

Abstract
A series of copper(I)-α-ketocarboxylate complexes have been prepared and shown to exhibit variable coordination modes of the α-ketocarboxylate ligand. Reaction with O2 induces decarboxylation of this ligand and the derived copper-oxygen intermediate(s) has been intercepted, resulting in hydroxylation of an arene substituent on the supporting N-donor ligand. Theoretical calculations have provided intriguing mechanistic notions for the process, notably implicating hydroxylation pathways that involve novel [CuI-OOC(O)R] and [CuII-O-· ↔ CuIII=O2-]+ species.
Understanding the properties of synthetic copper-oxygen intermediates is important for evaluating mechanisms of oxidations by enzymes and other catalysts.1,2 A growing class of reactive copper-oxygen complexes have been isolated or identified spectroscopically to date, including well-studied examples with [Cu2(O2)]2+, [Cu2(μ-O)2]2+, or [CuO2]+ cores.3 Mononuclear copper-oxo species ([CuII-O-· ↔ CuIII=O2-]+) are less well understood. Such species have been considered as a possible reactive intermediate in catalysis by copper enzymes (cf. peptidylglycine α-hydroxylating monooxygenase,4,5 particulate methane monooxygenase6) and in some synthetic reactions,7 but to our knowledge they have only been observed in the gas phase.8 Theory suggests that they should be powerful oxidants,4,8,9 perhaps even more reactive than the related [FeIV=O]2+ unit that has been extensively studied.10 Among the various routes by which [FeIV=O]2+ moieties may be accessed is that used by α-ketoglutarate-dependent nonheme iron enzymes (Scheme 1).14 Experimental and theoretical studies15 support a mechanism that involves reaction of O2 with a bidentate α-ketocarboxylate complex of FeII to yield an O2 adduct (A) that attacks the α-keto position to induce loss of CO2 and generate B. Formally a FeII-OOC(O)R species, B then undergoes O-O bond heterolysis to yield the [FeIV=O]2+ moiety (C) that is generally deemed to be responsible for attacking the C-H bond of substrate(s).
Scheme 1.
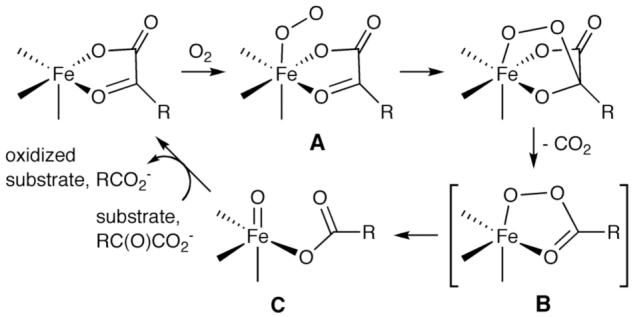
General mechanism for reactivity of FeII-α-ketocarboxylate sites in enzymes and model complexes.
Drawing an analogy to this chemistry, we envisioned that reaction of O2 with a CuI-α-ketocarboxylate could similarly generate novel [CuI-OOC(O)R] and/or derived [CuII-O-· ↔ CuIII=O2-]+ species. In view of their potentially high reactivity that could make these species difficult to observe directly, we postulated that they might be trapped by using a supporting ligand with an arene substituent appropriately positioned to be susceptible to intramolecular attack.16,17 Herein we report the synthesis and structural characterization of new CuI-α-ketocarboxylate complexes with such ligands, the discovery of decarboxylation and arene hydroxylation upon reaction of these complexes with O2, and theoretical calculations that provide provocative mechanistic insights into the reactions. These findings serve to illustrate a new pathway for the generation of novel copper-oxygen intermediates relevant to oxidation catalysis.
