glyceraldehyde-3-phosphate dehydrogenase expression) of five experiments.
Time Course of UVA Radiation-Induced AP-2 Activation and ICAM-1 mRNA Expression.
Human keratinocytes were exposed to UVA radiation, and, at the indicated time after UVA exposure, nuclear extracts were prepared and analyzed for AP-2 activation by GEMSA. UVA radiation-induced AP-2 activation exhibited a biphasic pattern (Fig. 2B). A first activation between 0.5 and 2 h postirradiation was followed by a second, more sustained activation that was detectable between 8 and 48 h after UVA irradiation. A similar biphasic activation pattern was observed when UVA radiation-induced up-regulation of ICAM-1 mRNA steady-state levels was analyzed in these cells (Fig. 2C). In this case, a first maximum was observed in UVA-irradiated cells 4–8 h after exposure, which was followed by a second increase after 24 h.
Effects of Singlet Oxygen Quenchers and Enhancers on UVA Radiation-Induced ICAM-1 Expression.
The generation of singlet oxygen was shown to play an important role in UVA radiation-induced expression of the human heme oxygenase and metalloproteinase-1 genes (13–15). Therefore, in the present study, reagents capable of quenching (sodium azide, α-tocopherol; ref. 32) or enhancing (deuterium oxide) singlet oxygen effects were screened for their capacity to affect UVA radiation-induced ICAM-1 expression using fluorescence-activated cell sorter analysis of ICAM-1 surface expression as a read-out system. As shown in Table 2, exposure of keratinocytes to UVA or UVB radiation within 24 h significantly induced ICAM-1 surface expression. Irradiation of cells in the presence of sodium azide completely inhibited UVA radiation-induced ICAM-1 expression whereas UVB radiation-induced ICAM-1 surface expression remained essentially unaltered (Table 2). A similar inhibition of UVA, but not UVB, radiation-induced ICAM-1 surface expression was observed when keratinocytes were treated with α-tocopheryl succinate 24 h before irradiation. Moreover, exposure of cells to UVA, but not UVB, radiation in the presence of deuterium oxide increased ICAM-1 surface expression (Table 2). In contrast, irradiation of cells in the presence of mannitol (quenching of hydroxyl radicals), catalase (dismutation of hydrogen peroxide), or superoxide dismutase (dismutation of superoxide anions) did not affect UVA or UVB radiation-induced ICAM-1 surface expression (Table 2).
Table 2.
Fluorescence-activated cell sorter analysis of ICAM-1 surface expression in cultured human keratinocytes after UVA irradiation
| Additive | ICAM-1 expression, mean fluorescence
intensity
|
||
|---|---|---|---|
| Sham-irradiated | UVA | UVB | |
| None | 6 ± 4 | 120 ± 14 | 140 ± 22 |
| Sodium azide | 20 ± 2 | 10 ± 2 | 140 ± 28 |
| α-Tocopheryl succinate | 15 ± 6 | 10 ± 1 | 130 ± 20 |
| Deuterium oxide | 7 ± 4 | 180 ± 10 | 125 ± 20 |
| Mannitol | 12 ± 6 | 115 ± 20 | 135 ± 8 |
| Catalase | 15 ± 5 | 125 ± 10 | 130 ± 15 |
| Superoxide dismutase | 5 ± 5 | 90 ± 20 | 100 ± 20 |
Keratinocytes were sham-irradiated, UVA-irradiated (30 J/cm2), or UVB-irradiated (100 J/m2) in the presence or absence of sodium azide (50 mM), deuterium oxide, mannitol (0-100 mM, shown for 100 mM), catalase (0-1600 units/ml, shown for 200 units/ml), or superoxide dismutase (0-750 units/ml, shown for 750 units/ml) or preincubated for 24 h with or without α-tocopheryl succinate (25 μM). Cells were harvested 24 h after exposure and were analyzed for ICAM surface expression as described. Data are given as mean ± SD of mean fluorescense intensity of four experiments.
Reagents that had been found to interfere with UVA radiation-induced ICAM-1 surface expression were tested for their capacity to modulate UVA radiation-induced ICAM-1 promoter activation (Table 3). Irradiation of 293 cells in the presence of sodium azide or pretreatment of cells with α-tocopheryl succinate diminished UVA radiation-induced ICAM-1 promoter activation to background levels whereas irradiation of cells in the presence of deuterium oxide resulted in a slight, but consistent, increase in UVA radiation-induced ICAM-1 promoter activation.
