

Abstract
A chimeric Lhcb gene encoding light-harvesting chlorophyll a/b-binding protein (LHCII) was expressed in transgenic tobacco plants. To separate native from recombinant LHCII, the protein was extended by six histidines at its C terminus. Recombinant LHCII was isolated by detergent-mediated monomerization of pure trimers followed by affinity-chromatography on Ni2+-NTA-agarose (NTA is nitrilotriacetic acid). Elution with imidazole yielded recombinant monomers that formed trimers readily after dilution of the detergent without further in vitro manipulations. LHCII subunits showed the typical chlorophyll a/b ratio at all steps of purification indicating no significant loss of pigments. Transgenic tobacco overexpressed amounts of recombinant protein that corresponded to about 0.7% of total LHCII. This yield suggested that expression in planta might be an alternative to the expression of eukaryotic membrane proteins in yeast. Recombinant LHCII was able to form two-dimensional crystals after addition of digalactolipids, which diffracted electrons to 3.6-Å resolution. LHCII carrying a replacement of Arg-21 with Gln accumulated to only 0.004% of total thylakoid proteins. This mutant was monomeric in the photosynthetic membrane probably due to the deletion of the phosphatidylglycerol binding site and was degraded by the plastidic proteolytic system. Exchange of Asn-183 with Leu impaired LHCII biogenesis in a similar way presumably due to the lack of a chlorophyll a binding site.
Keywords: light-harvesting complex, two-dimensional crystals, histidine tag
It is a challenging task to understand the relationships between the structure of photosynthetic proteins and their function at the atomic level. Crystallization of membrane proteins of the photosynthetic membrane led to the three-dimensional structures of bacterial reaction centers (1, 2) and bacterial photosystem I (3) by x-ray crystallography and to the structures of plant light-harvesting complex (LHCII; ref. 4), and at lower resolution, of bacterial antenna complex (LH2; ref. 5) and bacterial light-harvesting complex I (LH1; ref. 6) by cryoelectron crystallography. These structural studies have been performed with naturally abundant proteins that can be purified in large quantities. Genetics and physiology in purple bacteria allowed to introduce mutations in the reaction centers at residues that are involved in electron transfer and to accumulate sufficient quantities of the recombinant complexes. Several mutants have been analyzed crystallographically at resolutions around 3 Å (7). In contrast, it has been frequently observed that high-level accumulation of recombinant membrane proteins of both eukaryotic and prokaryotic origin is still a major obstacle for biochemical and structural studies. Many membrane proteins were expressed, in homologous and heterologous systems, using Escherichia coli, Saccharomyces cerevisiae, Schizosaccharomyces pombe, and insect or mammalian cells at low levels only and often in denatured form (8).
To study functional aspects of LHCII, it is necessary to produce quantities of recombinant antenna proteins that are sufficient for biophysical experiments. Only organisms synthesizing both chlorophyll a and b and carotenoids are able to correctly fold antenna protein–pigment complexes, as LHCII binds at least 12 chlorophylls, two carotenoid molecules, and two lipid species with different functions. In chlorophyll b-deficient plants, LHCII does not accumulate to normal levels as stabilization of the folded protein by chlorophyll b is missing (9). Alternative attempts to express recombinant LHCII in nonphotosynthetic organisms, therefore, led to denatured protein that needs to be refolded and reconstituted in vitro with all necessary ligands (10, 11).
It is expected that recombinant LHCII expressed in the nucleus of transgenic plants that are not depleted of LHCII will follow the same translocation and membrane integration events. However, endogenous LHCII proteins will accumulate as well. Therefore, recombinant LHCII must be modified in a way that makes it possible to separate native from recombinant complexes. We introduced a polyhistidine peptide at the C terminus of the LHCII monomer, designated LHCII-His6, which allows separation from native LHCII. By detergent-mediated dissociation of pure LHCII trimers, recombinant monomeric proteins were obtained from which only recombinant monomers were retained on a nickel-chelating resin by affinity chromatography. These monomers could be reassembled into fully recombinant trimeric complexes and yielded two-dimensional (2D) crystals that diffracted electrons to 3.6-Å resolution. Mutations effecting binding of phosphatidylglycerol at position 21 and chlorophyll a2 at position 183 had dramatic influences on the amount of recombinant LHCII complexes. In both cases, the protein accumulation dropped to 0.004% and 0.01%, respectively, in comparison to thylakoid proteins, indicating that the biogenesis of LHCII was impaired by these mutations.
MATERIALS AND METHODS
Genetic Manipulation and Production of Transgenic Plants.