Using modifications of known methods,18 we prepared bidentate LH,19 LMe, and Lm-OMe (Scheme 2), which were chosen because they would favor low coordination numbers in their complexes, provide a single appended arene in a position suited for intramolecular oxidation,7d,16,17,20 and contain a sterically bulky 2,6-diisopropylphenyl group to inhibit formation of bis-ligand complexes (e.g. L2Cu+) and dicopper-oxygen intermediates.3 Copper(I)-α-ketocarboxylate complexes 1-4 were synthesized by treatment of [Cu(Mes)]4 with benzoyl- or mesitoyl-formic acid, followed by addition of the N-donor ligand. For comparative O2 reactivity studies (see below), we also prepared 5 by treating LMe with CuCl and then AgO3SCF3. The compounds 1-5 were isolated as red-brown (1-4) or purple (5) solids in good yields (∼75-85%) and were fully characterized by NMR and UV-vis spectroscopy, CHN analysis, and X-ray crystallography.18
Scheme 2.
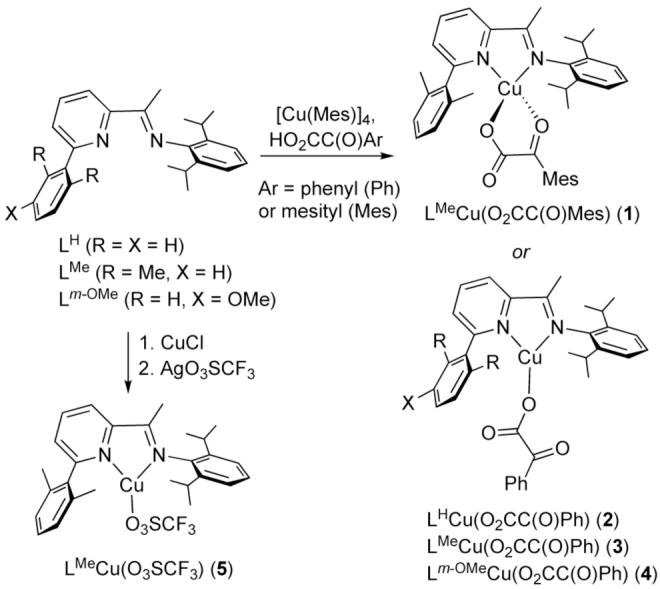
Synthesis of copper(I) complexes.
The structures of 1 and 2 are shown in Figure 1, while those of 3-5 appear in the supporting information (Figures S1-S3). The α-ketocarboxylate coordinates in chelating bidentate fashion via the carboxylate and the ketone O atoms in 1, but in 2-4 it binds solely through a carboxylate O atom with the keto group oriented approximately orthogonal to the carboxylate plane (torsional angle O1-C26-C27-O3 = -103.5(3)°). In 5, the triflate anion binds as a monodentate ligand to yield a 3-coordinate complex. All structures display metal-ligand bond distances typical for CuI complexes. For 1-4, similar 1H and 13C{1H} NMR spectra are observed in CD2Cl2 at ambient temperature which exhibit a single set of sharp peaks, indicating that in solution they either retain the structures observed by X-ray diffraction or undergo fluxional processes that are sufficiently rapid to result in an averaged spectrum (cf. bidentate/monodentate α-ketocarboxylate isomerization).18 Density functional calculations at the M06L level21 on model systems with unsubstituted aryl rings (hereafter 2′) found local minima having both monodentate and bidentate coordination of the ketoacid to copper, with the latter preferred by about 7 kcal/mol.
Figure 1.
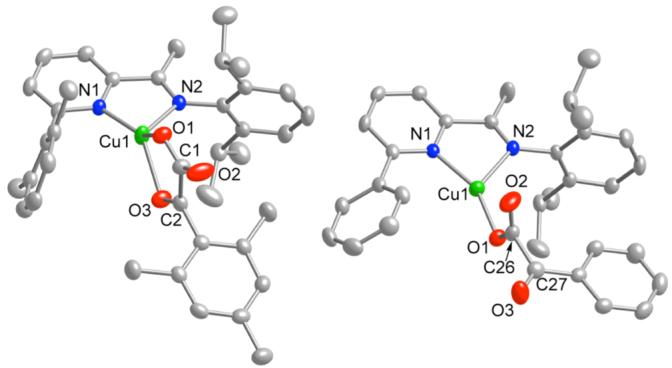
Representations of the X-ray structures of 1 (left) and 2 (right) as 50% thermal elliposoids and hydrogen atoms omitted for clarity. Selected interatomic distances (Å) and angles (deg) are as follows. 1: Cu1-N1, 1.9676(19); Cu1-O1, 1.9904(19); Cu1-N2, 2.107(2); Cu1-O3, 2.1815(19); O1-C1, 1.261(3); C2-O3, 1.223(3); C2-C1, 1.545(3); O2-C1, 1.230(3); N1-Cu1-O1, 137.52(8); N1-Cu1-N2, 79.99(8); O1-Cu1-N2, 115.31(8); N1-Cu1-O3, 134.09(8); O1-Cu1-O3, 79.18(7); N2-Cu1-O3, 112.07(8). 2: Cu1-O1, 1.9042(16); Cu1-N1, 1.9831(19); Cu1-N2, 2.1113(19); Cu1-O2, 2.888(2); C26-O2, 1.216(3); C26-O1, 1.279(3); C26-C27, 1.529(3); O3-C27, 1.216(3); O1-Cu1-N1, 154.80(8); O1-Cu1-N2, 123.60(8); N1-Cu1-N2, 80.74(7); O2-C26-O1, 127.2(2).
