

Abstract
The ob/ob mouse is genetically deficient in leptin and exhibits both an obese and a mild non-insulin-dependent diabetic phenotype. To test the hypothesis that correction of the obese phenotype by leptin gene therapy will lead to the spontaneous correction of the diabetic phenotype, the ob/ob mouse was treated with a recombinant adenovirus expressing the mouse leptin cDNA. Treatment resulted in dramatic reductions in both food intake and body weight, as well as the normalization of serum insulin levels and glucose tolerance. The subsequent diminishment in serum leptin levels resulted in the rapid resumption of food intake and a gradual gain of body weight, which correlated with the gradual return of hyperinsulinemia and insulin resistance. These results not only demonstrated that the obese and diabetic phenotypes in the adult ob/ob mice are corrected by leptin gene treatment but also provide confirming evidence that body weight control may be critical in the long-term management of non-insulin-dependent diabetes mellitus in obese patients.
Obesity is frequently characterized by a major peripheral hyperinsulinemia, resulting from both higher insulin secretion and reduced insulin clearance, as well as by marked insulin resistance (1–3). The impaired glucose tolerance seen in severely obese subjects can be explained by the presence of major insulin resistance that cannot be fully compensated for by adequate insulin secretion. With further intolerance to glucose, fasting hyperglycemia and diabetes occur (4).
These abnormalities in glucose–insulin metabolism may be improved after calorie restriction and weight loss in obese diabetic patients (5–7). These treatments are associated with improved insulin sensitivity, decreased glucose production, and increased insulin secretion. Exercise, often added to calorie restriction in treatment programs for obesity, also has preventive effects on the development of diabetes (8, 9). Although weight reduction can improve hyperglycemia, especially when treatment is initiated soon after diagnosis, normal glucose tolerance may be difficult to achieve or maintain (10).
The ob/ob mouse is genetically deficient in leptin and displays metabolic abnormalities similar to those seen in obese humans with non-insulin-dependent diabetes mellitus. These abnormalities include obesity, hyperglycemia, glucose intolerance, and hyperinsulinemia (11–13). The excess of body weight in ob/ob mice can be reduced by repeated injections of recombinant leptin (14–16).
Herein we report that ectopically produced leptin after in vivo gene delivery is efficacious in the control of food intake and body weight. To accomplish this, we constructed a recombinant adenovirus containing the mouse ob cDNA and tested the ability of this vector to express leptin in vitro and in vivo and to correct the obese phenotype of the adult ob/ob mice. Finally, we examined the possibility that the correction of the obese phenotype might lead to the spontaneous correction of the metabolic abnormalities associated with the diabetes in this genetically well-defined animal model.
MATERIALS AND METHODS
Animals.
Male C57BL/6J-ob/ob mice and male wild-type mice were obtained from The Jackson Laboratory and maintained under a 12-h light/12-h dark cycle at 22 ± 2°C until they were 14 weeks of age. At this time, they were caged individually and monitored for body weight and chow consumption every 3 days. Animals had free access to water and standard mouse chow. Recombinant adenovirus vectors were administered to anesthetized 18- to 22-week-old mice by tail-vein injection. Blood was collected from the tail vein at various intervals, and leptin, glucose, and insulin levels were determined as described below. All experiments were performed in accordance with the animal guidelines at Baylor College of Medicine.
Leptin cDNA and Expression in Escherichia coli.
Murine leptin cDNA was obtained by reverse transcription-coupled PCR from mouse white adipose tissue. RNA was extracted according to the method of Chomczynski (17). First-strand cDNA was synthesized from 5 μg of total RNA from mouse epididymal fat pad by the random-hexamers-primed reverse transcription (Superscript, Life Technologies, Gaithersburg, MD). The primers used for PCR span the region from bases 178 to 615 in the published sequence (18). The identity of the product was confirmed by DNA sequencing. The PCR product was inserted into the pQE-12 vector (Qiagen, Chatsworth, CA) for expression of a carboxyl-terminal hexahistidine-tagged leptin in E. coli. Recombinant proteins were purified under denaturing conditions with a Ni2+ affinity column and dialyzed in phosphate-buffered saline (PBS). The identity of mouse leptin was confirmed by its molecular weight and partial sequence determination of the amino terminus.