Table 3.
ICAM-1 promoter activation in UVA-irradiated 293 cells
| Exposure | Relative, specific luciferase activity
per microgram of protein
|
||
|---|---|---|---|
| pIC277 | pIC277ΔNFkB | pIC277ΔAP2 | |
| Sham | 1.0 ± 0.12 | 1.0 ± 0.15 | 1.0 ± 0.18 |
| UVA | 3.0 ± 0.59 | 3.0 ± 0.30 | 1.1 ± 0.22 |
| + Sodium azide | 1.3 ± 0.19 | 1.0 ± 0.10 | 1.3 ± 0.16 |
| + α-Tocopheryl succinate | 1.2 ± 0.24 | 0.9 ± 0.18 | 1.1 ± 0.23 |
| + Deuterium oxide | 3.6 ± 0.21 | 3.8 ± 0.15 | 1.0 ± 0.30 |
293 cells were transiently transfected with the indicated ICAM-1-based promoter constructs linked to the luciferase reporter gene, and ICAM-1 promoter activation was determined as described. Cells were either sham-irradiated or UVA-irradiated (30 J/cm2) in the presence or absence of sodium azide (50 mM) or deuterium oxide or after pretreatment with or without α-tocopheryl succinate (25 μM). Data are given as mean ± SD of relative, specific luciferase activity (per microgram of protein) and represent four experiments.
Moreover, in GEMSAs, UVA radiation-induced AP-2 activation was almost completely inhibited by sodium azide. In contrast, stimulation of unirradiated cells with H2O2 did not activate AP-2, and mannitol failed to suppress UVA radiation-induced AP-2 activation (Fig. 3A).
Figure 3.
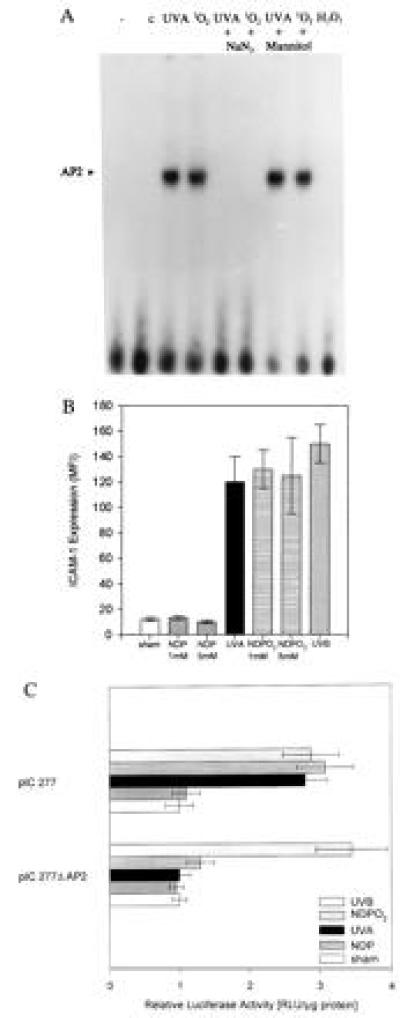
(A) GEMSAs of nuclear extracts from cultured human keratinocytes. Human keratinocytes were sham-irradiated (C) or stimulated with UVA irradiation (30 J/cm2), NDPO2 (1 mM for 60 min), H2O2 (250 mM) in the presence or absence of sodium azide (50 mM), mannitol (100 mM). Nuclear extracts were prepared 2 h after stimulation as described. Data represent one of two essentially identical experiments. (B) Fluorescence-activated cell sorter analysis of ICAM-1 surface expression in cultured human keratinocytes. Cells were either sham-irradiated, irradiated with 30 J/cm2 UVA radiation (UVA), or incubated in the presence of NDPO2 or NDP at a concentration of 1 or 5 mM for 60 min or irradiated with 100 J/m2 UVB radiation. Cells were harvested after 24 h and were analyzed for ICAM-1 surface expression as described. Data are given as mean fluorescence intensity (mean ± SD of four experiments). C. ICAM-1 promoter activation in 293 cells; 293 cells were transiently transfected with the indicated ICAM-1-based promoter constructs, and ICAM-1 promoter activation was determined as described. Cells were either sham-irradiated (open bars), incubated in the presence of NDPO2 (horizontally striped bars) or NDP (gray bars) at a concentration of 1 mM for 60 min, or exposed to 30 J/cm2 of UVA radiation (solid bars) or irradiated with 100 J/m2 of UVB radiation (diagonally striped bars). Data are given as mean ± SD of relative specific luciferase activity based on total protein contents and represent four experiments.
