

Abstract
As part of an ongoing effort to develop biocompatible, biodegradable conducting polymers, we report here the synthesis and characterization of a novel copolymer, 5,5"'bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene-co-adipic acid polyester (QAPE). This system was designed so as to incorporate alternating electroactive quaterthiophene units and biodegradable ester units into one macromolecular framework, while allowing for facile preparation of the polymer via a polycondensation reaction. In agreement with the design expectations, the ester groups were found to be incorporated into the polymer between the quaterthiophene subunits, as inferred from standard chemical and spectroscopic analyses. QAPE exhibited redox activity as detected by cyclic voltammetry and a new red-shifted absorption peak upon doping, providing support for the notion that the quaterthiophene units maintain electroactivity after incorporation into the QAPE polymer framework. The degradation, likely through surface erosion, of this polymer in the presence of cholesterol esterase was confirmed by the detection of a fluorescence signal at wavelengths corresponding to the quaterthiophene subunit and comparisons to appropriate controls. In vitro cytocompatability studies, carried out over 48 h, indicate that the QAPE polymer is nontoxic to Schwann cells.
Keywords: Conducting polymer, electroactive, biodegradable, surface erosion, biocompatible, cytocompatible, polyester, quaterthiophene
Introduction
Electrically conducting polymers (CPs), discovered in the late 1970's, have received tremendous attention because they combine the attractive properties of traditional polymers with those of metals and other inorganic conductors, including optical and electrical activity.1-5 They have been studied in the context of microelectronics applications (reviewed in Gurunathan et al., 19996) and increasingly have been explored for their use in the development of biomedical engineering applications.7-34 This latter research direction has led to a new focus on CPs.
One reason CPs have attracted the attention of biomedical engineers is the discovery that many cell types (e.g., neurons, osteoblasts, fibroblasts) respond to electrical currents in vitro and in vivo.35-39 This led to the consideration that rationally designed conducting scaffolds could play a role in tissue engineering. This interest was further bolstered when polypyrrole (PPy), an extensively studied CP, was found to be biocompatible in vitro and in vivo in rats,13,40,41 and when it was further demonstrated that adhesion, proliferation, and differentiation of endothelial cells34 and pheochromocytoma cells33 seeded on PPy could be controlled in vitro through electrical stimulation of PPy. Given these findings, other CPs, including polythiophene (PT), polyaniline (PANI), and polyethylenedioxythiophene (PEDOT), have been explored for their applicability to tissue regeneration and they too were found to possess properties similar to, and in some cases more favorable than, those of PPy.10,19,26,28,42-48 Despite the many positive attributes of CPs, including conductivity, biocompatibility, ability to entrap and controllably release biomolecules, capacity to undergo reversible doping, and their potential ease of modification, one inherent limitation remains their inability to undergo facile biodegradation in vivo.
Here it is worth mentioning that the term biodegradation, when referring to a biomaterial in vivo in common parlance, includes any process that results in dematerialization or biomaterial decomposition and which ultimately leads to loss of material integrity. Biodegradation can occur at the surface (i.e., erosion) or throughout the bulk of the material. Biodegradation mechanisms include chemical bond scission (e.g., hydrolysis, enzymatic degradation) to create smaller fragments of the original material and solubilization of the material at the material-tissue interface. In spite of the all-encompassing nature of this definition, few CP-based materials are biodegradable. There have only been a few attempts to generate biodegradable CPs. Two notable attempts include the synthesis of surface erodible oligopyrroles49 and PPy nanoparticle-polylactide (PLA) composites.41 In neither case is the CP backbone itself subject to chemical-based biodegradation. Rather, in the former example the CP is designed to be slightly water soluble and have a molecular weight that is low enough to allow renal clearance; this means that chemical degradation is necessarily less of an issue, although careful control over molecular weight distribution during synthesis is imperative. In contrast, in the PLA-based approach, the amount of PPy in the biomaterial is minimized obviating to some extent the need for degradability; nevertheless, the small amounts of PPy introduced into the body as a result of using this material would be expected to remain in vivo indefinitely.