Recombinant Adenovirus.
A mouse leptin cDNA spanning the region from bases 105 to 621 in the published sequence (18) was inserted in pADL.1/RSV plasmid (19) to generate pAd.RSV-mLeptin. This resulting plasmid contains the leptin cDNA under the transcriptional control of the Rous sarcoma virus (RSV) long terminal repeat and the bovine growth hormone polyadenylylation signal sequence. PJM17, a plasmid containing a replication-defective copy of the adenovirus genome (20), and the pAd.RSV-mLeptin plasmid were cotransfected into 293 cells. The recombinant adenovirus from a single plaque was expanded and twice purified by cesium chloride gradient ultracentrifugation. The purified virus was dialyzed in 10 mM Tris·HCl, pH 7.4/1 mM MgCl2/10% glycerol. Viral titer was determined by plaque assay (21).
In Vitro Leptin Expression.
Thirty plaque-forming units (pfu) per cell were used to infect cultured Hep G2 cells. The cells were incubated with the recombinant adenovirus for 1 h and replaced in a fresh culture medium for 24 h. Then, the cells were washed with PBS and incubated in serum-free medium for 12 h. Proteins in the medium were precipitated with 10% trichloroacetic acid and centrifuged. The pellet was washed and dissolved in 1% SDS/60 mM Tris·HCl, pH 6.8. Leptin was detected by Western blot analysis.
Western Blot Analysis.
Recombinant leptin was sent to Research Genetics (Huntsville, AL) for the antibodies production in rabbits. IgG fraction from rabbit antisera was purified using a serum IgG purification kit (Bio-Rad). Western blots with the purified rabbit IgG fraction identified a 16-kDa band in the medium of Hep G2 cells infected with the recombinant adenovirus containing the mouse leptin cDNA. The specificity of the antibody was confirmed by the absence of the 16-kDa band when rabbit preimmune serum or ob/ob mouse serum was used. Sera from treated mice were electrophoresed on 10–20% SDS/polyacrylamide gels under reducing conditions and electroblotted onto nitrocellulose membranes. Blots were blocked with 5% nonfat dry milk and then exposed to rabbit anti-leptin antibodies for 1 h, washed in PBS/0.05% Tween 20, and then incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase for 1 h. After a second wash with PBS/0.05% Tween, the immunocomplexes were visualized by chemiluminescence (ECL kit, Amersham). To quantify the amount of leptin in serum samples, serial dilutions of purified recombinant leptin were electrophoresed on the same gel, and band intensities were compared by densitometry.
Glucose Tolerance Tests.
After a 16-h fast, glucose at 1 mg/g of body weight was injected intraperitoneally in awake mice. Blood was sampled from tail vein before injection (time zero) and 15, 30, 60, 120, and 180 min after glucose injection.
Blood Glucose and Serum Insulin Determinations.
Blood glucose was measured with a One Touch II glucose meter (Lifescan, Mountain View, CA). Serum insulin was measured with a rat insulin radioimmunoassay kit (Linco Research Immunoassay, St. Charles, MO).
Statistical Analysis.
Statistical analysis was done using the unpaired t test calculated with the sigmastat program.
RESULTS
Leptin Expression in Adenovirus-Transduced Cells in Vitro.
A replication-deficient recombinant adenoviral vector was constructed that contains the mouse leptin cDNA under the transcriptional control of a RSV long terminal repeat promoter. The ability of the recombinant adenovirus to produce the leptin was tested in HepG2 cells. As shown in Fig. 1A, leptin was present in conditioned medium from cells transduced with the recombinant adenoviral vector Ad.RSV-mLeptin but not from cells transduced with either a control recombinant adenovirus expressing human α1-antitrypsin (hAAT; named Ad.RSV-hAAT) or PBS. The immunoreactive protein exhibited a molecular mass of 16 kDa, consistent with the known molecular mass of leptin. For comparison, a hexahistidine-tagged leptin, which was expressed in E. coli and purified with a Ni2+ affinity column, is also shown in Fig. 1A. The slight difference in size reflects the inclusion of 14 amino acids in the hexahistidine-tagged leptin construct.