For Agrobacterium-mediated transformation a binary plasmid (based on pBIN19; ref. 12) was assembled carrying the cauliflower mosaic virus (CaMV) 35S promoter, the transit sequence of a pea rbcS gene, and a chimeric Lhcb1-2 gene with an affinity tag and a polyadenylylation site. The promoter, transit sequence, and polyadenylylation site were derived from pJIT117 (13). The N-terminal half of the Lhcb gene was constructed in frame from the following synthetic oligonucleotide primers according to (14): A, 5′-AAAGTGCATGCGTAAGTCCGCAACAACCAAAAAGGTTGCCAGCAGCGGATCTCCTTGGTACGGACCAGATAGAGTTAAATATTTAGGAC-3′; B, 5′-GTAGTCCCCGGGAAATTCTCCTGTGAGATAAGAAGGGCTTTCTCCGCTAAAAGGTCCTAAATATTTAACTC-3′; C, 5′-ATTTCCCGGGGACTACGGTTGGGATACAGCCGGTTTGAGCGCCGACCCAGAGACCTTCAGTAAAAATAGAGAACTCGAAGTTATTCATTCTC-3′; D, 5′-GTTTCTGCTAAGAAGCTCAGGAAAAACGCAGCCCAAGGCGCCCAACATAGCCCAACGAGAATGAATAACTTC-3′; E, 5′-GCTTCTTAGCAGAAACGGAGTTAAATTTGGAGAGGCTGTTTGGTTTAAAGCCGGTAGCCAGATCTTCTCTG-3′; F, 5′-GACCAAGCTTGGGTTACCAAGATAGTCCAAGCCGCCCTCAGAGAAGATCTGGCTAC-3′. The C-terminal part (3′ of the second HindIII site, Fig. 1) was obtained from pscabIIC of pea (GenBank accession no. X56538X56538) and its histidine tag was introduced via PCR using the primers A and His6 5′-TTTGTCGACTTAATGGTGATGGTGATGGTGTTTTCCGGGAACAAAGTTGG-3′. Sequencing of the chimeric Lhcb gene ensured the absence of undesired mutations. Transgenic plants were obtained according to (15). Plants were analyzed for transgene expression using the rbcS transit sequence via PCR amplification with primers 5′-S (5′-ATGGCTTCTATGATATCC-3′) and 3′-S (5′-TTCACCTGCATGCACTTTACTCTTCCACC-3′) in the presence of [32P]dATP. Mutations were introduced via PCR mutagenesis (16). Arg-21 was replaced with Gln using primers R21Q (5′-GGACCAGATCAAGTTAAATAT-3′) and R21Q-rev (5′-ATATTTAACTTGATCTGGTC-3′). Asn-183 was replaced with Leu using the primers N183L (5′-GGAACTCAAGTTGGGTAGATTAGC-3′) and N183L-rev (5′-GCTAATCTACCCAACTTGAGTTCC-3′).
Figure 1.
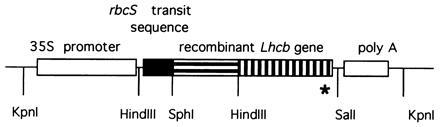
Structure of recombinant Lhcb transgene. A cassette containing transcriptional regulatory elements (open boxes) and a recombinant Lhcb gene that carries an affinity tag of six histidines (asterisk) was inserted into the binary vector pBIN19. To direct the precursor protein into plastids, the endogenous Lhcb transit sequence was replaced by the pea rbcS transit sequence (solid box). In frame the Lhcb gene was constructed from synthetic oligonucleotides (horizontal stripes) and from the C-terminal half of a pea Lhcb gene (vertical stripes).
Gel Electrophoresis and Immunodetection.
SDS/PAGE was performed as described (17). Partially denaturing gels contained 0.1% SDS in the stacking gel and no detergent in the running gel (18). Unless otherwise stated, green unstained gels are shown. Western blot analysis was performed as described (15) with the monoclonal anti-LHCII antibody MLH1 using ECL reagents (Amersham).
Preparation of Recombinant Trimeric LHCII-His6.
LHCII was purified from tobacco plants as described (19) with the modification that membranes were solubilized in 2% Triton X-100 at a chlorophyll (Chl) concentration of 1 mg/ml. Purified trimeric complexes were precipitated from sucrose gradients and dissolved at 3.5 mg of Chl per ml in 0.37% Triton X-100. Recombinant complexes were obtained by separation from endogenous native complexes on Ni2+-NTA-agarose (Quiagen, Chatsworth, CA; NTA is nitrilotriacetic acid). Trimeric LHCII was resuspended in loading buffer [0.8% n-octyl β,d-glucopyranoside (OG)/100 mM Na2HPO4/10 mM imidazole) at a Chl concentration of 1.5–2.0 mg/ml and incubated with the NTA-agarose for 1 h at 4°C. Unbound native complexes were removed with 10–20 vol of washing buffer (100 mM Na2HPO4/0.8% OG) and 2 vol of washing buffer supplemented with 40 mM imidazole. Retained complexes were eluted with 100–200 mM imidazole in 100 mM Na2HPO4/0.8% OG and precipitated (5 min, 14,000 × g) by the addition of KCl to a final concentration of 300 mM. For quantitative experiments with mutant LHCII, solubilized thylakoid membranes were passed over the Ni2+-NTA-resin and LHCII was isolated with the same purity.
Pigment Determination.
Chlorophyll a (Chla) and chlorophyll b (Chlb) values were determined in 80% acetone (20). HPLC separation and quantitation of pigments extracted from LHCII in 100% acetone was performed (21) using the solvent system described as program I. The chromatographic system was a Waters model 626 system equipped with Waters model 996 photodiode array detector.
Disassembly and Oligomerization of LHCII.
Monomerization of trimeric complex was achieved by detergent treatment. LHCII was incubated with Chl at 1 mg/ml in the presence of 1.8–2% OG for 4–6 days at 29°C. Recombinant monomers were bound to Ni2+-NTA-agarose, washed, and eluted with 100–200 mM imidazole as described above. Trimerization of monomeric proteins was achieved by diluting OG below its critical micelle concentration and precipitation of the protein (10 min, 14,000 × g) by the addition of KCl to a final concentration of 300 mM.
Lipid Determination.
Lipids bound to isolated LHCII complexes were identified by 2D thin layer chromatography (TLC) on precoated silica plates (Silica Gel 60, Merck). Chromatograms were developed as described (22). About 15–25 μg of LHCII was usually enough to detect lipids and detergent by iodine vapor staining. Phospholipids were specifically detected by the molybdenum blue stain (ref. 23; Sigma spray reagent M3389). Galactolipids were identified by α-naphthol.
Crystallization and Structural Analysis.