Exposure of solutions of 2 or 4 to O2 in acetone at -80 °C resulted in gradual (∼2-4 h) formation of brown solutions. After warming and removal of copper under basic conditions, a mixture of unperturbed ligand (LH or Lm-OMe) and hydroxylated versions (LOH or Lm-OMe-OH) were identified by 1H NMR spectroscopy (>95% recovery, ratios indicated in Scheme 3). Acidic work-up led to the isolation (>98% recovery, NMR) of benzoyl formic acid and benzoic acid (Scheme 3). The different extents of formation of the hydroxylated ligand and BA for 2 vs. 4 indicate differing extents of decarboxylation and more efficient arene hydroxylation for 4, which contains the more electron rich arene group that would be expected to more rapidly trap an electrophilic copper-oxygen species. For the reaction of 4, positive ion ESI-MS data were acquired on the crude brown reaction solution and the recovered ligand residue. Intense peaks with correct m/z and isotope patterns for [(Lm-OMe-O)2Cu2(benzoate)]+ and [Lm-OMe-OCu]+ for the reaction solution and [Lm-OMe-OH + X+] (X = H or Na) for the ligand residue were observed. Critically, when 18O2 was used, the ESI-MS data for these species showed that one 18O atom was incorporated into the ligand and one into the benzoate,18 consistent with expectation for a reaction pathway akin to that drawn for Fe in Scheme 1.22
Scheme 3.
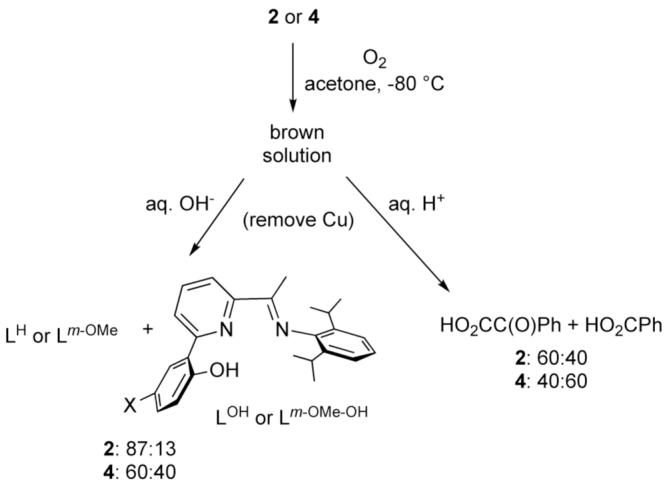
Reactivity of 2 and 4 with O2.
Computed mechanisms at the M06L density functional level and including singlet-state energy corrections based on CASPT2 calculations for the reaction of model system 2′ with oxygen are shown in Scheme 4 and, more fully, Figures S11 and S12.21 Addition of O2 delivers a complex (“O2 adduct”) that adopts either of two energetically similar structures: a singlet structure with a side-on bound O2 fragment, analogous in character to Cu(III)-peroxo species supported by diketiminate ligands3e or a triplet state featuring end-on O2 coordination (shown). After migration of one O atom of the O2 fragment to the keto carbon of the ketoacid, loss of CO2 to generate a singlet Cu(I)-peracid complex (“Peracid”) is predicted to occur with a gas-phase free energy of activation of 15.4 kcal/mol. Decarboxylation is predicted overall to be exergonic by about 34 kcal/mol.