Singlet Oxygen Generation Induces ICAM-1 Expression and AP-2 Activity.
The capacity of sodium azide and α-tocopheryl succinate (32) to suppress, and of deuterium oxide to enhance, UVA radiation-induced ICAM-1 surface and promoter expression as well as AP-2 activation indicated a prominent role for singlet oxygen in this system. We therefore next assessed whether these UVA radiation-induced effects could be mimicked by stimulating unirradiated cells with singlet oxygen. Singlet oxygen was generated by thermal decomposition of NDPO2 (14, 15, 24). As shown in Fig. 3B, singlet oxygen generation increased ICAM-1 surface expression in unirradiated keratinocytes to an extent similar to that observed in UVA- or UVB-irradiated cells. Similar to ICAM-1 surface expression, singlet oxygen generation also induced ICAM-1 promoter activation in unirradiated cells (Fig. 3C). Significant induction of ICAM-1 promoter activation was observed in 293 cells, which had been transfected with the pIC277 construct and had been irradiated with UVB or UVA radiation or left unirradiated but stimulated with singlet oxygen. Deletion of the AP-2 consensus sequence from the pIC277 construct (pIC 277ΔAP-2) completely abrogated UVA radiation- or singlet oxygen-induced, but not UVB radiation-induced, ICAM-1 promoter activation (Fig. 3C). Stimulation of cells with NDP did not induce ICAM-1 surface expression (Fig. 3B) or ICAM-1 promoter activation (Fig. 3C).
These experiments indicated the involvement of AP-2 in singlet oxygen-induced ICAM-1 promoter activation. Accordingly, when nuclear extracts from human keratinocytes were analyzed by GEMSA, activation of AP-2 was observed in cells that had been stimulated with singlet oxygen as generated by NDPO2 (Fig. 3A).
DISCUSSION
Abrogation of the UVA Effect by Deletion of the AP-2-Binding Site.
Deletion of the putative AP-2-binding site of the 5′-regulatory region of the human ICAM-1 promoter resulted in loss of the UVA radiation-induced response in ICAM-1 promoter-based luciferase reporter gene experiments. Moreover, UVA radiation was shown to induce AP-2 activity in nuclear extracts of irradiated cells, and kinetic experiments revealed that UVA radiation-induced AP-2 activation followed a biphasic pattern that strongly resembled that observed for UVA radiation-induced ICAM-1 mRNA expression in these cells. These observations identified the AP-2-binding site as the UVA radiation-responsive element of the human ICAM-1 gene.
Abrogation of UVA radiation-induced ICAM-1 promoter activation by deletion of the AP-2-binding site was observed regardless of whether human embryonal kidney cells (293 cells) or normal human keratinocytes were used. The latter cell type forms the outermost frontier of the human body to the environment and thus constitutes one of the primary cellular targets for UVA radiation from sunlight or artificial irradiation devices. Deletion of the AP-2-binding site prevented activation of the pIC277 ICAM-1 promoter construct by UVA irradiation, but these constructs were still found to be activated in the same cells by UVB irradiation. This indicates that (i) the putative AP-2 binding site is specifically involved in UVA radiation-induced ICAM-1 promoter activation, and (ii) UVA radiation and UVB radiation induce ICAM-1 promoter activation by different molecular mechanisms.