Experiments to form 2D crystals of fully recombinant trimeric LHCII-His6 were based on method B (24) with some modifications (25). For preparing the crystallization mixture LHCII-His6 was reconstituted with lipids. Lipid concentrations ranged from 2.6 to 3.5 mg/ml for digalactosyl-diacylglycerol (DGDG). A combination of DGDG and monogalactosyl-diacylglycerol (MGDG) was used at a ratio of 1:1 at around 2.6 mg/ml. The final Chl concentration was adjusted to 0.85–1.0 mg/ml; the glycerol concentration was 50%. Crystals formed in a volume of 40–50 μl, and crystalline arrays merged into a larger lattice during incubation at 37°C for 3–5 h. Specimen preparation, recording of electron diffraction patterns, and cryoelectron microscopy were performed as described (24).
RESULTS
Overexpression of LHCII in Transgenic Plants.
The homologous expression of LHCII in plants that produce naturally all necessary ligands like pigments and lipids is an approach that has not been attempted, to our knowledge. To understand structure–function relationships and to prove the tentative assignment of Chla and Chlb to the identidfied ligand binding sites as proposed (4), we expressed in tobacco LHCII from pea for which the atomic model has been presented. The strong 35S promoter was used to drive the expression of a chimeric Lhcb gene. The endogenous Lhcb transit sequence was replaced with that of rbcS from pea (Fig. 1). This peptide was shown to direct LHCII into chloroplasts and allowed correct assembly into LHCII and with PSII (26). Transgenic plants were propagated on kanamycin-containing medium. To select transgenic plants expressing the chimeric Lhcb gene at high level, Northern blot analysis with the radioactively labeled rbcS probe was performed. Hybridization signals for 50 transgenic plants were quantitated directly from filter-bound RNA. Wild-type tobacco didn’t show cross-reaction with the rbcS probe. Several plants were characterized by high steady-state levels of transgene mRNA showing similar intensities (Fig. 2). All transgenic plants used in Fig. 2 were selected for further experiments, selfed for seed production, and grown under greenhouse conditions for 4–5 weeks prior to their harvest for LHCII isolation.
Figure 2.
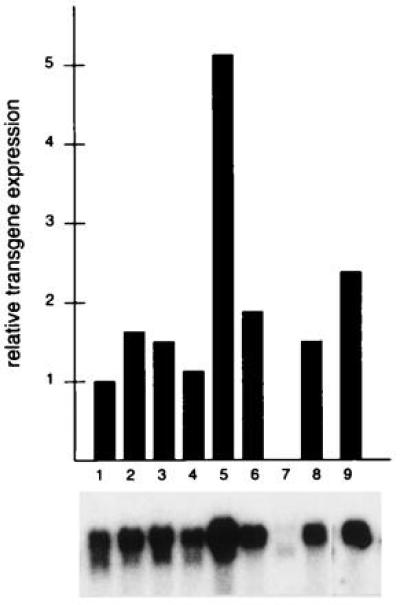
Expression of the Lhcb transgene. Ten micrograms of total RNA was blotted onto nitrocellulose membranes and analyzed for expression of the recombinant transgene by Northern blot hybridization with the radioactively labeled rbcS probe. Radioactivity was determined directly from the filters by using a PhosphorImager. Plants that were selected for high-level expression of the Lhcb transgene showed similar hybridization signals (lanes 1–6 and 8–9). RNA from wild-type tobacco did not hybridize with rbcS (lane 7).
Purification of Histidine-Tagged LHCII by Affinity Chromatography.
Leaves with a fresh weight of up to 450 g were harvested. Roughly 100 mg of LHCII was isolated from detergent-solubilized thylakoid membranes by sucrose-gradient centrifugation and salt precipitation. Pure trimeric LHCII was characterized by a Chla/Chlb ratio of 1.43–1.48. This ratio was indicative of seven molecules of Chla and five molecules of Chlb, consistent with the atomic model (4). Purified LHCII consisted of a mixture of native and histidine-tagged LHCII. Trimeric LHCII that contained at least one recombinant monomer was isolated via affinity chromatography on Ni2+-NTA-agarose. Chromatography with Ni2+-NTA resin is characterized by a remarkable specificity for binding selectively proteins that have as a structural element neighboring histidines on the surface (27). LHCII contains only three histidines that are not adjacent to each other but located in transmembrane helix B (His-68), at the N-terminal end of helix C (His-120), and at the C-terminal end of helix D (His-212). LHCII was bound to the resin with continuous agitation. The resin was then packed into a column that was extensively washed. As LHCII is a green chlorophyll-binding protein, its isolation and purification could be followed visually (Fig. 3). As predicted by the absence of nonadjacent histidines, native LHCII did not bind to the resin (Fig. 4). This was also true for the largest amount of LHCII isolated from thylakoids of transgenic plants (Fig. 3). Bound material was washed extensively with 40 mM imidazole-containing buffer prior to elution. Elution of the recombinant complex was observed with 100–150 mM imidazole, which structurally mimics the histidine side chain and competes with proteins for binding to the resin. The eluted complexes, in total roughly 1 mg of Chl, represented a mixture of native and recombinant monomers in a trimer. In a control experiment, solubilized thylakoids from wild-type tobacco were passed over the same column. No protein was retained after the wash. This system is therefore suitable for the purification of other membrane proteins expressed in plants.
Figure 3.

Isolation of histidine-tagged LHCII trimers. Pure LHCII trimers were obtained by detergent solubilization of thylakoids and sucrose gradient centrifugation. Histidine-tagged proteins were separated by affinity chromatography. Elution was monitored visually by the green Chl color characteristic of LHCII. Endogenous native LHCII trimers did not bind to Ni2+-NTA-agarose as adjacent histidines are missing (Upper). Recombinant LHCII-His6 was retained selectively and eluted with imidazole (Lower) (5). Lanes: 1 and 2, flow through of unbound native LHCII; 3 and 4, column wash; 5, wash with 40 mM imidazole; 6–8, elution with increasing imidazole concentration (100, 150, and 200 mM).
Figure 4.
