

Abstract
Apolipoprotein (apo) A-II is the second most abundant apolipoprotein in high density lipoprotein (HDL). To study its role in lipoprotein metabolism and atherosclerosis susceptibility, apo A-II knockout mice were created. Homozygous knockout mice had 67% and 52% reductions in HDL cholesterol levels in the fasted and fed states, respectively, and HDL particle size was reduced. Metabolic turnover studies revealed the HDL decrease to be due to both decreased HDL cholesterol ester and apo A-I transport rate and increased HDL cholesterol ester and apo A-I fractional catabolic rate. The apo A-II deficiency trait was bred onto the atherosclerosis-prone apo E-deficient background, which resulted in a surprising 66% decrease in cholesterol levels due primarily to decreased atherogenic lipoprotein remnant particles. Metabolic turnover studies indicated increased remnant clearance in the absence of apo A-II. Finally, apo A-II deficiency was associated with lower free fatty acid, glucose, and insulin levels, suggesting an insulin hypersensitivity state. In summary, apo A-II plays a complex role in lipoprotein metabolism, with some antiatherogenic properties such as the maintenance of a stable HDL pool, and other proatherogenic properties such as decreasing clearance of atherogenic lipoprotein remnants and promotion of insulin resistance.
Low levels of high density lipoproteins (HDL) are strongly associated with coronary heart disease risk (1–3), with each 1 mg/dl decrease in HDL accompanied by a 2–3% increase in risk (4). HDL are macromolecular complexes containing equal amounts of lipid and protein. The major HDL proteins are apolipoprotein (apo) A-I and apo A-II, comprising 70% and 20% of the total, respectively (5). Although apo A-II is an abundant plasma protein, relatively little is known about its role in HDL metabolism and how this might relate to the anti-atherogenic properties of HDL. In vitro studies have shown apo A-II can displace apo A-I from HDL particles (6), stimulate or inhibit hepatic lipase (HL) (7–9), inhibit lecithin cholesterol acyl transferase (LCAT) (10), and inhibit cholesterol ester transfer protein (CETP) activities (11–13). In human studies apo A-II levels do not correlate with HDL cholesterol levels (14), yet in some epidemiological studies apo A-II levels are inversely correlated with coronary heart disease susceptibility, as are HDL cholesterol and apo A-I levels (1). In contrast, clinical observations and tissue culture studies suggest that increased levels of apo A-II may be proatherogenic by increasing the concentration of HDL containing apo A-I and apo A-II, a particle with properties thought to be less favorable than HDL containing only apo A-I (15, 16). In the only known cases of human apo A-II deficiency, two Japanese sisters ages 62 and 59 were homozygous for an intron 3 splice donor site mutation in the apo A-II gene and had HDL cholesterol levels of 47 and 43 mg/dl (17). Based on these in vitro and human epidemiological and clinical studies, no clear answer has emerged as to the true physiological role of apo A-II.
To shed further light on this subject, in the last few years transgenic mice have been created with both the human and the mouse apo A-II transgenes (18–20). In different studies, apo A-II overexpression has left unchanged, increased, or decreased HDL cholesterol levels. With regard to mechanism, data have been presented that apo A-II can act as either an inhibitor of endogenous HL (21) or LCAT (20). Finally, in several studies apo A-II transgene expression was associated with increased susceptibility to atherosclerosis (22, 23). Thus, particularly with regard to its effects on the lipoprotein system, the apo A-II transgenic mouse studies have not provided a clear picture of the physiological role of this apolipoprotein.
The current study was designed to further investigate the role of apo A-II by using the gene knockout technology to create apo A-II-deficient mice. These studies revealed a major role for apo A-II in the maintenance of the plasma HDL pool and, in crossbreeding experiments with apo E-deficient mice, indicated that apo A-II is a regulator of lipoprotein remnant clearance. They also showed an effect of apo A-II on free fatty acid (FFA), glucose, and insulin levels.
METHODS
Construction of Targeting Vector.
The apo A-II gene was cloned from a BALB/c λ phage genomic library (Stratagene). The DNA probe was a PCR product amplified by two primers which covered most of the mouse apo A-II gene (24). Two overlapping clones were obtained. These clones were mapped in detail and shown to contain 20 kb of mouse DNA extending from 5 kb 5′ to 14 kb 3′ of the apo A-II gene (Fig. 1A). The targeting vector was constructed by cloning a 1.2-kb BamHI–SphI fragment located 5′ to the gene (short arm), and a 9.5-kb SpeI–EcoRI fragment which includes parts of exon 4 and sequences located 3′ to the gene (long arm) into vector pPNT (a gift from Mario Capecchi, University of Utah) (Fig. 1B). This strategy almost replaces the entire apo A-II gene with an intact neo gene (Fig. 1C).
Figure 1.
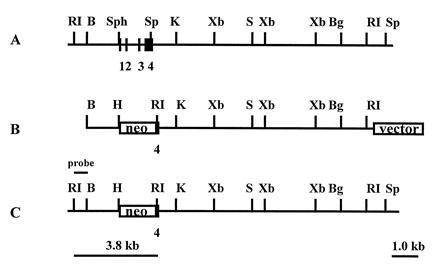
Targeted disruption of the mouse apolipoprotein A-II gene. (A) The mouse apo A-II gene locus. Exons 1–4 are represented by black boxes. (B) The gene targeting construction. The neomycin resistant gene (neo) cassette replaced the entire mouse apo A-II gene, including the promoter region. (C) The gene locus after DNA homologous recombination. The 12-kb EcoRI fragment of the endogenous locus was modified to 3.8 kb in the targeted locus. RI, EcoRI; B, BamHI; Sp, SpeI; Sph, SphI; K, KpnI; Xb, XbaI; S, SalI, Bg, BglII.
Generation of Apo A-II Knockout Mice.