Figure 1.
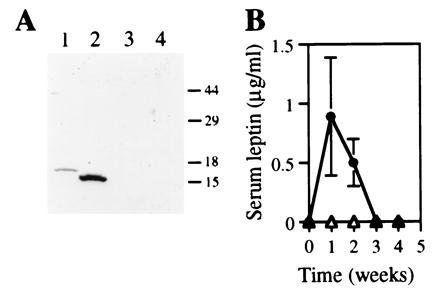
(A) Expression of leptin by HepG2 cells transduced with Ad.RSV-mLeptin (lane 2) or a recombinant adenovirus expressing the hAAT, Ad.RSV-hAAT (lane 3), or the buffer (lane 4). Recombinant leptin produced in E. coli is also shown (lane 1). Numbers at right indicate molecular mass standards (in kDa). (B) Serum leptin levels in ob/ob mice after recombinant adenovirus administration. The ob/ob mice were injected with either Ad.RSV-mLeptin (•) or Ad.RSV-hAAT (▵) at 3 × 109 pfu per mouse. Serum leptin concentrations were measured by Western blot analysis. Two microliters of sera from treated mice was electrophoresed on SDS/PAGE gels. Each point represents the mean ± SEM of seven mice. The limit of detection was 0.05 μg/ml. Three of 10 mice injected with Ad.RSV-mLeptin died within the first 3 weeks for unknown reasons.
Serum Leptin Levels in Recombinant Virus-Treated ob/ob Mice.
To test the functionality of the recombinant adenoviral vector in vivo, ob/ob mice were injected via the tail vein with either Ad.RSV-mLeptin or the control virus Ad.RSV-hAAT at 3 × 109 pfu per mouse. After recombinant adenovirus injection, the mice were bled weekly and serum leptin levels were determined. As shown in Fig. 1B, ob/ob mice that received Ad.RSV-mLeptin had a serum leptin level of 0.9 ± 0.5 μg/ml (n = 7) after 1 week. Serum samples from ob/ob mice that received either Ad.RSV-hAAT or PBS had no detectable leptin. After 3 week however, the serum leptin concentration in the leptin gene-treated mice fell to undetectable level as expected.
Effects of de Novo Leptin Gene Expression on Food Intake and Body Weight in ob/ob Mice.
Administration of 3 × 109 pfu of Ad.RSV-mLeptin to 18- to 22-week-old ob/ob mice resulted in a dramatic decrease in food intake and body weight. As shown in Fig. 2A, food intake of the leptin gene-treated mice had declined by 93 ± 4% below the pretreatment level at 1 week after viral injection. Five of seven leptin gene-treated mice refused feeding for 2 weeks. At week 4, food intake started to resume, which coincided with the reduction of serum leptin levels. By week 5, leptin gene-treated mice had a food intake similar to that of age-matched C57BL/6J mice (10.7 ± 2.3 g per mouse per 3 days versus 12.0 ± 0.7 g per mouse per 3 days). By week 7, food consumption of leptin gene-treated mice was similar to that of PBS- or hAAT-treated mice (18.1 ± 1.7 g per mouse per 3 days versus 19.0 ± 2.0 or 20.5 ± 1.3 g per mouse per 3 days, respectively).
Figure 2.
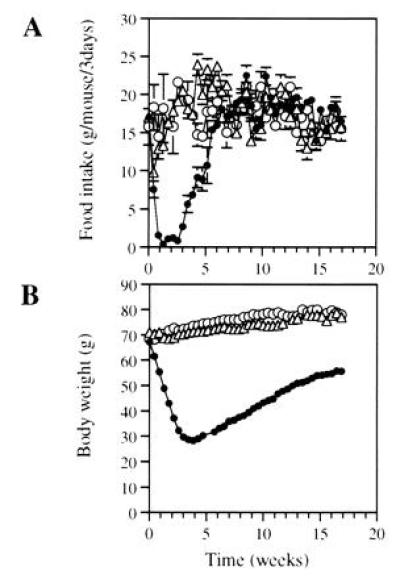
Reduction of food intake (A) and body weight (B) in ob/ob mice after leptin gene administration. The ob/ob mice were injected with either Ad.RSV-mLeptin (•) or Ad.RSV-hAAT (▵) at 3 × 109 pfu per mouse or PBS only (○). The mice were caged individually. Food intake and body weight were measured every 3 days. Food intake data are expressed as number of grams consumed every 3 days. All points represent the mean ± SEM of seven mice (some error bars are hidden within symbols).