By using GEMSAs, we have demonstrated that UVA radiation is capable of activating transcription factor AP-2. This conclusion is based on the observations that (i) UVA-irradiated cells expressed a protein capable of binding to oligonucleotides containing AP-2 consensus sequences, which were either deduced from the human ICAM-1 promoter or from the simian virus 40 promoter, and (ii) this binding could be competed with specific competitors in molar excess but not with consensus oligonucleotides containing an AP1 site. In addition to AP-2, UVA radiation recently has been demonstrated to activate transcription factor NF-κB (33). Activation of NF-κB, in contrast to AP-2 activation, was not of functional relevance for UVA radiation-induced ICAM-1 promoter activation; deletion of putative NF-κB binding sites from the human ICAM-1 promoter did not alter UVA radiation-induced ICAM-1 promoter activation. These results are in agreement with the notion that singlet oxygen generation, which has been shown here to mediate UVA radiation-induced ICAM-1 promoter and AP-2 activation, does not activate transcription factor NF-κB (34). In conclusion, our data demonstrate that AP-2 activity is an indispensable prerequisite for UVA radiation-induced ICAM-1 promoter activation, although it presently may not be excluded that additional transcription factors may be activated by UVA radiation and, together with AP-2, may form a functional active transcription factor complex necessary for ICAM-1 promoter activation (3, 35). Addition of dithiothreitol (0–200 mM) did not prevent retardation of AP-2 as detected by GEMSA (data not shown). Further studies are required to define the precise mechanism by which transcription factor AP-2 is activated by UVA radiation.
The functional relevance of UVA radiation-induced AP-2 activation for UVA radiation-induced ICAM-1 expression is further supported by the observation that up-regulation of ICAM-1 mRNA expression and AP-2 binding is partially overlapping (e.g., at 2 h), but maximal AP-2 binding precedes maximal ICAM-1 mRNA expression. In addition, AP-2 activation and ICAM-1 mRNA expression follow a biphasic time course, further suggesting a cause–effect relationship. Biphasic activation of gene expression has previously been reported for UVB radiation-induced expression of c-fos mRNA (8), and, more recently, for UVA radiation-induced expression of selected cytokine genes (23), and it has been proposed that the second, later activation may be due to cytokine-mediated, autocrine loops in UV-irradiated cells.
Abrogation of the Singlet Oxygen Effect by Deletion of the AP-2-Binding Site.
Singlet oxygen is produced by a variety of biological systems, including human cells, and is a significant biochemical intermediate in several biological processes (36). Singlet oxygen has been shown to serve as a primary effector in UVA radiation-induced transcriptional activation of the human heme oxygenase and of the matrix metalloproteinase-1 genes (13–15). No direct methods exist to detect singlet oxygen in irradiated cells. Evidence for the involvement of singlet oxygen in gene induction is therefore circumstantial and is based on indirect methods, including the use of singlet oxygen quenchers, enhancers of singlet oxygen life-time, and effects induced by singlet oxygen-generating systems (13–15). In the present work, a similar experimental approach was used to demonstrate that singlet oxygen is involved in UVA radiation-induced ICAM-1 promoter activation (Fig. 4). Our studies indicate that singlet oxygen generation in human cells serves an important function in transcription factor activation and regulation of gene expression, in particular in response to UVA radiation. Similarly, H2O2 previously has been shown to induce gene expression, including ICAM-1 (37), and to activate transcription factors such as NF-κB (34). In UVA radiation-induced ICAM-1 promoter activation, however, no evidence for the involvement of H2O2 could be obtained because the hydroxyl radical quencher mannitol failed to inhibit UVA radiation-induced ICAM-1 promoter activation, and H2O2 stimulation of unirradiated cells did not activate transcription factor AP-2 at concentrations capable of inducing ICAM-1 expression (ref. 37; Fig. 3A). We propose that UVA radiation-induced ICAM-1 promoter activation is primarily mediated through the generation of singlet oxygen and that UVA radiation/singlet oxygen-induced ICAM-1 promoter activation critically depends on the activation of transcription factor AP-2 (Fig. 4). Our studies indicate that transcription factor AP-2 is not only involved in gene regulation as it relates to differentiation and morphogenesis of cells but also is involved in the transcriptional response induced in mammalian cells by long wavelength UV radiation.
Figure 4.
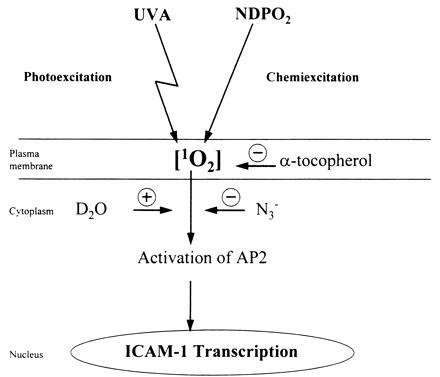
Scheme of UVA radiation- and singlet oxygen-induced, AP-2-mediated ICAM-1 transcription.