Ten micrograms of targeting vector was linearized by NotI and then transfected by electroporation (BRL cell-porator) into 20 × 106 J1 embryonic stem (ES) cells in 1.0 ml of ES medium at 200 V and 800 μF (25). After selection in G418 (400 μg/ml) for 7 days, colonies were picked and grown in 96-well plates. Wells were then expanded by growth in 24-well plates. After 3 days, a portion of the cells in each well were used for DNA isolation, and PCR analysis was performed to identify clones that had undergone homologous recombination. PCR was done using primer pairs MAII8 and Neo1. Primer MAII8 is located directly outside the short arm of the targeting vector with a sequence of 5′-GGAATCTGATGTACCCTTCTGTTGTT-3′, and primer Neo1 is located in the 5′-promoter region of the neo gene cassette with a sequence of 5′-TGCGAGGCCAGAGGCCACTTGTGTAGC-3′. In this assay, DNA from the clones that had undergone homologous recombination gave a PCR band of 1.4 kb. Homologous recombination was confirmed by Southern blot analysis after overnight EcoRI digestion of ES cell DNA by using a probe 5′ to the short arm which gave a band of 3.8 kb compared with a 12-kb EcoRI band identified with the same probe in the unmodified locus. (The ES cell PCR and Southern blotting data are not shown.) The homologous recombination rate was ≈4 in 100 neo resistant clones, which is relatively high without a thymidine kinase-positive selection. One correctly targeted ES cell line was microinjected into C57BL/6J and BALB/cJ host blastocysts. This produced seven chimerical male mice which contained 50–100% agouti coat color contributed by the ES cell line. Four of these chimerical males gave germ-line transmission, passing the disrupted apo A-II gene to the next generation. DNA was isolated from the tail tips of the mice and a PCR based assay developed to follow the wild-type and mutant alleles in the colony. The wild-type allele was identified by a PCR primer pair MAII6u and MAII4l. Primer MAII6u is located in the third intron of apo A-II gene with a sequence of 5′-TCCAATCTGCAGAGTCTGATCC-3′, and primer MAII4l is located in the end of exon 4 of the apo A-II gene with a sequence of 5′-TCTTGGTCTAGAAGCAGCTGGGGTGGGAAGACTG-3′. With these primers the wild-type allele gave a PCR band of 0.35 kb. The mutant allele was identified by a PCR primer pair MAII4l, as above, and Neo901, located in the end portion of the neo gene cassette with a sequence of 5′-CGCCGCTCCCGATTCGCAGCGCATCGC-3′. With these primers the mutant allele gave a PCR band of 0.7 kb.
Animal Maintenance.
Animals were housed at the Rockefeller University Laboratory Animal Research Center in rooms maintained on 12-h light (7 a.m. to 7 p.m.)/dark cycles. Animals were fed a low fat–low cholesterol diet (chow diet) (no. 5001; Ralston Purina. The chow diet contained (wt/wt) 4.5% fat, 59.8% carbohydrate, 23.4% protein, 5.0% fiber, 7.3% minerals, and 0.02% cholesterol. In this diet fat provided 9% of calories, which were divided equally among saturated, monounsaturated, and polyunsaturated fats.
Plasma Lipid and Lipoprotein Analysis.
Blood was drawn from the retroorbital venous plexus into EDTA tubes. Fed specimens were taken between 8 and 9 a.m. For fasted specimens, food was removed from cages at 9 a.m. and the animals bled between 4 and 5 p.m. Cholesterol and triglyceride levels were determined enzymatically using Boehringer Mannheim reagents. The distribution of cholesterol among the lipoprotein size classes was analyzed by fast protein liquid chromatography (FPLC). For this analysis, 500 μl of pooled mouse plasma from littermates of the same genotype was first filtered through a 0.45-μm filter and then through two Superose 6 columns connected in series (Pharmacia). Forty-five fractions each of 0.5 ml were collected, and cholesterol in each fraction was determined as above. Pooled plasmas were also used to isolate very low density lipoprotein (VLDL), low density lipoprotien (LDL), and HDL by preparative ultracentrifugation.
HDL particle size distribution was determined by 4–20% nondenaturing gradient gel electrophoresis (26, 27). The HDL isolated by preparative ultracentrifugation was electrophoresed for 20 min at 70 V and then overnight at 150 V. The gel was then stained with 0.05% Coomassie blue and destained with 9% acetic acid and 20% methanol. HDL particle size was judged in comparison to the size of known protein standards (high molecular weight calibration kit; Pharmacia). Human HDL subpopulations and particle diameters determined by this technique are as follows: HDL2b, 12.9–9.8 nm; HDL2a, 9.8–8.8 nm; HDL3a, 8.8–8.2 nm; HDL 3b, 8.2–7.8 nm; and HDL 3c, 7.8–7.2 nm. SDS/PAGE was used to analyze lipoprotein–apolipoprotein content. HDL protein, cholesterol, cholesterol ester, and triglyceride content were determined by using the kits from Boehringer Mannheim.
HDL Turnover Study.
Mouse HDL, isolated by preparative ultracentrifugation, was delipidated and chromatographed on Mono Q column to isolate apo A-I (28). Apo A-I was radiolabeled with 125I by the iodine monochloride method. The cholesterol ester in HDL was radiolabeled as [3H]cholesterol oleoyl ether as follows: 10 μCi (1 Ci = 37 GBq) of [3H]cholesterol oleoyl ether was dried under nitrogen gas for 20 sec. Subsequently, 1 ml of 0.9% NaCl and 1.5 μl of 10–20% intralipid (KabiVitrum, Stockholm) were added. After sonication, either 500 μl of plasma from control mice or 1500 μl of plasma from apo A-II knockout mice were added along with 2.0 μg of purified human CETP (a gift from Alan Tall, Columbia University). The mixture was incubated for 8 h at 37°C, and radiolabeled HDL was then isolated by preparative ultracentrifugation. Double-label HDL turnovers were done by injecting 2–4 μg of 125I-labeled mouse apo A-I and 100,000–200,000 dpm of 3H-labeled HDL. Serial blood samples were obtained from the retroorbital plexus at 10 and 90 min and 3, 8, and 24 h. The apo A-I and HDL cholesterol ester fractional catabolic rates were calculated by the Matthew’s two pool model (29).