Scheme 4.
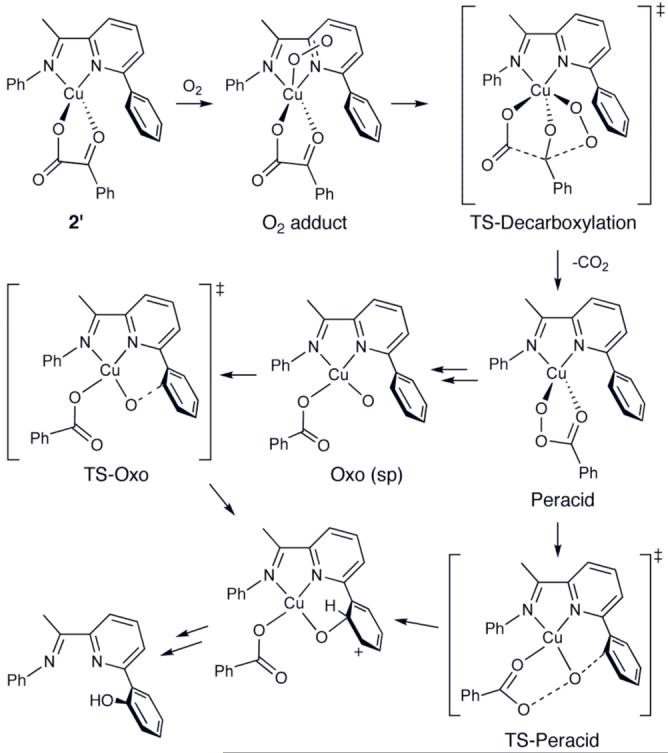
Calculated mechanisms for O2-induced decarboxylation and arene substituent hydroxylation of model 2′.
Two pathways leading to oxygenation of the 2-pyridyl benzene ring from this peracid complex were characterized. In the first, a singlet transition-state structure involving oxidation of the ligand by the peracid itself was located (“TS-Peracid”); this structure features O-O and O-C bond distances of 2.47 and 2.38 Å, respectively, and a Cu-O bond distance of 1.72 Å, so it appears very “oxo-like” in character. In the second pathway, initial cleavage of the peracid O-O bond led to a stable trigonal bipyramidal copper-oxo intermediate (shown in Figure S11), which isomerizes to a square planar isomer (“Oxo (sp)”), both of which are best described as triplet Cu(II) oxyl species.1e,8 The square planar intermediate is the reactive species in the hydroxylation reaction, which proceeds through a triplet transition-state structure (“TS-Oxo”) having Cu-O and O-C bond lengths of 1.84 and 1.93 Å, respectively. Computationally, both paths lead to the same triplet oxygenated cyclohexadienyl cation product, with the latter pathways predicted to be more accessible than the former (highest activation free energies of 10.4 and 17.8 kcal/mol relative to the peracid intermediate, respectively).21 We note, however, that additional kinetic constraints associated with spin crossings between the singlet and triplet surfaces may permit either or both pathways to contribute to the observed reactivity. Such crossings have been noted in prior studies of Cu(II) oxyl reactivity8 and α-ketoglutarate-dependent nonheme iron reactivity15 and their general relevance to iron-based oxidations has been demonstrated.10d-g Accounting for spin-crossing barriers within the context of transition-state theory has been discussed,23 and while a detailed kinetic analysis of this effect is of interest, it goes beyond the scope of this communication. [We note that we have computed the relevant spin-orbit coupling matrix elements between the singlet and triplet states for the two TS structures originating from the singlet peracid intermediate. They have values of about 350 cm-1 in each case; values of this magnitude indicate that spin crossing should be reasonably efficient along either of the two paths.]
