
Figure 1.
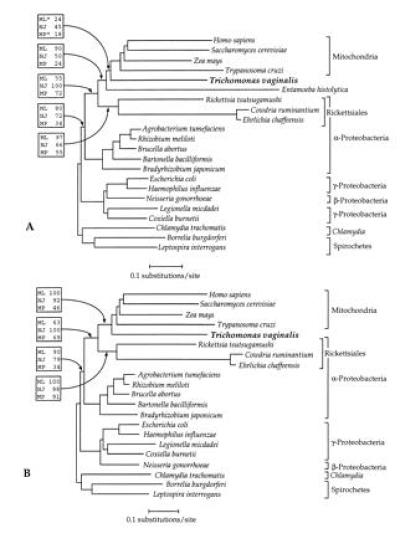
Phylogenies of cpn60 homologs. Sequences were selected from the database for organisms previously shown to branch in the region of the mitochondrial cpn60 clade (11, 12). The trees shown are derived from neighbor-joining analysis of a accepted point mutation corrected distance matrix. Percentage bootstrap support is shown above selected branches in boxes, from bootstrap analyses employing the protein maximum likelihood (ML), neighbor-joining distance (NJ), and maximum parsimony (MP) methods. (A) Cpn60 tree derived from the full alignment. The maximum likelihood tree (ln likelihood = −8933.6) differed from the neighbor-joining tree by the placement of the T. vaginalis and E. histolytica sequences as sister groups. Parsimony generated five trees of length = 2369 all of which differed principally from the tree shown by the placement of T. vaginalis as a sister group of the Rickettsiales species. Asterisks (∗) indicate that the method used did not recover this node in the majority of bootstrap replicates. (B) Cpn60 tree with the E. histolytica sequence excluded. Maximum likelihood yielded a tree of identical topology (ln likelihood = −12181.7), while parsimony generated three trees of length = 2209. Two of these differed from the neighbor-joining tree by the placement of the α-Protebacteria (excluding the Rickettsiales) as a sister group to the γ- and β-Proteobacteria. The third differed by placing T. vaginalis as an immediate relative to the Rickettsiales (see Results).