β-VLDL Turnover Study.
The VLDL fraction was isolated from fasted apo E knockout mice (25) by overnight ultracentrifugation. Thirty μCi of [3H]cholesteryl oleoyl ether was dried in a glass tube under a stream of nitrogen gas. One hundred microliters of the VLDL, which can be used for a β-VLDL turnover study in two mice, was added into the glass tube and well mixed. To start the labeling reaction, 1.0 μg of purified human CETP was added and incubated overnight at 37°C. The labeled mixture was then passed through a 1.0 ml G-50 column to remove the nonlipid-associated [3H]cholesteryl oleoyl ether. Fractions containing 3H-labeled VLDL were collected. Approximately 1,000,000 dpm was injected into each mouse for the β-VLDL turnover study. Serial blood samples were obtained from the retroorbital plexus at 2, 10, and 25 min and at 1, 2, and 3 h.
FFA, Glucose, and Insulin Determinations.
Retroorbital blood was collected from mice in the fed and fasted state into EDTA-coated capillary tubes. Blood was kept on ice, plasma was separated by centrifugation, and free acids were quantitated immediately by using a commercial kit following the instructions of the manufacturer (NEFA CCC; Wako Pure Chemical, Osaka). Glucose levels were measured by using the Autokit Glucose provided by Wako Pure Chemical, and insulin levels were obtain using commercial available kit provided from Linco Research Immunoassay (St. Charles, MO).
Statistical Analysis.
Results are given as mean ± SD if not otherwise indicated. The Student t test was used to compare differences between groups. Statistical significance was defined as P < 0.05.
RESULTS
The apo A-II gene was knocked out by homologous recombination in ES cells as described in Methods. Mice carrying the mutant apo A-II genome were crossed in brother–sister matings, and the resultant animals were genotyped using the PCR-based assays for wild-type and mutant alleles as described. As shown in Fig. 2A, the three genotypes, wild-type, heterozygous, and homozygous knockout, are easily distinguished. Based on the offspring of such matings the Mendelian ratio of 1:2:1 was observed for the three genotypes, indicating that apo A-II deficiency confers no selective disadvantage. Apo A-II deficiency also did not confer any gross phenotypic abnormality.
Figure 2.
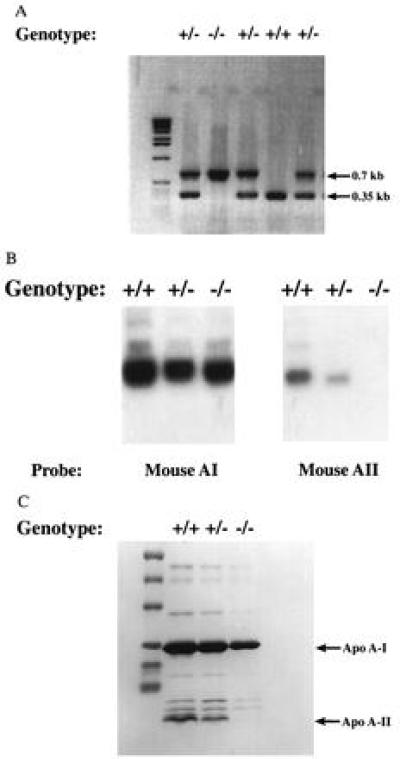
PCR identification of apo A-II-deficient mice, Northern blot analysis of mouse liver RNA, and SDS/PAGE analysis of mouse HDL. (A) The wild-type allele was identified by the PCR primer pair MAII6u and MAII2l, which gave a 0.35-kb band. The mutant allele was identified by the PCR primer pair MAII4l and Neo901, which gave a 0.7-kb band. (B) RNA was isolated from mouse liver. The Northern blot on the left was hybridized with mouse apo A-I cDNA probe; the Northern blot on he right was hybridized with mouse apo A-II cDNA probe. Apo A-II is completely absent from the apo A-II knockout lane. The mRNA levels of heterozygous are only half of that of controls. Apo A-I mRNA levels were unchanged among the three genotypes. +/+, +/−, and −/− represent control littermates, heterozygous-deficient mice, and homozygous apo A-II-deficient mice, respectively. (C) HDL was isolated by sequential ultracentrifugation and HDL from equal amounts of plasma was subjected onto SDS/PAGE (10–25% gradient). The gel was stained with Coomassie blue R-250. There was no detectable apo A-II in the homozygous-deficient mouse HDL.
To confirm the biochemical phenotype associated with the disruption of the apo A-II gene, liver RNA and plasma were obtained from wild-type, heterozygous, and homozygous apo A-II knockout mice. As shown in Fig. 2B, apo A-II knockout homozygotes were devoid of hepatic apo A-II mRNA, which is normally the major site of apo A-II synthesis in the body, with a gene dosage effect observed in heterozygotes. HDL was isolated by ultracentrifugation of plasma from wild-type, heterozygous, and homozygous mice and equal amounts of plasma run on SDS/PAGE. As shown in Fig. 2C, the apo A-II band was absent in HDL from homozygotes and diminished in HDL from heterozygotes.
The effect of the apo A-II knockout on the plasma lipid and lipoprotein profile was determined as shown in Table 1. In the fasted state, compared with wild-type littermates, cholesterol was reduced 21% and 67% in heterozygous and homozygous apo A-II knockout mice, respectively. This was primarily due to HDL cholesterol reductions of 17% and 73%, respectively, although non-HDL cholesterol was also significantly reduced. No significant differences in triglyceride concentrations between animals of the different genotypes were observed. In the fed state, compared with wild-type littermates, cholesterol was reduced 14% and 52% in heterozygous and homozygous knockout mice, respectively, due to HDL cholesterol reductions of 16% and 64%, respectively, but without significant changes in non-HDL cholesterol levels. These data indicate that a lack of apo A-II can cause a very large decrease in HDL cholesterol levels.
Table 1.