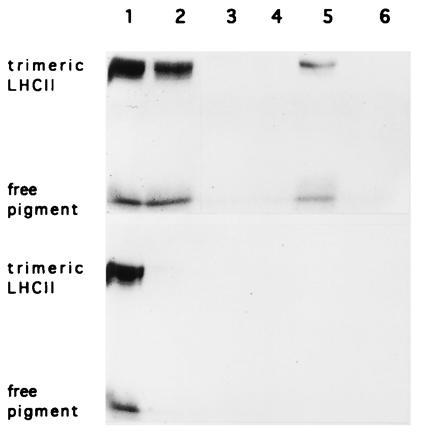
Coomassie-stained gels of complexes isolated on Ni2+-NTA-agarose. LHCII retained on Ni2+-NTA-resin (see Fig. 3) was separated electrophoretically by SDS/PAGE. Native LHCII did not bind to the resin (Lower) whereas LHCII from transgenic plants was retained. The recombinant complex was eluted with 100 mM imidazole. Lanes: 1, unbound LHCII; 2 and 3, column wash; 4, wash with 40 mM imidazole; 5 and 6, elution with 100 and 200 mM imidazole, respectively.
Controlled Monomerization and Trimerization of LHCII.
The isolated trimers consisted only of one-third of the recombinant protein, the remainder being native tobacco LHCII. In a crystallographic analysis, any differences between recombinant and native LHCII would thus be obscured by the preponderance of wild-type protein. Trimeric LHCII can be dissociated with elevated concentrations of nonionic detergent and reassembled at reduced detergent levels (25). Incubation with OG at a concentration of 1.8–2% for 4–6 days led to complete dissociation of trimeric into monomeric LHCII. Detergent concentrations above 2% eventually caused a gradual breakdown of the complex, as observed by loss of pigments. The monomers were characterized by a Chla/Chlb ratio of 1.41–1.43 that was identical to that of wild-type trimers. Monomerized LHCII was bound to Ni2+-NTA-agarose using the conditions established for the isolation of trimeric complexes. The resin was washed with 20 bed vol of buffer and eluted with 150 mM imidazole (Fig. 5). As expected, recombinant monomers represented one-third of the starting material, yielding 0.3 mg of Chl or roughly 0.6 mg of LHCII. The monomeric form reassociated readily into trimers upon dilution of the protein/detergent mixture below the critical micellar concentration of the detergent, followed by salt precipitation (Fig. 6). As judged by native gel electrophoresis the disassembly of oligomeric LHCII was almost fully reversible. A Chla/Chlb ratio of 1.38–1.40 was measured with the reassociated trimers indicating no significant loss of Chl during extended incubation in high concentrations of OG. HPLC separation of pigments extracted from native and recombinant LHCII did not show significant differences in content. The lutein/Chla ratio in native LHCII was 0.27 mol/mol and 0.26 mol/mol in LHCII-His6.
Figure 5.
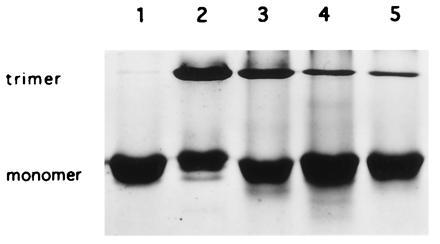
Monomerization of LHCII trimers. Trimers containing recombinant monomers were separated by chromatography on Ni2+-NTA-agarose. Trimers were disassembled by incubation in 1.8–2% OG. Lanes: 1, monomeric LHCII after detergent-mediated dissociation (6 days); 2, trimeric LHCII purified on Ni2+-NTA-agarose (Monomeric native LHCII is observed due to the presence of 0.1% SDS in the native gel system.) 3–5, time-dependent dissociation of trimeric complexes into monomeric protein over approximately 4 days.
Figure 6.
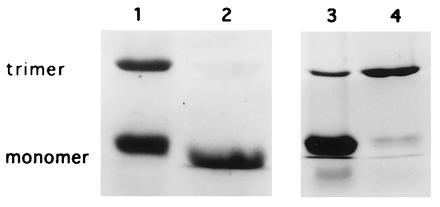
Assembly of recombinant monomers into trimeric LHCII-His6 complexes. Recombinant monomers were isolated by affinity chromatography on Ni2+-NTA-agarose. Trimers of LHCII-His6 formed by dilution of the detergent below its critical micelle concentration and salt precipitation of the protein. Lanes: 1, native LHCII as marker; 2, monomeric LHCII after affinity-chromatography; 3, monomeric LHCII-His6 after addition of KCl; 4, trimeric LHCII-His6 after dilution of detergent and salt precipitation.
Lipid Content of LHCII-His6.
LHCII-His6 was distinguishable from native LHCII by its different composition of lipids. Native LHCII binds different concentrations of various lipids that fulfill different functions for two- and three-dimensional crystallization (25). In LHCII-His6 the glycolipids DGDG and MGDG were undetectable after 2D TLC (Fig. 7) because they were lost during detergent-mediated dissociation and affinity chromatography. Phosphatidylcholine was also stripped off the recombinant complexes. This lipid is believed to be located specifically in the chloroplast envelope. It was probably a contaminant that bound unspecifically to LHCII when thylakoid membranes were solubilized in detergent, as observed previously with native LHCII. Phosphatidylglycerol (PG) was the only lipid that remained tighly attached to the protein, even at high concentrations of non-ionic detergent. PG is thought to function as molecular glue between LHCII monomers (4), and reconstitution of completely delipidated LHCII monomers with PG is difficult to achieve (11).
Figure 7.