In summary, a series of copper(I)-α-ketocarboxylate complexes have been prepared and shown to exhibit variable coordination modes of the α-ketocarboxylate ligand. Reaction with O2 induces decarboxylation of this ligand and the derived copper-oxygen intermediate(s) has(have) been intercepted, resulting in hydroxylation of an arene substituent on the supporting N-donor ligand. The results show that the oxidative decarboxylation pathway for substrate oxidations by iron(II)-α-ketocarboxylate species in enzymes and model complexes14,15 can be extended to copper analogs. Theoretical calculations have yielded intriguing mechanistic notions for the process, notably implicating hydroxylation pathways that involve novel [CuI-OOC(O)R] and [CuII-O-· ↔ CuIII=O2-]+ species. Future research will focus on identifying these species experimentally, using them to oxidize exogenous substrates, and differentiating between the postulated hydroxylation reaction pathways using theoretical and experimental methods.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM47365 to W. B. T.), the National Science Foundation (CHE06-10183 to C. J. C.), the Swiss National Science Foundation (200021-111645/1 to L. G.), and the DAAD (fellowship to S. H.) for financial support of this research.
REFERENCES
- (1)(a).Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]; (b) Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (c) Klinman JP. Chem. Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; (d) Smirnov VV, Brinkley DW, Lanci MP, Karlin KD, Roth JP. J. Mol. Cat. A. 2006;251:100–107. [Google Scholar]; (e) Decker A, Solomon EI. Curr. Opin. Chem. Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]; (f) Solomon E, Sarangi R, Woertink J, Augustine A, Yoon J, Ghosh S. Acc. Chem. Res. 2007;40:581–591. doi: 10.1021/ar600060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Arends I, Gamez P, Sheldon RA. Adv. Inorg. Chem. 2006;58:235–279. [Google Scholar]
- (3)(a).Selected recent reviews:Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z.Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r.Hatcher L, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4.Itoh S. Curr. Opin. Chem. Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012.Cramer CJ, Tolman WB. Acc. Chem. Res. 2007;40:601–608. doi: 10.1021/ar700008c.Suzuki M. Acc. Chem. Res. 2007;40:609–617. doi: 10.1021/ar600048g.
- (4)(a).Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J. Am. Chem. Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]; (b) Kamachi T, Kihara N, Shiota Y, Yoshizawa K. Inorg. Chem. 2005;44:4226–4236. doi: 10.1021/ic048477p. [DOI] [PubMed] [Google Scholar]; (c) Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg. Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]; (d) Evans JP, Ahn K, Klinman JP. J. Biol. Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]; (e) Chen P, Solomon EI. J. Am. Chem. Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- (5).Arguments against a [CuO]+ acting as the oxidant in this enzyme are summarized in ref. 4d and the following:Klinman JP. J. Biol. Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200.
- (6).Balasubramanian R, Rosenzweig A. Acc. Chem. Res. 2007 doi: 10.1021/ar700004s. ASAP. [DOI] [PubMed] [Google Scholar]
- (7)(a).For examples, see:Kitajima N, Koda T, Iwata Y, Moro-oka Y. J. Am. Chem. Soc. 1990;112:8833–8839.Capdevielle P, Sparfel D, Baranne-Lafont J, Cuong NK, Maumy M. J. Chem. Soc., Chem. Commun. 1990:565–566.Reinaud O, Capdevielle P, Maumy M. J. Chem. Soc., Chem. Commun. 1990:566–568.Maiti D, Lucas HR, Sarjeant AAN, Karlin KD. J. Am. Chem. Soc. 2007;129:6998–6999. doi: 10.1021/ja071704c.