The plasma cholesterol and triglyceride levels in apo A-II-deficient mice in fed and fasting conditions on chow diet
| Mouse | n | TC, mg/dl | TG, mg/dl | Non- HDL-C, mg/dl | HDL, mg/dl |
|---|---|---|---|---|---|
| Fasted | |||||
| +/+ | 9 | 85 ± 12 | 56 ± 13 | 19 ± 6 | 66 ± 7 |
| +/− | 9 | 67 ± 8** | 44 ± 13 | 12 ± 5* | 55 ± 5** |
| −/− | 9 | 28 ± 7** | 46 ± 13 | 10 ± 4** | 18 ± 4** |
| Fed | |||||
| +/+ | 9 | 80 ± 10 | ND | 24 ± 7 | 56 ± 9 |
| +/− | 10 | 69 ± 8* | ND | 22 ± 9 | 47 ± 6* |
| −/− | 9 | 38 ± 8** | ND | 17 ± 10 | 20 ± 8** |
Plasma cholesterol and triglyceride analyses were performed on littermates. For the fed condition blood was drawn from the retroorbital venous plexus at 9 a.m. For the fasting condition, food was removed at 9 a.m. with the animal having ad lib access to water and blood drawn from the retroorbital venous plexus at 5 p.m. The values of VLDL–LDL were obtained by subtracting total cholesterol with HDL cholesterol. TC, total cholesterol; TG, triglyceride; C, cholesterol. Lipid values are shown as mean ± SD. +/+, +/−, and −/− represent control littermates, heterozygous-deficient mice, and apo A-II-deficient mice, respectively. ND, not determined.
P < 0.05 as compared with control mice.
P < 0.01 as compared with control mice.
P < 0.001 as compared with control mice.
The size distribution of lipoprotein particles in the apo A-II knockout mice was studied by FPLC analysis as shown in Fig. 3. In wild-type mice most of the cholesterol is in the HDL fraction as expected. This is also true for the apo A-II knockout mice; however, it appears that the HDL are smaller in these mice than in controls. To confirm this result, HDL from mice of different genotypes was subjected to non denaturing gradient gel electrophoresis. As shown in Fig. 4, independent of genotype, there is a single size distribution of HDL particles. However, wild-type HDL has a mean particle diameter of 9.7 nm, considerably larger than the 8.9 nm in the apo A-II knockout HDL. Heterozygote HDL is similar in size to wild-type HDL. Thus the absence of apo A-II from HDL results in particles of smaller size.
Figure 3.
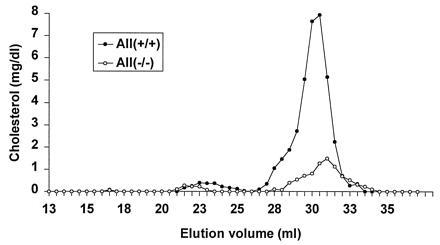
FPLC fractionation of plasma from apo A-II-deficient and control mice on chow diet. Two hundred microliters of fasted, pooled, and filtered mouse plasma was applied to two Superose 6 column (Pharmacia) connected in series. The lipoproteins were eluted at a constant flow rate of 0.3 ml/min with 1 mM sodium EDTA and 0.15 M NaCl. Fractions of 0.5 ml were shown as mg/dl in each fraction.
Figure 4.
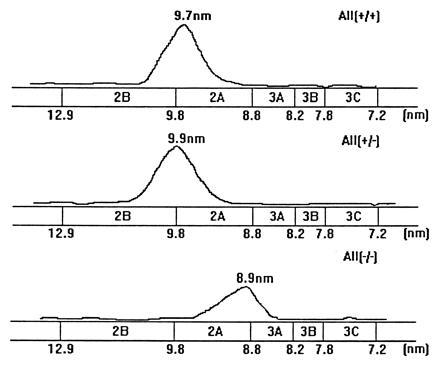
The distribution of HDL particle size diameters. HDL was isolated by sequential ultracentrifugation, and HDL from equal amount of plasma was subjected onto nondenaturing gradient gel electrophoresis (4–20% gradient). The gel was then stained and scanned by LKB Laser Densitomer. +/+. +/−, and −/− represent control littermates, heterozygous apo A-II-deficient mice, and homozygous apo A-II-deficient mice, respectively.
The mechanism underlying the decrease in HDL cholesterol levels in the apo A-II knockout mice was examined by in vivo turnover studies using HDL doubly labeled with 125I apo A-I and [3H]cholesteryl oleoyl ether. HDL was isolated from wild-type and apo A-II knockout mice, radiolabeled, and injected intravenously into both wild-type and apo A-II knockout mice. The decay of radioactivity in plasma was measured and HDL cholesterol ester (CE), and apo A-I turnover parameters were determined by the Matthew’s method. The results for control HDL and apo A-II knockout HDL injected into wild-type and knockout mice are shown in Tables 2 and 3, respectively. If we specifically compare the turnover parameters for control HDL injected into wild-type mice and apo A-II knockout HDL injected into knockout mice we see a 68% increase in HDL CE (fractional catabolic rate; FCR) and 57% decrease in HDL CE (transport rate; TR) and an 80% increase in apo A-I FCR and a 41% decrease in apo A-I TR. This indicates that the decreases in HDL CE and apo A-I in the knockout mice are due to both increased FCR and decreased TR.
Table 2.
Control HDL metabolism in apo A-II knockout mice and controls
| Mouse | n | HDL-C, mg/dl | HDL CE FCR, pools/h | HDL CE TR, units | Apo A-I, mg/dl | Apo A-I FCR, pools/h | Apo A-I TR, units |
|---|---|---|---|---|---|---|---|
| +/+ | 5 | 61 ± 9 | 0.19 ± 0.01 | 8.6 ± 0.9 | 184 ± 16 | 0.10 ± 0.01 | 18.5 ± 3.0 |
| −/− | 6 | 22 ± 5*** | 0.26 ± 0.03*** | 4.3 ± 0.7*** | 74 ± 13*** | 0.14 ± 0.02** | 10.4 ± 1.6*** |
HDL isolated from controls was double labeled with 125I and 3H and injected into controls and apo A-II knockout mice; ∗∗, P < 0.01 as compared to controls; and ∗∗∗, P < 0.001 as compared to controls.