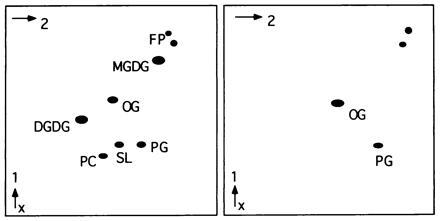
Lipid composition of native LHCII and recombinant LHCII-His6. LHCII-His6 (Right) has lost some lipids compared with native LHCII (Left) during column purification. Only PG remained tightly bound to LHCII-His6. SQ, sulfoquinovosylglycerol; PC, phosphatidylcholine.
Crystallization and Electron Diffraction of Recombinant LHCII-His6.
Recombinant LHCII-His6 lost most of its lipids but retained all pigments. Crystal formation of LHCII-His6 required first trimeric protein and second a minimal set of lipids. Only trimers of LHCII could be crystallized, and PG was the essential mediator for the oligomerization of monomeric LHCII (25). Additionally, DGDG was needed for the formation of 2D crystals (25). To mimic the lipid environment of the thylakoid membrane, DGDG was added to the protein/detergent mixture. The addition of DGDG only was sufficient to form crystals. Crystals from all protein preparations formed readily. However, without further optimization of growth conditions, crystals often stacked together and did not form lattices larger than 3 μm in diameter. The following conditions yielded crystals reproducibly: LHCII-His6 was used at a concentration of 0.9 ± 0.1 mg of Chl per ml, the detergents NG and Triton X-100 were used at a final concentration of 0.1% and 0.22%, respectively, and the lipids DGDG and MGDG were added in a 1:1 ratio of 2.6 mg/ml. The glycerol concentration was 50%, while the concentration of glycine were kept unchanged at 100 mM. This mixture was incubated for 2 days at 25°C and then at 37°C for at least 3–5 h.
These conditions yielded crystals that were large enough for recording electron diffraction patterns as one-third of all crystals formed were 5 μm in size. For diffraction studies, crystals were embedded in 0.5% tannin and diffraction patterns recorded at −130°C. LHCII-His6 diffracted to 3.6-Å resolution. Clearly, the histidine-tag did not interfere with 2D lattice formation. A typical electron diffraction pattern is shown in Fig. 8. This resolution was close to the resolution of 3.2–3.4 Å, which was achieved routinely with native LHCII complexes.
Figure 8.

Electron diffaction pattern of recombinant LHCII-His6. The electron diffraction pattern was recorded from a 2D crystal of LHCII-His6 preserved with 0.5% tannin. The partially lipid-depleted trimer was reconstitued with a mixture of DGDG and MGDG (1:1 ratio) and diffracted to 3.6-Å resolution.
Expression of PG-Free LHCII in Tobacco.
There has been a long-standing debate whether monomeric or trimeric LHCII is the functional form in the thylakoid membrane. To provide in vivo data, we expressed mutant LHCII in tobacco. Based on biochemical evidence (25) and the results of in vitro reconstruction (28), it is thought that residue 21 is involved in binding of the phosphatidyl head group of PG. Accordingly, we changed Arg-21 to Gln (R21Q) using site-directed mutagenesis. The mutated Lhcb gene was integrated into the tobacco genome. Northern blot hybridizations with the radioactively labeled rbcS transit sequence showed transgene expression (data not shown) that was comparable with expression observed earlier (see Fig. 2). LHCII was isolated from thylakoids and LHCII-R21Q separated on Ni2+-NTA-resin. Although expression of the transgene was high, protein accumulation of LHCII-R21Q dropped to low levels in comparison to LHCII-His6. To quantitate the accumulation of LHCII-R21Q, equal amounts of thylakoid membranes from plants expressing either LHCII-His6 or LHCII-R21Q were passed over Ni2+-NTA-agarose. In comparison to thylakoids with LHCII-His6, less than 0.004% of LHCII-R21Q complexes accumulated in the membrane. Was LHCII-R21Q assembled with native LHCII monomers to form trimers despite its lack of PG? Isolated LHCII-R21Q was separated on native gels by electrophoresis and blotted onto nitrocellulose filters. Due to its low abundance LHCII-R21Q was detected immunologically. The Western blot showed that the amount of LHCII-R21Q that accumulated in the thylakoid membrane was not forming trimers but remained unassembled as monomeric LHCII (Fig. 9). This result was confirmed with native stained gels (Fig. 9) after isolating LHCII-R21Q from about 300 mg of a pure LHCII mixture. These results provide additional evidence for the involvement of Arg-21 in PG binding, and for the importance of this lipid in trimer formation.
Figure 9.
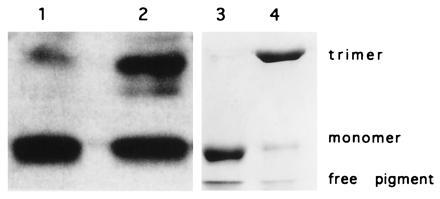
Assembly defect of LHCII-R21Q. LHCII-R21Q was isolated by affinity chromatography and subjected to nondenaturing gel electrophoresis. When blotted onto nitrocellulose filters, monomeric LHCII-R21Q was detected immunologically with anti-LHCII antibodies (lane 1). Additionally, in green gels LHCII-R21Q also behaved as a monomer (lane 3). Lanes 2 and 4 show native LHCII as a marker.
Expression of LHCII Devoid of One Chla Binding Site.
The above mentioned expression studies demonstrated the value of in vivo manipulation of LHCII in transgenic plants. We introduced another mutation in helix A to test experimentally the proposed atomic model of LHCII. As the assignment of Chla and Chlb in this model is tentative (4) the binding site for Chla2 at Asn-183 was mutated to Leu (LHCII-N183L). Transformants with high expression levels for the Lhcb transgene mRNA were obtained (data not shown). LHCII-N183L was isolated from thylakoid membranes of transgenic plants by Ni2+-NTA-affinity chromatography. Surprisingly, only very low amounts of LHCII-N183L accumulated in the thylakoid membrane. This result was also confirmed when solubilized thylakoid membranes instead of purified LHCII complexes were passed over the chelating-agarose column. The amount of LHCII-N183L accumulating in the membrane was less than 10% in comparison to LHCII-His6, accounting for only about 0.07% of native LHCII or 0.01% of thylakoid proteins. Nondenaturing gel electrophoresis demonstrated that LHCII-N183L was associated with native LHCII molecules forming trimeric complexes (Fig. 10).