- (8).Schroder D, Holthausen MC, Schwarz H. J. Phys. Chem. B. 2004;108:14407–14416. [Google Scholar]
- (9).Gherman BF, Tolman WB, Cramer CJ. J. Comp. Chem. 2006;27:1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]
- (10)(a).For example, see:Que L., Jr. Acc. Chem. Res. 2007;40:493–500. doi: 10.1021/ar700024g.Krebs C, Galonic DF, Fujimori D, Walsh C, Bollinger J. Acc. Chem. Res. 2007;40:484–492. doi: 10.1021/ar700066p.Nam W. Acc. Chem. Res. 2007;40:522–531. doi: 10.1021/ar700027f.Shaik S, Hirao H, Kumar D. Acc. Chem. Res. 2007;40:532–542. doi: 10.1021/ar600042c.Schroder D, Shaik S, Schwarz H. Acc. Chem. Res. 2000;33:139–145. doi: 10.1021/ar990028j.Shaik S, Kumar D, deVisser SP, Altun A, Thiel W. Chem. Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j.Harvey JN, Poli R, Smith KM. Coord. Chem. Rev. 2003;238-239:347–361.Anastasi AE, Comba P, McGrady J, Lienke A, Rohwer H. Inorg. Chem. 2007;46:6420–6426. doi: 10.1021/ic700429x.Bautz J, Comba P, de Laorden CL, Menzel M, Rajaraman G. Angew. Chem. Int. Ed. 2007;46:8067–8070. doi: 10.1002/anie.200701681.Kryatov SV, Rybak-Akimova EV, Schindler S. Chem. Rev. 2005;105:2175–2226. doi: 10.1021/cr030709z.
- (14)(a).Costas M, Mehn MP, Jensen MP, Que L., Jr. Chem. Rev. 2005:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (b) Abu-Omar MM, Loaiza A, Hontzeas N. Chem. Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]; (c) Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem. Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]; (d) Purpero V, Moran GR. J. Biol. Inorg. Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- (15)(a).Borowski T, Bassan A, Siegbahn PEM. Inorg. Chem. 2004;43:3277–3291. doi: 10.1021/ic035395c. [DOI] [PubMed] [Google Scholar]; (b) Bassan A, Borowski T, Siegbahn PEM. Dalton Transactions. 2004:3153–3162. doi: 10.1039/B408340G. [DOI] [PubMed] [Google Scholar]
- (16)(a).Examples of related arene substituent hydroxylations using mononuclear complexes have been reported. For cases proposed to involve a [CuO]+ intermediate, see refs. 7c and d, and for examples involving other types of intermediates, see:Kunishita A, Teraoka J, Scanlon JD, Matsumoto T, Suzuki M, Cramer CJ, Itoh S. J. Am. Chem. Soc. 2007;129:7248–7249. doi: 10.1021/ja071623g.Mehn MP, Fujisawa K, Hegg EL, Que L. J. Am. Chem. Soc. 2003;125:7828–7842. doi: 10.1021/ja028867f.Jensen MP, Lange SJ, Mehn MP, Que EL, Que L., Jr. J. Am. Chem. Soc. 2003;125:2113–2128. doi: 10.1021/ja028478l.
- (17).For a copper(II)-promoted ortho-hydroxylation of 2-phenylpyridine proposed to involve a single electron transfer mechanism, see:Chen X, Hao XS, Goodhue CE, Yu JQ. J. Am. Chem. Soc. 2006;128:6790–6791. doi: 10.1021/ja061715q.
- (18).For details, see supporting information.
- (19)(a).Bianchini C, Mantovani G, Meli A, Migliacci F, Laschi F. Organometallics. 2003;22:2545–2547. [Google Scholar]; (b) Bianchini C, Lenoble G, Oberhauser W, Parisel S, Zanobini F. Eur. J. Inorg. Chem. 2005:4794–4800. [Google Scholar]
- (20).Holland PL, Rodgers KR, Tolman WB. Angew. Chem. Int. Ed. 1999;38:1139–1142. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1139::AID-ANIE1139>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- (21).(a) M06L:Zhao Y, Truhlar DG. J. Chem. Phys. 2006;125:194101. doi: 10.1063/1.2370993.(b) CASPT2:Andersson K, Malmqvist P-Å, Roos BO. J. Chem. Phys. 1992;96:1218–1226.(c) See supporting information for additional details and references.
- (22).Compounds 1 and 3 react with O2 only upon warming, and only unperturbed ligand and the α-ketoacid were isolated (no oxidative decarboxylation). Compound 5 also was unreactive with O2, suggesting that involvement of a peroxo- or bis(oxo)dicopper species derived from reaction of the LCu(I) fragment of 2 or 4 with O2 is unlikely to be involved in the process(es) that yields hydroxylated ligand.
- (23).Harvey JN. Phys. Chem. Chem. Phys. 2007;9:331–343. doi: 10.1039/b614390c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.