Table 3.
A-II knockout HDL metabolism in apo A-II knockout mice and controls
| Mouse | n | HDL-C, mg/dl | HDL CE FCR, pools/h | HDL CE TR, units | Apo A-I, mg/dl | Apo A-I FCR, pools/h | Apo A-I TR, units |
|---|---|---|---|---|---|---|---|
| +/+ | 6 | 52 ± 13 | 0.21 ± 0.01 | 8.3 ± 2.2 | 156 ± 22 | 0.10 ± 0.01 | 15.9 ± 2.4 |
| −/− | 4 | 15 ± 3*** | 0.32 ± 0.02*** | 3.7 ± 0.8** | 62 ± 15*** | 0.18 ± 0.01*** | 11.0 ± 2.7* |
HDL isolated from apo A-II knockout mice was double labeled with 125I and 3H and injected into controls and apo A-II knockout mice: ∗, P < 0.05 as compared to controls; ∗∗, P < 0.01 as compared to controls; and ∗∗∗, P < 0.001 as compared to controls.
To determine whether apo A-II deficiency influences atherosclerosis susceptibility, the apo A-II knockout trait was bred onto the apo E-deficient atherosclerosis-prone background. Surprisingly, there was a dramatic effect of the apo A-II genotype on total cholesterol levels. Compared with apo E-deficient mice, apo E-deficient apo A-II heterozygous knockouts had a 34% reduction and apo E-deficient apo A-II homozygous knockouts had a 66% reduction in cholesterol levels. FPLC analysis of plasma lipoproteins shown in Fig. 5 confirms the significant decrease in non-HDL lipoprotein levels in the double knockout mice and also reveals the expected decrease in HDL levels. To assess the metabolic basis for the severe reduction in non-HDL cholesterol in the double knockout mice, the VLDL fraction from apo E knockout mice was isolated and radiolabeled with cholesterol oleoyl ether and turnover was compared in apo E knockout and double knockout mice. As shown in Fig. 6, in the absence of apo A-II, these particles had faster FCR. Since plasma cholesterol in the apo E-deficient mice is comprised mainly of cholesterol ester rich remnants of intestinally derived particles (25, 30), it appears that remnant clearance is accelerated when apo A-II levels diminish, implying a role for apo A-II in the regulation of remnant lipoprotein clearance.
Figure 5.
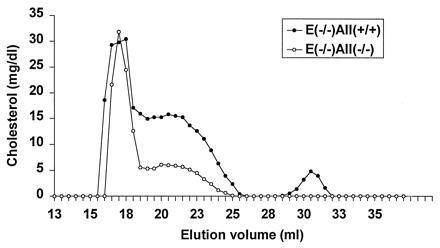
FPLC fractionation of plasma from apo E and apo A-II double knockout mice and apo E knockout only littermates on chow diet. Two hundred microliters of fasted, pooled, and filtered mouse plasma was applied to two Superose 6 columns (Pharmacia) connected in series. The lipoproteins were eluted at a constant flow rate of 0.3 ml/min with 1 mM sodium EDTA and 0.15 M NaCl. Fractions of 0.5 ml were collected, and cholesterol concentrations were measured enzymatically. Cholesterol valued are shown as mg/dl in each fraction.
Figure 6.
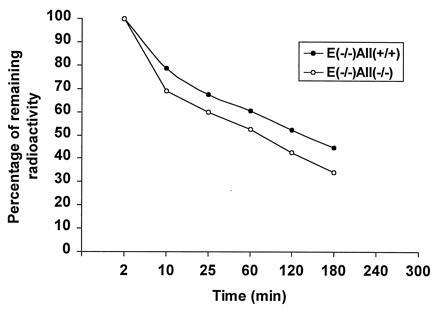
Apo E-deficient β-VLDL turnover study in apo E and apo A-II double knockout mice and apo E knockout only littermates. Five mice are in each group. β-VLDL was isolated from fasted apo E-deficient mice and in vitro labeled with [3H]cholesterol oleoyl ether. Mice were anesthetized with sodium pentobarbital. About 1,000,000 dpm of β-VLDL was injected through the femoral vein and serial blood samples were collected from the retroorbital plexus at 2, 15, and 25 min and at 1, 2, and 3 h. The [3H]radioactivity was measured. The remaining radioactivity (%) represents the average value of [3H]radioactivity at each time point divided by the [3H]radioactivity obtained at 2 min after injection.
Warden et al. (31) have provided evidence for a direct relationship between apo A-II and FFA levels in mice and humans. To confirm this relationship, FFA levels were measured in both the fasted and fed states in apo A-II knockout mice and controls; the results are shown in Table 4. In both metabolic states FFA levels were the same in heterozygous knockout mice and controls; however, homozygous knockout mice had 58% and 32% reductions in FFA levels in the fasted and fed states, respectively, compared with controls. Given the apo A-II effect on FFA levels, other metabolic parameters such as glucose and insulin levels were measured. Compared with control mice, the apo A-II knockout mice had 11% and 32% reductions in fasting glucose and insulin levels, respectively, as shown in Table 5.
Table 4.
The plasma concentration of FFA (mM) in fed and fasting conditions in apo A-II-deficient mouse plasma
| Mouse | n | Fasted condition | Fed condition |
|---|---|---|---|
| +/+ | 10 | 0.515 ± 0.181 | 0.355 ± 0.092 |
| +/− | 10 | 0.517 ± 0.132 | 0.356 ± 0.096 |
| −/− | 10 | 0.216 ± 0.071** | 0.242 ± 0.051** |
FFA analyses were performed on littermates. For the fed condition, blood was drawn from the retroorbital venous plexus at 9 a.m. For the fasting condition, food was removed at 9 a.m. with the animal having ad lib access to water and blood drawn from the retroorbital venous plexus at 5 p.m. FFA values are shown as mean ± SD. +/+, +/−, and −/− represent control littermates, heterozygous apo AII-deficient mice, and homozygous apo A-II-deficient mice, respectively. FFA analyses were performed immediately after blood was taken.