Figure 10.
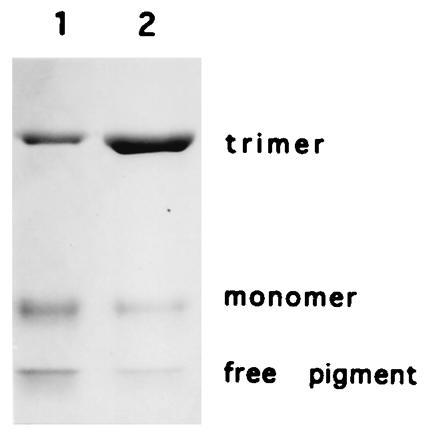
Association of LHCII-N183L into trimers. LHCII-N183L was isolated by affinity chromatography and subjected to nondenaturing gel electrophoresis. In green gels, LHCII-N183L assembled into trimeric complexes (lane 1) when compared with native LHCII (lane 2).
DISCUSSION
Homologous Expression of LHCII.
The functional status of light-harvesting complexes is dependent on the correctly folded protein structure that is formed and maintained by the interaction of all available pigment binding sites with the protein moiety. Pigment complexes like the D1 protein of photosystem (PS) II can be expressed in E. coli as insoluble inclusion bodies and reconstituted in vitro with pigments to retain their native structures (29). In vitro reconstituted LHCII has been analyzed by 2D crystallization and found to form 2D crystals with the same crystal symmetry as the native LHCII (11). Due to the small amount of material recovered, it is difficult to optimize crystallization conditions for high-resolution electron diffraction. High resolution, however, is needed to identify chlorophylls and conformational changes due to mutations. Expression in E. coli and in vitro reconstitution was also achieved for bacteriorhodopsin (bR, ref. 30) and porin (31). However, the properties of some mutant bR were dependent on whether bR was expressed in the native Halobacterium salinarium or in E. coli (32). In vitro reconstituted mutant bR did not assemble readily into purple membrane patches (8). To avoid these difficulties, we attempted to express LHCII in transgenic plants.
A chimeric Lhcb gene encoding LHCII from pea was transcribed from the strong 35S CaMV promoter that allowed high level expression in wild-type tobacco (Fig. 2), as LHCII-depleted mutants that could be used for transformation were not available. Expressed recombinant LHCII needed to be separated from native LHCII. Therefore, the apoprotein was extended genetically by a sequence encoding six histidines. Pure LHCII was obtained from transgenic plants by a standard protocol. Recombinant LHCII was isolated via affinity chromatography and dissocation/reassociation of the oligomeric form of LHCII. Including all manipulations, 0.7% of total pure LHCII was recombinant. In vivo expression of LHCII in plants and in vitro manipulation of oligomeric LHCII complexes produced recombinant trimeric protein that formed well-ordered crystals even with its histidine-tag at the C terminus.
The established green expression system yielded recombinant LHCII to 0.21% of thylakoid proteins. This is still lower than the expression levels of some membrane proteins reported for yeast. For example, the plastidic triose phosphate translocator was expressed to a level of 1% of internal cell membrane proteins in S. pombe (33). However, recombinant LHCII-His6 might be expressed at a higher level in plants grown under conditions that enhance thylakoid grana formation or with LHCII-depleted mutants. Studies with bR demonstrated the value of deletion mutants for homologous expression. The level of bR expression in those cells reached about 40% (34) or even wild-type level (35) for several mutants. We therefore aimed for the expression of Lhcb genes in plants deficient of LHCII. However, mutants described in the literature miss either Chlb (9) or lack LHCII integration factors (36), both essential factors for the correct biogenesis of LHCII. Lhcb antisense approaches in tobacco (15) demonstrated the problem of repressing accumulation of LHCII.
Essential N-Terminal Arginine for LHCII Assembly.
It has been widely proposed that the unusual fatty acid composition that characterizes chloroplast membranes is critical to the maintenance of photosynthetic function. It has been shown clearly that LHCII binds PG and in particular δ3-trans-hexadecenoic acid (trans-C16:1), which is always found esterified to the second position of PG (37). In this report the importance of PG in the biogenesis of LHCII was challenged in vivo for the first time. We exchanged Arg-21 with the uncharged hydrophilic residue glutamine and integrated the mutant Lhcb gene into tobacco. Immunological analysis of affinity-selected LHCII-R21Q demonstrated that the mutation blocked the assembly of monomeric LHCII into trimeric LHCII (Fig. 9). The low accumulation of LHCII-R21Q in thylakoids was, therefore, not unexpected and is consistent with other findings. The fadA mutant of Arabidopsis thaliana lacked detectable levels of the trans-C16:1 fatty acid and had a compensating increase in palmitic acid (C16:0) without influencing thylakoid structure or energy transfer from LHCII to PSII (38). The A. thaliana mutant act1 was characterized by a 15–20% reduction in PG and minor decreases in MGDG, DGDG, and sulfoquinovosylglycerol (SQ). This mutant, however, showed altered thylakoid organization and altered excitation energy transfer from antenna chlorophyll to PSII and PSI (39). Although PG containing the specific trans-C16:1 is apparently not required for thylakoid organization, a 15–20% reduction in the total amount of PG caused ultrastructural changes and reduced the accumulation of the PG-binding protein LHCII by 13% (39, 40). trans-C16:1 significantly protects trimeric LHCII against dissociation by SDS in vitro. Its exact role in vivo remains to be explored since the lack of trans-C16:1 is easily compensated by palmitic acid. Even in vitro, trimeric LHCII can be reconstituted with dipalmitoyl-PG (11) instead of trans-C16:1. However, in vitro reconstitution of LHCII from its apoprotein and isolated pigments fails unless dipalmitoyl-PG is present (11). Replacement of Arg-21 with Gln abolished trimerization of this mutant probably due to the deletion of the PG binding site (28). LHCII-R21Q of this report had the same exchange and did not accumulate in the membrane at high level. PG could not be detected by 2D TLC. This provides strong evidence for trimeric LHCII being the only functional form for energy transfer from antenna to reaction center Chl. The nonfunctional monomer is probably degraded by the plastidic proteolytic system. Degradation in plastids was shown to take place for an assembly-deficient ribulose bisphosphate carboxylase (Rubisco) small subunit (41, 42). Additionally, in Chlamydomonas mutants with 50% of wild-type PG (43), trimeric LHCII was absent. LHCII underlies progressive degradation that was illustrated by the continued accumulation of low molecular mass polypeptides that are immunologically related to LHCII (44). The in planta expression of histidine-tagged recombinant LHCII highlighted the functional significance of PG and supported the data in greening plants (45) that suggested that monomeric LHCII is only a transient form during the assembly of the PSII antenna.