Relation to in Vivo Conditions.
The present observation that an environmental stimulus such as UVA radiation may activate transcription factor AP-2 in vitro in human keratinocytes raises questions about the in vivo relevance of these effects. In this regard, it is of interest that UVA radiation doses used in this study were equivalent to the amount of UVA radiation human skin may be exposed to on a summer day at approximately noon at the Northern latitude of 30°–35° during a 1- to 2-h period (16). In addition, UVA radiation doses identical to those used in the present study are capable of inducing skin lesions in patients suffering from polymorphous light eruption (38). The development of skin lesions in these patients after UVA radiation exposure is thought to be due to the induction of proinflammatory genes such as ICAM-1 in keratinocytes (38). Further studies therefore will have to assess whether UVA radiation is capable of inducing AP-2 activation in vivo in human skin and whether differences in UVA radiation-induced AP-2 activation may constitute the molecular basis for the abnormal UVA response observed in the skin of patients with polymorphous light eruption.
Acknowledgments
We thank T. Ruzicka (Düsseldorf, Germany) for continuous support. This study was supported by a grant from the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 503, Projects B2 and B1.
Footnotes
Abbreviations: ICAM-1, intercellular adhesion molecule 1; GEMSA, gel electrophoretic mobility-shift assay; NDP, 3,3′-(1, 4-naphthylidene) dipropionate; NDPO2, endoperoxide of 3,3′-(1, 4-naphthylidene) dipropionate.
References
- 1.Karin M, Herrlich P. In: Genes and Signal Transduction in Multistage Carcinogenesis. Colburn N H, editor. New York: Dekker; 1989. pp. 415–440. [Google Scholar]
- 2.Mitchell P J, Wang C, Tjian R. Cell. 1987;50:847–861. doi: 10.1016/0092-8674(87)90512-5. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell P J, Timmons P M, Hiebert J M, Rigby P S J, Tjian R. Genes Dev. 1991;5:105–119. doi: 10.1101/gad.5.1.105. [DOI] [PubMed] [Google Scholar]
- 4.Leask A, Byrne C, Fuchs E. Proc Natl Acad Sci USA. 1991;88:7948–7952. doi: 10.1073/pnas.88.18.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imagawa M, Chiu R, Karin M. Cell. 1987;51:251–260. doi: 10.1016/0092-8674(87)90152-8. [DOI] [PubMed] [Google Scholar]
- 6.Lüscher B, Mitchell P J, Williams T, Tjian R. Genes Dev. 1989;3:1507–1517. doi: 10.1101/gad.3.10.1507. [DOI] [PubMed] [Google Scholar]
- 7.Baeuerle P A. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 8.Shah G, Ghosh R, Amstad P A, Cerutti P A. Cancer Res. 1993;53:38–45. [PubMed] [Google Scholar]
- 9.Grewe M, Gyufko K, Krutmann J. J Invest Dermatol. 1995;104:3–6. doi: 10.1111/1523-1747.ep12613446. [DOI] [PubMed] [Google Scholar]
- 10.Ullrich S E. Photochem Photobiol. 1995;62:389–401. doi: 10.1111/j.1751-1097.1995.tb02359.x. [DOI] [PubMed] [Google Scholar]
- 11.Krutmann J, Grewe M. J Invest Dermatol. 1995;105:67S–70S. doi: 10.1111/1523-1747.ep12316095. [DOI] [PubMed] [Google Scholar]
- 12.Kochevar I E. In: Photoimmunology. Krutmann J, Elmets C A, editors. Oxford: Blackwell Scientific; 1995. pp. 19–33. [Google Scholar]
- 13.Basu–Modak S, Tyrrell R M. Cancer Res. 1993;53:4505–4510. [PubMed] [Google Scholar]
- 14.Scharffetter–Kochanek K, Wlaschek M, Briviba K, Sies H. FEBS Lett. 1993;331:304–306. doi: 10.1016/0014-5793(93)80357-z. [DOI] [PubMed] [Google Scholar]