P < 0.01 as compared with control mice.
P < 0.001 as compared with control mice.
Table 5.
The plasma concentration of fasting glucose and insulin in apo A-II-deficient mouse plasma
Glucose and insulin analyses were performed on littermates in the fasted state. Food was removed at 9 a.m. with the animal having ad lib access to water. Blood was drawn from the retroorbital venous plexus at 5 p.m. Glucose and insulin values are shown as mean ± SD. +/+ and −/− represent control littermates and apo A-II-deficient mice, respectively. Analyses were performed immediately after blood was taken.
P < 0.05 as compared with control mice.
DISCUSSION
The current study used gene knockout technology to create apo A-II-deficient mice to further investigate the role of apo A-II in the body. Apo A-II-deficient mice had a 70% decrease in HDL cholesterol levels, and their HDL particles were significantly diminished in size. Metabolic turnover studies indicated that the reduced HDL cholesterol level was associated with both increased HDL CE and apo A-I FCR and decreased HDL CE and apo A-I TR. Compared with apo E knockout mice, double apo E/apo A-II knockout mice had markedly lower levels of intestinally derived lipoprotein remnant particles, implying a role for apo A-II in regulating clearance of these atherogenic particles. The apo A-II-deficient state was also associated with decreased plasma FFA, glucose, and insulin levels, suggesting a metabolic disturbance compatible with hypersensitivity to insulin. These studies have revealed multifaceted roles for apo A-II.
In human epidemiological and clinical studies plasma concentrations of apo A-II, in contrast to apo A-I, do not correlate with HDL cholesterol levels (14). Moreover, transgenic mouse studies have been inconsistent with regard to the effect of apo A-II expression on HDL cholesterol levels. In initial reports human apo A-II transgenic mice had normal HDL cholesterol levels (18), whereas mouse apo A-II transgenic mice had elevated levels (19). In a recent report human apo A-II transgenic mice with high levels of apo A-II expression actually had decreased HDL cholesterol levels (20). In light of all these previous studies, the profound decrease in HDL cholesterol levels observed in the apo A-II-deficient mice was unexpected. This decrease was comparable in magnitude to that seen in the apo A-I knockout mice (32, 33), and implies a major function of apo A-II in maintaining the plasma HDL pool.
The metabolic basis for the decrease in HDL was studied and found to be due to both increased FCR and decreased TR of HDL cholesterol ester and apo A-I. This was accompanied by decreased HDL size. In previous human metabolic turnover studies we showed that the differences between people in HDL cholesterol levels were mainly inversely correlated with apo A-I FCR, and that apo A-I FCR was mainly inversely correlated with HDL size (14). This study suggested that HDL size was a major determinant of apo A-I FCR and thereby of HDL cholesterol levels. In the apo A-II knockout mice the decrease in HDL size could cause increased HDL particle FCR. The additional finding of decreased HDL CE and apo A-I TR implies that in apo A-II-deficient mice there is a decrease in particles entering the HDL pool.
HDL is formed in plasma by lipoprotein processing reactions (5, 34, 35). Nascent HDL, which consists of apo A-I phospholipid discs attract free cholesterol from membranes. The cholesterol is esterified by LCAT and the particle becomes spherical. HDL particle size can be enlarged by acquisition of surface components liberated as lipoprotein lipase hydrolyzes triglyceride- rich particles and by further acquisition of membrane cholesterol and its esterification by LCAT. HDL size can be diminished by a two-step process whereby HDL cholesterol ester is exchanged via CETP for triglycerides from triglyceride-rich lipoproteins, followed by HDL triglyceride and phospholipid hydrolysis by hepatic lipase.
How then does apo A-II influence HDL size and metabolism? We have previously shown, by crossbreeding transgenic mouse lines that express various human lipoprotein transport genes, that apo A-II does not inhibit the CETP-mediated exchange of HDL cholesterol ester for triglycerides (21). However, apo A-II does inhibit the decrease in HDL size normally induced by CETP in the presence of hypertriglyceridemia, resulting in enlarged triglyceride-enriched HDL in human apo A-II transgenic mice. Since the decrease in HDL size is thought to be mediated by HL, we have suggested that apo A-II is an endogenous HL inhibitor. Supporting data for this role of apo A-II was provided showing that HDL prepared from mice of various genotypes showed inhibition of emulsion-based hepatic lipase activity in proportion to the apo A-II/apo A-I ratio of HDL. In addition, we found that the largely apo A-II only HDL present in the plasma of apo A-I knockout mice were 8-fold enriched in triglycerides (A. S. Plump and J.L.B., unpublished data). Together these studies clearly show that apo A-II is an endogenous inhibitor of HL and suggest that in apo A-II deficiency there may be unopposed and therefore excessive HL activity. Normally HDL is not triglyceride enriched; therefore, the main overactivity of HL may be toward phospholipid, which fluxes through the HDL compartment. Excessive HL activity may shift the balance severely diminishing the HDL phospholipid pool. This could effect both the synthesis of new HDL particles and the size and clearance of the particles that have formed. This dual effect is compatible with what we observed in the HDL turnover studies.
Recently, Marzal-Casacuberta et al. (20) studied human apo A-II transgenic mice with high levels of expression and found they had decreased HDL levels in association with decreased endogenous LCAT activity. In our studies of apo A-I knockout mice the remaining apo A-II-enriched HDL particles had a 1.9-fold increase in free cholesterol and a 2.2-fold decrease in cholesterol ester content (A. S. Plump and J.L.B., unpublished data), also suggesting that apo A-II can act as an endogenous inhibitor of LCAT. However, if this were a major mechanism, apo A-II deficiency might result in increased endogenous LCAT activity and increased HDL levels, not the profound decrease in levels we observed in the apo A-II knockout mice.