How Susceptible Is LHCII to Mutations in Vivo?
Due to the lack of LHCII-deficient photosynthetic organisms that could be complemented with altered LHCIIs in planta experiments with mutant antenna complexes have not yet been performed. The result of in planta expression of LHCII-N183L was surprising to us. The yield of LHCII-N183L was unexpectedly low, even though LHCII-N183L accumulated in the thylakoid membrane and assembled into trimers. The effect of this mutation on the LHCII biogenesis remains unclear. Is Chla2 initiating/supporting protein–pigment interactions or were conformational changes induced that prevented normal accumulation? The three transmembrane helices of LHCII provide the scaffold around which Chl and carotenoids are arranged. This scaffold was probed by chloroplast import assays with mutated precursor proteins of LHCII. His-120 and His-212, outside the central helix pair in a lumenal loop and in the amphipathic helix D, respectively, were not essential as the mutants LHCII-H120A and LHCII-H212A assembled with LHCII. His-68, located in helix B, was deleterious to LHCII assembly and rapid degradation was discussed (46). Proteins in which charged residues were exchanged (K203E, E83R, and K203R) assembled into LHCII to almost wild-type levels (47). However, when similar replacements were introduced into helix B [Arg-70 with Glu (R70E), Glu-83 with Lys (E83K)], R70E did not accumulate and E83K assembled in the complex to significantly reduced amounts only (46). Interestingly, Arg-70 is essential for holding helices A and B together by interacting with Glu-180 whereas for Glu-83 no specific function has been assigned yet (4). It seems that plants do not tolerate mutations in LHCII effecting either structurally essential or less important residues in its central helices. This might explain the extremely high conservation of the LHCII sequence among photosynthetic organisms. It remains to be established whether the membrane insertion, oligomerization, LHCII interactions, or stability of LHCII-N183L were impaired in the LHCII biogenesis.
Footnotes
Abbreviations: LHCII, light-harvesting chlorophyll a/b-binding protein; 2D, two dimensional; Chl, chlorophyll; OG, n-octyl β,d-glucopyranoside; DGDG, digalactosyl-diacylglycerol; MGDG, monogalactosyl-diacylglycerol; NTA, nitrilotriacetic acid; PG, phosphoglycerol; PS, photosystem; bR, bacteriorhodopsin.
References
- 1.Deisenhofer J, Michel H. EMBO J. 1989;8:2149–2170. doi: 10.1002/j.1460-2075.1989.tb08338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ermler U, Fritzsch G, Buchanan S K, Michel H. Structure. 1994;2:925–936. doi: 10.1016/s0969-2126(94)00094-8. [DOI] [PubMed] [Google Scholar]
- 3.Krauss N, Hinrichs W, Witt I, Fromme P, Pritzkow W, Dauter Z, Betzel C, Wilson K S, Witt H T, Saenger W. Nature (London) 1993;361:326–331. [Google Scholar]
- 4.Kühlbrandt W, Wang D N, Fujiyoshi Y. Nature (London) 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 5.Savage H, Cyrklaff M, Montoya G, Kühlbrandt W, Sinning I. Structure. 1996;4:243–252. doi: 10.1016/s0969-2126(96)00029-9. [DOI] [PubMed] [Google Scholar]
- 6.Karrasch S, Bullough P A, Ghosh R. EMBO J. 1995;14:631–638. doi: 10.1002/j.1460-2075.1995.tb07041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chirino A J, Lous E J, Huber M, Allen J P, Schenck C C, Paddock M L, Feher G, Rees D C. Biochemistry. 1994;33:4584–4593. doi: 10.1021/bi00181a020. [DOI] [PubMed] [Google Scholar]
- 8.Grisshammer R, Tate C G. Q Rev Biophys. 1995;28:315–422. doi: 10.1017/s0033583500003504. [DOI] [PubMed] [Google Scholar]
- 9.Knoetzel J, Simpson D. Planta. 1991;185:111–123. doi: 10.1007/BF00194522. [DOI] [PubMed] [Google Scholar]
- 10.Paulsen H, Rümler U, Rüdiger W. Planta. 1990;181:204–211. doi: 10.1007/BF02411539. [DOI] [PubMed] [Google Scholar]
- 11.Hobe S, Prytulla S, Kühlbrandt W, Paulsen H. EMBO J. 1994;13:3423–3429. doi: 10.1002/j.1460-2075.1994.tb06647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevan M, Barnes W M, Chilton M-D. Nucleic Acids Res. 1983;11:369–385. doi: 10.1093/nar/11.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerineau F, Woolston S, Brooks L, Mullineaux Nucleic Acids Res. 1988;16:11380. doi: 10.1093/nar/16.23.11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strauss M, Bartsch F O, Stollwerk J, Trstenjak M, Böhning A, Gassen H G, Machleidt W, Turk V. Biol Chem Hoppe-Seyler. 1988;369:209–218. [PubMed] [Google Scholar]