- 15.Wlaschek M, Briviba K, Stricklin G P, Sies H, Scharffetter-Kochanek K. J Invest Dermatol. 1995;104:194–198. doi: 10.1111/1523-1747.ep12612751. [DOI] [PubMed] [Google Scholar]
- 16.Frederick J E, Alberts A D. In: Biological Responses to Ultraviolet A Radiation. Urbach F, editor. Overland Park, KS: Valdenmar; 1992. pp. 7–18. [Google Scholar]
- 17.Roza L, Baan R A, Van der Leun J C, Kligman L, Young A R. J Photochem Photobiol B. 1989;3:281–287. doi: 10.1016/1011-1344(89)80069-7. [DOI] [PubMed] [Google Scholar]
- 18.Krutmann J, Czech W, Diepgen T, Niedner R, Kapp A, Schöpf E. J Am Acad Dermatol. 1992;26:225–230. doi: 10.1016/0190-9622(92)70031-a. [DOI] [PubMed] [Google Scholar]
- 19.Stege H, Schöpf E, Ruzicka T, Krutmann J. Lancet. 1996;347:64. doi: 10.1016/s0140-6736(96)91600-1. [DOI] [PubMed] [Google Scholar]
- 20.Stade B G, Messer G, Riethmüller G, Johnson J P. Immunobiology. 1990;182:79–87. doi: 10.1016/S0171-2985(11)80585-1. [DOI] [PubMed] [Google Scholar]
- 21.Degitz K, Lian–Jie L, Caughman S W. J Biol Chem. 1991;266:14024–14029. [PubMed] [Google Scholar]
- 22.Graham F L, Smiley J S. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 23.Morita, A., Grewe, M., Grether–Beck, S., Olaizola–Horn, S. & Krutmann, J. (1997) Photochem. Photobiol. 64, in press. [DOI] [PubMed]
- 24.Di Mascio P, Sies H. J Am Chem Soc. 1989;111:2909–2914. [Google Scholar]
- 25.Jiang Ch–K, Connolly D, Blumenberg M. J Invest Dermatol. 1991;94:969–973. doi: 10.1111/1523-1747.ep12491889. [DOI] [PubMed] [Google Scholar]
- 26.van der Stolpe A, Caldenhoven E, Stade B G, Koendermann L, Raaijmakers J A M, Johnson J P, van der Saag P T. J Biol Chem. 1994;269:6185–6196. [PubMed] [Google Scholar]
- 27.Dignam J D, Martin P L, Skastry B S, Roeder R G G. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 28.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Goodwin G H. In: Gel Electrophoresis of Nucleic Acids: A Practical Approach. Rickwood D, Hames B D, editors. Oxford: IRL; 1990. pp. 225–247. [Google Scholar]
- 30.Henninger H P, Hoffmann R, Grewe M, Schulze–Specking A, Decker K. Biol Chem Hoppe-Seyler. 1993;374:625–634. doi: 10.1515/bchm3.1993.374.7-12.625. [DOI] [PubMed] [Google Scholar]
- 31.Krutmann J, Köck A, Schauer E, Parlow F, Möller A, Kapp A, Förster E, Schöpf E, Luger T A. J Invest Dermatol. 1990;95:127–131. doi: 10.1111/1523-1747.ep12477839. [DOI] [PubMed] [Google Scholar]
- 32.Kaiser S, DiMascio P, Murphy M E, Sies H. Arch Biochem Biophys. 1990;277:101–108. doi: 10.1016/0003-9861(90)90556-e. [DOI] [PubMed] [Google Scholar]
- 33.Vile G F, Tanew–Iliitschew A, Tyrrell R M. Photochem Photobiol. 1995;62:463–468. doi: 10.1111/j.1751-1097.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]
- 34.Schreck R, Albermann K, Baeuerle P. Free Radical Res Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 35.Magnaldo T, Vidal R G, Ohtsuki M, Freedberg I M, Blumenberg M. Gene Exp. 1993;3:307–314. [PMC free article] [PubMed] [Google Scholar]
- 36.Kanofsky J R. Chem Biol Interact. 1989;70:1–28. doi: 10.1016/0009-2797(89)90059-8. [DOI] [PubMed] [Google Scholar]
- 37.Ikeda M, Schroeder K K, Mosher L B, Woods C W, Akeson A L. J Invest Dermatol. 1994;103:791–796. doi: 10.1111/1523-1747.ep12413176. [DOI] [PubMed] [Google Scholar]
- 38.Norris P G. Curr Opin Dermatol. 1993;1:185–190. [Google Scholar]