As shown in Fig. 2B, leptin-gene-treated mice lost 56 ± 3% of their body weight after 3 weeks. At this time, the body weight of leptin-gene-treated mice were the same as that of age-matched C57BL/6J mice (29.7 ± 1.7 g versus 29.9 ± 0.4 g, n = 7). As the serum leptin level fell and food consumption increased after 3 weeks, the animals gradually regained weight and reached 83 ± 5% of their pretreatment weight by week 17.
Effects of Leptin Gene Expression on Glycemia, Insulinemia, and Insulin Resistance.
The ob/ob mouse exhibits an excessive deposition of body fat as well as a moderate degree of hyperglycemia in the presence of hyperinsulinemia. Serum insulin and glucose levels were measured after Ad.RSV-mLeptin administration to determine whether the correction of the obese phenotype will spontaneously lead to the correction of the hyperglycemia and hyperinsulinemia in this well-characterized genetic model. Both fasting blood glucose (Fig. 3A) and fasting serum insulin (Fig. 3B) levels decreased significantly 1 week after Ad.RSV-mLeptin administration in the ob/ob mice but remained elevated in hAAT- and PBS-treated mice. At this time, fasting glucose and insulin concentrations in leptin-gene-treated mice were similar to those observed in age-matched C57BL/6J mice (82 ± 8 mg/dl and 0.3 ± 0.1 ng/ml in leptin-gene-treated mice versus 88 ± 5 mg/dl and 0.4 ± 0.1 ng/ml in C57BL/6J mice, respectively). While serum insulin level was stably maintained for 2 weeks more, blood glucose level was further reduced during the same period. At week 3, the blood glucose level of the leptin-gene-treated mice was only 55 ± 9% (P < 0.001) of the age-matched C57BL/6J mice. Fasting glucose level gradually returned to normal values by week 13 and was maintained at 17 weeks. This level was significantly lower than those of the hAAT- and PBS- treated mice [85 ± 6 mg/dl versus 122 ± 7 mg/dl (P < 0.005) and 109 ± 9 mg/dl (P < 0.05), respectively]. After leptin concentrations fell to undetectable levels in 3 weeks, serum insulin also gradually increased in leptin-gene-treated mice, albeit at levels that were still significantly reduced (68 ± 6%; P < 0.01) as compared with hAAT-treated mice after 17 weeks. No significant changes in fasting levels of glucose and insulin were observed in the control mice. Compared with glucose levels of C57BL/6J mice, the control ob/ob mice maintained hyperglycemia during the entire observation period. Thus, the administration of Ad.RSV-mLeptin resulted in a significant reduction of serum insulin and normalization of blood glucose levels in the ob/ob mice.
Figure 3.
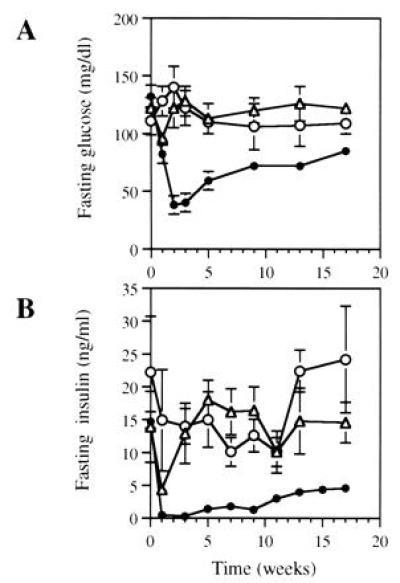
Fasting glucose (A) and insulin (B) levels in ob/ob mice after leptin gene administration. The ob/ob mice were injected with Ad.RSV-mLeptin (•) or Ad.RSV-hAAT (▵) at 3 × 109 pfu per mouse or PBS only (○) and periodically bled for serum insulin and blood glucose measurements. These parameters were determined in 16-h fasted mice. Each value represents mean ± SEM of six to eight mice. The mean values of serum insulin and blood glucose levels in age-matched C57BL/6J mice were 0.4 ± 0.1 ng/ml and 88 ± 5 mg/dl, respectively.