- 15.Flachmann R, Kühlbrandt W. Plant Cell. 1995;7:149–160. doi: 10.1105/tpc.7.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higuchi R. In: PCR Technology. Erlich H A, editor. New York: Stockton; 1989. pp. 61–70. [Google Scholar]
- 17.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Anderson J M. Biochim Biphys Acta. 1980;591:113–126. doi: 10.1016/0005-2728(80)90225-x. [DOI] [PubMed] [Google Scholar]
- 19.Kühlbrandt W, Thaler T H, Wehrli E. J Cell Biol. 1983;96:1414–1424. doi: 10.1083/jcb.96.5.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porra W, Thompson W A, Kriedemann P E. Biochim Biophys Acta. 1989;975:384–394. [Google Scholar]
- 21.Gilmore A M, Yamamoto H Y. J Chromatogr. 1991;543:137–145. [Google Scholar]
- 22.Allen C F, Good P. Methods Enzymol. 1971;23:523–547. [Google Scholar]
- 23.Kates M. Techniques of lipidology. Amsterdam: North–Holland; 1972. [Google Scholar]
- 24.Wang D N, Kühlbrandt W. J Mol Biol. 1991;217:691–699. doi: 10.1016/0022-2836(91)90526-c. [DOI] [PubMed] [Google Scholar]
- 25.Nuβberger S, Dörr K, Wang D N, Kühlbrandt W. J Mol Biol. 1993;234:347–356. doi: 10.1006/jmbi.1993.1591. [DOI] [PubMed] [Google Scholar]
- 26.Hand J M, Szabo L J, Vasconcelos A C, Cashmore A R. EMBO J. 1989;8:3195–3206. doi: 10.1002/j.1460-2075.1989.tb08478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hochuli E, Döbeli H, Schacher A. J Chromatogr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 28.Hobe S, Förster R, Klingler J, Paulsen H. Biochemistry. 1995;34:10224–10228. doi: 10.1021/bi00032a016. [DOI] [PubMed] [Google Scholar]
- 29.Efimov V A, Fradkov A F, Raskind A B, Khristin M S, Klimov V V, Chakmakhcheva O G. FEBS Lett. 1994;348:153–157. doi: 10.1016/0014-5793(94)00586-9. [DOI] [PubMed] [Google Scholar]
- 30.Miercke L J W, Betlach M C, Mitra A K, Shand R F, Fong S K, Stroud R M. Biochemistry. 1991;30:3088–3098. doi: 10.1021/bi00226a016. [DOI] [PubMed] [Google Scholar]
- 31.Qi H L, Tai J Y, Blake M S. Infect Immun. 1994;62:2432–2439. doi: 10.1128/iai.62.6.2432-2439.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanyi J K. J Bioenerg Biomembr. 1992;24:169–179. doi: 10.1007/BF00762675. [DOI] [PubMed] [Google Scholar]
- 33.Loddenkötter B, Kammerer B, Fischer K, Flügge U I. Proc Natl Acad Aci USA. 1993;90:2155–2159. doi: 10.1073/pnas.90.6.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krebs M P, Hauss T, Heyn M P, Rajbhandary U L, Khorana H G. Proc Natl Acad Sci USA. 1991;88:859–863. doi: 10.1073/pnas.88.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krebs M P, Mollaaghababa R, Khorana H G. Proc Natl Acad Sci USA. 1993;90:1987–1991. doi: 10.1073/pnas.90.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawata E E, Cheung A Y. EMBO J. 1990;9:4197–4203. doi: 10.1002/j.1460-2075.1990.tb07644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trémolières A, Dubacq J-P, Ambard-Bretteville F, Rémy R. FEBS Lett. 1981;130:27–30. [Google Scholar]
- 38.Browse J, McCourt P, Somerville C. Science. 1985;227:763–765. doi: 10.1126/science.227.4688.763. [DOI] [PubMed] [Google Scholar]
- 39.Kunst L, Browse J, Somerville C. Plant Physiol. 1989;90:846–853. doi: 10.1104/pp.90.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Knowles P F, Murphy D L, Nishida I, Marsh D. Biochemistry. 1989;28:7446–7452. doi: 10.1021/bi00444a044. [DOI] [PubMed] [Google Scholar]
- 41.Flachmann R, Bohnert H J. J Biol Chem. 1992;267:10576–10582. [PubMed] [Google Scholar]
- 42.Adam Z. Photosynth Res. 1995;43:143–147. doi: 10.1007/BF00042971. [DOI] [PubMed] [Google Scholar]
- 43.Maroc J, Trémolières A, Garnier J, Guyon D. Biochim Biophys Acta. 1987;893:91–99. [Google Scholar]
- 44.Dubertret G, Mirshahi A, Mirshahi M, Gerard-Hirne C, Trémolières A. Eur J Biochem. 1994;226:473–482. doi: 10.1111/j.1432-1033.1994.tb20072.x. [DOI] [PubMed] [Google Scholar]
- 45.Dreyfuss B W, Thornber J P. Plant Physiol. 1994;106:829–839. doi: 10.1104/pp.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tobin E M, Kohorn B D. J Biol Chem. 1987;262:12897–12899. [PubMed] [Google Scholar]
- 47.Kohorn B D. Plant Physiol. 1989;93:339–342. doi: 10.1104/pp.93.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]