In the current study we set out to explore the role of apo A-II in atherosclerosis susceptibility. Epidemiological studies have shown a weak correlation of apo A-II levels with protection against coronary heart disease (1) In contrast, clinical observations and tissue culture studies imply that HDL containing apo A-I plus apo A-II is less antiatherogenic than HDL containing only apo A-I, suggesting that increased plasma levels of apo A-II are proatherogenic (15, 16). Transgenic mouse studies have also suggested a proatherogenic role for apo A-II. When human apo A-II transgenic mice were bred to human apo A-I transgenic mice, there was less protection against atherosclerosis than provided by the human apo A-I transgene alone (22). This study was based on the mouse atherosclerosis model in which C57/Bl6 mice are fed a toxic, unphysiological very high cholesterol/cholic acid diet and develop limited foam cell lesions and the findings must be interpreted cautiously. In another study, high level expression of a mouse apo A-II transgene caused increased HDL as well as non-HDL cholesterol and triglyceride levels, and these mice developed aortic foam cell lesions on a chow diet (23). Both of these studies were interpreted to mean that changing HDL composition from apo A-I only to apo A-I plus apo A-II particles was proatherogenic.
In the current study we set out to test the role of apo A-II in atherosclerosis susceptibility by crossbreeding the apo A-II knockout trait onto the apo E knockout background. Apo E knockout mice develop fibroproliferative atherosclerotic lesions at the branch points of vessels, similar to human lesions (25, 36). Lesions develop on a low cholesterol/low fat chow diet and are increased in size and progress faster when the mice are fed a Western-type diet (25). As presented, apo A-II deficiency caused a decrease in HDL levels in the apo E knockout mice; however, there was also a profound, gene dosage-dependent decrease in non-HDL cholesterol levels. Metabolic turnover studies indicated that in the absence of apo A-II there was more rapid catabolism of the intestinally derived atherogenic lipoprotein remnants that accumulate in the apo E knockout mice. This suggests a role for apo A-II in blocking the clearance of remnants through a non-apo E-dependent pathway. This observation suggests that in the mouse apo A-II transgenic mice, mentioned above, the increase in non-HDL cholesterol and triglyceride levels was due to apo A-II excess blocking remnant clearance (19). This raises the rather strong possibility that the accumulation of atherogenic remnant lipoproteins rather than or in addition to the conversion of HDL to mostly apo A-I plus apo A-II particles was responsible for the atherosclerosis seen in these mice.
Warden et al. (31) have provided evidence for a relationship between apo A-II and FFA levels in two species. In the mouse they used a backcross between Mus spretus and C57BL6/J to show that apo A-II and FFA levels correlated. In humans they used sib-pair methodology to show linkage of the apo A-II gene with both apo A-II and FFA levels. In this study there was also a significant correlation between apo A-II and FFA levels. Finally, they provided evidence for a direct causal relationship between apo A-II production and levels and FFA levels by showing a significant 25% increase in plasma FFA levels in mouse apo A-II transgenic mice compared with controls. Compatible with these observations, we observed that apo A-II knockout mice had lower FFA levels than control mice in both the fasted and the fed state. Moreover, there was no decrease in FFA levels in the apo A-II knockout mice in the fed compared with the fasted state as there was in control mice. Finally, in the fasted state the apo A-II knockout state was associated with lower glucose and insulin levels. Since FFA levels are regulated primarily by hormone sensitive lipase in adipose tissue which is inhibited by insulin, the finding of low FFA and for that matter low glucose in the presence of low insulin levels suggests an insulin hypersensitivity state in the absence of apo A-II. In addition, the low FFA levels in the fasted state in the knockout mice suggests evidence of insulin action even in low insulin states. These data combined with Warden’s suggest that apo A-II, by a mechanism not yet known, can cause insulin resistance. Insulin resistance is the hallmark of type II diabetes and is also associated with syndrome X (37), both atherosclerosis susceptibility conditions. These findings suggest yet another mechanism whereby apo A-II may be proatherogenic.
In summary, apo A-II appears to play a complex role in lipoprotein metabolism and atherosclerosis susceptibility. On the one hand sufficient apo A-II must be made to provide adequate amounts of a stable HDL pool, and on the other excess apo A-II can cause the accumulation of atherogenic remnant lipoproteins and perhaps also create a less antiatherogenic HDL. Excess apo A-II could also induce an insulin resistant state. This implies that there is an optimal amount of apo A-II and too much or too little may be detrimental.
Acknowledgments
We thank our colleagues, Drs. Jonathan Smith and Li-Shin Huang, for their helpful comments. This manuscript was supported by Grants HL54591, HL32435, and HL33714 from the National Institutes of Health.
Footnotes
Abbreviations: HDL, high density lipoprotein; apo, apolipoprotein; FFA, free fatty acid; HL, hepatic lipase; LCAT, lecithin cholesterol acyl transferase; CETP, cholesterol ester transfer protein; ES cell, embryonic stem cell; FPLC, fast protein liquid chromatography; VLDL, very low density lipoprotein; FCR, fractional catabolic rate; CE, cholesterol ester; TR, transport rate.