To determine whether reduced fasting blood glucose in leptin-gene-treated mice was accompanied with correction of glucose intolerance, glucose tolerance tests were performed on ob/ob mice at 5 and 17 weeks after administration of Ad.RSV-mLeptin, Ad.RSV-hAAT, or PBS. The complete glucose tolerance curves of treated mice and age-matched C57BL/6J mice are shown in Fig. 4. Compared with C57BL/6J mice, the blood glucose of hAAT- or PBS-treated mice attained much greater peak concentrations and failed to return to the fasting values at 2 h after glucose injection. At week 5 (Fig. 4A), response in leptin-gene-treated mice was similar to that of C57BL/6J mice. As shown in Fig. 4B, the normalization of glucose tolerance in the leptin-gene-treated ob/ob mice was maintained at 17 weeks.
Figure 4.
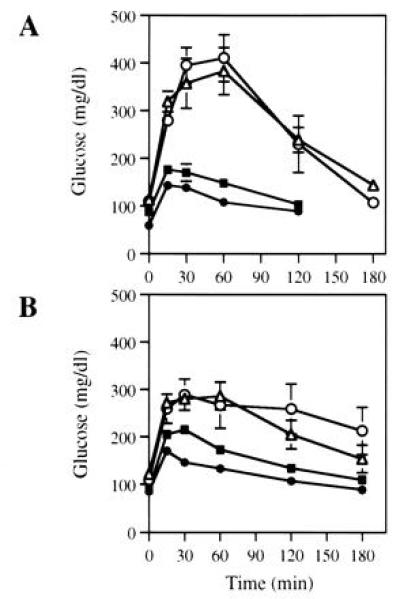
Glucose tolerance test in ob/ob mice after leptin gene treatment. The ob/ob mice were injected with Ad.RSV-mLeptin (•) or Ad.RSV-hAAT (▵) at 3 × 109 pfu/mouse or PBS only (○). Five weeks (A) and 17 weeks (B) after injection, glucose tolerance test were performed on treated mice. Blood glucose were measured prior to and at indicated times after intraperitoneal glucose injection. Glucose tolerance test was also performed with age-matched C57BL/6J mice (▪) for comparison. Each value represents mean ± SEM of six to eight mice.
Normalization of glucose tolerance with significantly higher serum insulin levels than that of the normal mice suggests the presence of an insulin resistance state in the leptin-gene-treated ob/ob mice. As shown in Fig. 5, the insulin-to-glucose molar ratios in the ob/ob mice before treatment were between 350 × 10−6 and 640 × 10−6, which were 27- to 49-fold higher than that in C57BL/6J mice (13 × 10−6). Administration of Ad.RSV-mLeptin in the ob/ob mice reduced the ratio significantly by 95 ± 2% (P < 0.001) during the first 3 weeks, which was similar to that of the C57BL/6J mice. However, at week 5, this ratio increased significantly by approximately 6-fold (P < 0.05) as compared with that of the C57BL/6J mice. After 5 weeks, the increase in insulin resistance paralleled the gradual gain in body weight. Neither the administration of Ad.RSV-hAAT or PBS produced significant changes on insulin resistance in ob/ob mice.
Figure 5.
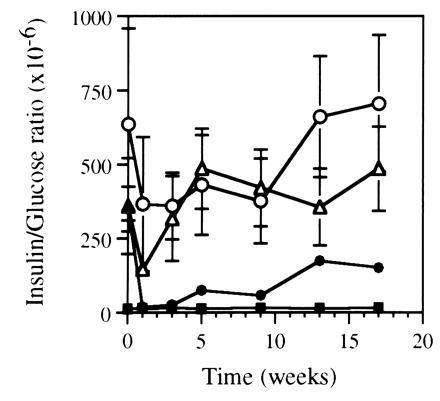
Insulin resistance in ob/ob mice after leptin gene treatment. After administration of Ad.RSV-mLeptin (•) or Ad.RSV-hAAT (⋄) at 3 × 109 pfu per mouse or PBS only (○), the mice were periodically bled for serum insulin and blood glucose measurements. The insulin-to-glucose molar ratio was determined in fasted mice. The same parameters were also measured in age-matched C57BL/6J mice (▪) for comparison. Each value represents mean ± SEM of six to eight mice.
DISCUSSION
Adenoviral vectors have been used to transfer genes in various animal models of metabolic disorders (22). We have administered through tail-vein injection a recombinant adenovirus expressing mouse leptin to treat a genetically defined mouse model of obesity and non-insulin-dependent diabetes. This treatment led to the production of serum leptin levels in the ob/ob mice that were 70 times higher than those in the C57BL/6J mice (23), which resulted in a rapid reduction in food consumption and a drastic weight loss.
The serum leptin levels in treated animals did not persist beyond 2–3 weeks. While the reason for the loss has not been examined directly, several previous studies have suggested that adenovirally transduced cells are destroyed by a host immune-mediated response in vivo (24, 25). In addition, recent studies have demonstrated that cellular and humoral responses to the transgene-encoded product may also play an important role in limiting the duration of transgene expression (26, 27). Despite of the lack of persistence of leptin gene expression, a complete correction of the obese phenotype was achieved after leptin gene transfer in the ob/ob mice. The rapid resumption of food intake to pretreatment values and the gradual body weight gain after serum leptin fell to undetectable levels are indicative that the treatment produced no long-lasting ill effects.
Our data showed that hyperinsulinemia and insulin resistance were also corrected after leptin gene administration in the ob/ob mice. While the effect on hyperinsulinemia has been observed previously by administration of recombinant leptin in the ob/ob mice (14, 28), the present study showed that serum insulin and insulin resistance were completely normalized after only 1 week of leptin gene treatment, even when the mice were still severely obese. It is not clear at this time if the correction of hyperinsulinemia and insulin resistance are the result of fasting induced by leptin gene treatment or if high levels of serum leptin have direct effects on circulating insulin levels and its actions.
Fasting blood glucose was also reduced after leptin gene administration. The persistence of the effects of leptin gene treatment on glucose levels after resumption of food intake suggests that body weight reduction improves insulin action. However, it is also possible that this effect may be caused by serum leptin at concentrations below the limit of sensitivity of our assay. It has been shown that low doses of recombinant leptin normalized glucose levels in ob/ob mice without any significant change of body weight and food intake (14).
Glucose tolerance test revealed sustained high glucose levels in the control ob/ob mice. This exaggerated hyperglycemia in response to a glucose load was corrected after leptin gene treatment. The long-term normalization of glucose tolerance in the leptin-gene-treated ob/ob mice suggests that insulin resistance in these animals can be compensated for by elevated insulin secretion after glucose challenge.
Thus, leptin gene administration led to a total correction of the obese phenotype of the ob/ob mice. This correction was accompanied with the normalization of several metabolic abnormalities, including hyperinsulinemia, insulin resistance, and impaired glucose tolerance. Therefore, the treatment also resulted in the correction of the non-insulin-dependent diabetic phenotype in the ob/ob mouse.
The diminution of leptin expression led to the rapid resumption of food consumption to pretreatment level and a gradual gain of body weight. It also led to a gradual increase of serum insulin and the recurrence of insulin resistance. The results suggest that body weight is a critical parameter in the return of hyperinsulinemia and insulin resistance in this wel-defined genetic mouse model. These data also provide substantiating evidence that body weight control needs to be emphasized in the long-term management of obese patients with non-insulin-dependent diabetes mellitus.
Acknowledgments
We thank Milton Pyron for technical assistance. P.M. was a Postdoctoral Fellow of the Swiss National Science Foundation. S.L.C.W. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviations: hAAT, human α1-antitrypsin; RSV, Rous sarcoma virus; pfu, plaque-forming unit(s).