References
- 1.Miller N E. Am Heart J. 1987;113:589–597. doi: 10.1016/0002-8703(87)90638-7. [DOI] [PubMed] [Google Scholar]
- 2.Castelli W P, Garrison R J, Wilson P W F, Abbott R D, Kalousdian S, Kannel W B. J Am Med Assoc. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 3.Gordon T, Castelli W P, Hjortland M C, Kannel W B, Dawber T R. Am J Med. 1977;62:701–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 4.Gordon D J, Probstfield J L, Garrison R J, Neaton J D, Castelli W P, Knoke J D, Jacobs D R, Jr, Bangdiwala S, Tyroler H A. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg S. J Lipid Res. 1984;25:1017–1058. [PubMed] [Google Scholar]
- 6.Lagocki P A, Scanu A M. J Biol Chem. 1980;255:3701–3706. [PubMed] [Google Scholar]
- 7.Thuren T, Wilcox R W, Sisson P, Waite M. J Biol Chem. 1991;266:4853–4861. [PubMed] [Google Scholar]
- 8.Kubo M, Matsuzawa Y, Yokoyama S, Tajima S, Ishikama K, Yamamoto A, Tarui S. J Biochem (Tokyo) 1982;92:865–870. doi: 10.1093/oxfordjournals.jbchem.a134000. [DOI] [PubMed] [Google Scholar]
- 9.Jahn C E, Osborne J C, Schaffer E J, Brewe H B. Eur J Biochem. 1983;131:25–29. doi: 10.1111/j.1432-1033.1983.tb07227.x. [DOI] [PubMed] [Google Scholar]
- 10.Chung J, Abano D A, Fless G M, Scanu A M. J Biol Chem. 1979;254:7456–7464. [PubMed] [Google Scholar]
- 11.Rye K A, Carrety K H, Barter P J. J Lipid Res. 1992;33:215–224. [PubMed] [Google Scholar]
- 12.Lagrost L, Persegol L, Lallemant C, Gambert P. J Biol Chem. 1994;269:3189–3197. [PubMed] [Google Scholar]
- 13.Ikewaki K, Rader D J, Sakamoto T, Nishiwaki M, Wakimoto N, Schaefer J R, Ishikawa T, Fairwell T, Zech L A, Nakamura H, Nagano M, Brewer H B. J Clin Invest. 1993;92:1650–1658. doi: 10.1172/JCI116750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinton E A, Eisenberg S, Breslow J L. Arterioscler Thromb. 1994;14:707–720. doi: 10.1161/01.atv.14.5.707. [DOI] [PubMed] [Google Scholar]
- 15.Puchois P, Kandoussi A, Fievet P, Fourrier J L, Bertrand M, Koren E, Fruchart J C. Atherosclerosis. 1987;68:35–40. doi: 10.1016/0021-9150(87)90091-8. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y D, von Eckardstein A, Wu S, Assmann G. Arterioscler Thromb Vasc Biol. 1995;15:1412–1418. doi: 10.1161/01.atv.15.9.1412. [DOI] [PubMed] [Google Scholar]
- 17.Deeb S S, Takata K, Peng R L, Kajiyama G, Albers J J. Am J Hum Genet. 1990;46:822–827. [PMC free article] [PubMed] [Google Scholar]
- 18.Schultz J R, Gong E L, McCall M R, Nichols A V, Clift S M, Rubin E M. J Biol Chem. 1993;267:21630–21636. [PubMed] [Google Scholar]
- 19.Hedrick C C, Castellani L W, Warden C H, Puppione D L, Lusis A J. J Biol Chem. 1993;268:20676–20682. [PubMed] [Google Scholar]
- 20.Marzal-Casacuberta A, Blanco-Vaca F, Ishida B Y, Julve-Gil J, Shen J, Calvet-Marquez S, Gonzalez-Sastre F, Chan L. J Biol Chem. 1996;271:6720–6728. doi: 10.1074/jbc.271.12.6720. [DOI] [PubMed] [Google Scholar]
- 21.Zhong S B, Goldberg I J, Bruce C, Rubin E M, Breslow J L, Tall A R. J Clin Invest. 1994;94:2457–2467. doi: 10.1172/JCI117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz J L, Verstuyft J G, Gong E L, Nichols A V, Rubin E M. Nature (London) 1993;365:762–764. doi: 10.1038/365762a0. [DOI] [PubMed] [Google Scholar]
- 23.Warden C H, Hedrick C C, Qiao J-H, Castellani L W, Lusis A J. Science. 1993;261:469–472. doi: 10.1126/science.8332912. [DOI] [PubMed] [Google Scholar]
- 24.Yonezu T, Toda M, Yamagishi H, Higuchi K, Takeda T. Gene. 1989;84:187–191. doi: 10.1016/0378-1119(89)90154-6. [DOI] [PubMed] [Google Scholar]
- 25.Plump A S, Smith J D, Hayek T, Aalto-Setala K, Walsh A, Verstuyft J G, Rubin E M, Breslow J L. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 26.Blanche P J, Gong E L, Forte T M, Nichols A V. Biochim Biophys Acta. 1981;665:408–419. doi: 10.1016/0005-2760(81)90253-8. [DOI] [PubMed] [Google Scholar]
- 27.Nichols A V, Krauss R M, Musliner T A. Methods Enzymol. 1986;128:417–431. doi: 10.1016/0076-6879(86)28084-2. [DOI] [PubMed] [Google Scholar]
- 28.Hayek T, Azrolan N, Verdery R B, Walsh A, Chajek-Shaul T, Agellon L B, Tall A R, Breslow J L. J Clin Invest. 1993;92:1143–1152. doi: 10.1172/JCI116683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthews C. Phys Med Biol. 1957;2:36–53. doi: 10.1088/0031-9155/2/1/305. [DOI] [PubMed] [Google Scholar]
- 30.Plump A S, Forte T M, Eisenberg S, Breslow J L. Circulation. 1993;88:I-2. (abstr.) [Google Scholar]
- 31.Warden C H, Daluiski A, Bu X, Purcell-Huynh D A, De Meester C, Shieh B-H, Puppione D L, Gray R M, Reaven G M, Chen Y-D I, Rotter J I, Lusis A J. Proc Natl Acad Sci USA. 1993;90:10886–10890. doi: 10.1073/pnas.90.22.10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plump A S, Erickson S K, Weng W, Partin J S, Breslow J L, Williams D L. J Clin Invest. 1996;97:2660–2671. doi: 10.1172/JCI118716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson R, Lee D, Hagaman J, Maeda N. Proc Natl Acad Sci USA. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tall A R. J Clin Invest. 1990;86:379–384. doi: 10.1172/JCI114722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslow J L. In: The Metabolic Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 2031–2052. [Google Scholar]
- 36.Nakashima Y, Plump A S, Raines E W, Breslow J L, Ross R. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 37.Reaven G M. Annu Rev Med. 1993;44:121–131. doi: 10.1146/annurev.me.44.020193.001005. [DOI] [PubMed] [Google Scholar]