References
- 1.Olefsky J M, Kolterman O G, Scarlett J A. Am J Physiol. 1982;243:E15–E30. doi: 10.1152/ajpendo.1982.243.1.E15. [DOI] [PubMed] [Google Scholar]
- 2.Zuniga-Guajardo S, Jimenez J, Angel A, Zinman B. Metabolism. 1986;35:278–282. doi: 10.1016/0026-0495(86)90214-3. [DOI] [PubMed] [Google Scholar]
- 3.Jahr H, Ratzman K P, Beckert R, Besch W, Hahn H J. Metabolism. 1983;32:1101–1106. doi: 10.1016/0026-0495(83)90055-0. [DOI] [PubMed] [Google Scholar]
- 4.Felber J P. Int J Obesity. 1992;16:937–952. [PubMed] [Google Scholar]
- 5.Wing R R, Koeske R, Epstein L H, Nowalk M P, Gooding W, Becker D. Arch Intern Med (Moscow) 1987;147:1749–1753. [PubMed] [Google Scholar]
- 6.Henry R R, Scheaffer L, Olefsky J M. J Clin Endocrinol Metab. 1985;61:917–925. doi: 10.1210/jcem-61-5-917. [DOI] [PubMed] [Google Scholar]
- 7.Hughes T A, Gwynne J T, Switzer B R, Herbst C, White G. Am J Med. 1984;77:7–17. doi: 10.1016/0002-9343(84)90429-7. [DOI] [PubMed] [Google Scholar]
- 8.Bogardus C, Ravussin E, Robbins D C, Wolfe R R, Horton E S, Sims E A H. Diabetes. 1984;33:311–318. doi: 10.2337/diab.33.4.311. [DOI] [PubMed] [Google Scholar]
- 9.Eriksson K F. Diabetologia. 1991;34:891–898. doi: 10.1007/BF00400196. [DOI] [PubMed] [Google Scholar]
- 10.Knowler W C, Narayan K M V, Hanson R L, Nelson R G, Bennett P H, Tuomilehto J T, Schersten B, Pettitt D J. Diabetes. 1995;44:483–488. doi: 10.2337/diab.44.5.483. [DOI] [PubMed] [Google Scholar]
- 11.Herberg L, Coleman D L. Metabolism. 1977;26:59–99. doi: 10.1016/0026-0495(77)90128-7. [DOI] [PubMed] [Google Scholar]
- 12.Bray G A, York D A. Physiol Rev. 1979;59:719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- 13.Bailey C J, Flatt P R. In: Recent Advances in Diabetes II. Nattrass M, editor. Edinburgh: Churchill Livingstone; 1986. pp. 71–89. [Google Scholar]
- 14.Pelleymounter M A, Cullen M J, Baker M B, Hecht R, Winters D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 15.Halaas J L, Gajiwala K S, Maffei M, Cohen S L, Chait B T, Rabinowitz D, Lallone R L, Burley S K, Friedman J M. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 16.Campfield L A, Smith F J, Guisez Y, Devos R, Burn P. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J M. Nature (London) 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 19.Fang B, Eisensmith R C, Li X H C, Finegold M J, Shedlovsky A, Dove W, Woo S L C. Gene Ther. 1994;1:247–254. [PubMed] [Google Scholar]
- 20.McGrory W J, Bautista D S, Graham F L. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 21.Graham F L, Prevec L. In: Methods in Molecular Biology: Gene Transfer and Expression Protocols. Murray E J, editor. Vol. 7. Clifton, NJ: Humana; 1991. pp. 109–128. [DOI] [PubMed] [Google Scholar]
- 22.Kay M A, Woo S L C. Trends Genet. 1994;10:253–257. doi: 10.1016/0168-9525(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 23.Maffei M, Halaas J, Ravussin E, Pratley R E, Lee G H, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern P A, Friedman J M. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Ertl H C J, Wilson J M. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 26.Tripathy S K, Black H B, Goldwasser E, Leiden J M. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Jooss K U, Su Q, Ertl H C J, Wilson J M. Gene Ther. 1996;3:137–144. [PubMed] [Google Scholar]
- 28.Levin N, Nelson C, Gurney A, Vandlen R, De Sauvage F. Proc Natl Acad Sci USA. 1996;93:1726–1730. doi: 10.1073/pnas.93.4.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]