Rivers et al.50 explored an alternative approach to achieving degradability. In this study, the polymer backbone was itself designed to be both chemically degradable and conductive. The particular system prepared by these researchers, a biodegradable electrically conducting polymer (BECP), consists of a copolymer of pyrrole-thiophene-pyrrole oligomers with alternating ester linkers. BECP was found to undergo degradation in vitro through esterase cleavage of the ester bonds.50 However, it proved impossible to dope this particular polymer with anything but iodine, which is toxic to cells, and which resulted in a very low conductivity. In a study geared towards understanding oligothiophene conductivity for electronic applications, Miller et al. provided evidence suggesting that it is possible to generate oligothiophene-based polyester systems that have higher conductivities.51 However, these polymers were also doped with iodine. Further, their biodegradability was never explored. Thus, there remains a need for copolymers that are optimized to be conducting and biodegradable, as well as biocompatible.
In this paper we present the design, synthesis, and characterization of 5,5"'-bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene-co-adipic acid polyester (QAPE). We present evidence that suggests this new system is surface erodible and electroactive, as inferred from the finding that it can undergo redox events to generate doped species. We also show that this material is cytocompatible with Schwann cells. We therefore believe it marks an important step forward in the search for biodegradable electroactive materials suitable for tissue repair applications.
Experimental Section
Materials
All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise specified. Organic solvents were anhydrous and were used immediately following their collection from a catalyst-based drying device, except dichloroethane (DCE) and dimethylsulfoxide (DMSO). These latter solvents were stored over molecular sieves and used when needed.
Methods
The final polymer, consisting of desired conductive and degradable subunits units, was generated in a five-step synthetic sequence as detailed below. All reactions were performed in flame-dried glassware under dry argon atmosphere. Reaction solutions were taken to dryness on a rotary evaporator under reduced pressure. The purification methods employed varied depending on the intermediate product and are specified accordingly. Intermediate products were characterized by 1H NMR (400 MHz Varian Mercury spectrometer), chemical ionization mass spectroscopy (triple quadrupole mass spectrometer) or electrospray ionization mass spectroscopy (ESI MS) (LCQ), UV-vis spectroscopy (UV-vis-NIR 5000 spectrophotometer Varian, Cary series), and, in the case of dimethyl quaterthiophene and bishydroxymethyl-dimethyl quaterthiophene (bMdMQT), Fourier transform infrared (FT-IR) spectroscopy (Perkin Elmer Spectrum BX). Polymer synthesis was confirmed using ESI MS, FT-IR spectroscopy, UV-vis spectroscopy, and 1H and 13C NMR spectroscopy (500 mHz Varian Mercury spectrometer). Characterization of the degradation temperature (Td) and melting temperature (Tm) of the bMdMQT and QAPE were performed under an atmosphere of nitrogen using thermogravimetric analysis (Mettler Toledo TGA/SDTA851e) at a scan rate of 20°C/min from 25 - 450°C and differential scanning calorimetry (Mettler Toledo, DSC823e) at a scan rate of 10°C/min cycling between -50 - 200°C five times, respectively. Elemental analyses were performed by Midwest Microlabs, LLC, Indianapolis, IN. The electroactivity of QAPE was assessed using UV-vis spectroscopy and cyclic volatammetry (CV). Degradation studies were performed in vitro in the presence of cholesterol esterase (CE), whereas cytocompatibility was determined in vitro in the presence of Schwann cells using the Live/Dead® viability/cytotoxicity assay and CellTiter-Blue® cell viability assay.
2-Bromo-magnesio-3-methylthiophene (1)
This compound was synthesized according to literature procedures.52,53 In brief, a solution of 2 bromo-3-methylthiophene (785 mg, 4.43 mmol) in anhydrous diethyl ether (1 ml) under argon was added drop-wise to a solution containing one chip of I2, magnesium (108 mg, 4.43 mol), and diethyl ether (1 mL). The mixture was left to stir overnight at room temperature under argon. The resulting Grignard reagent was used immediately without further purification.
3,3"'-Dimethyl-2,2':5',2":5",2"'-quaterthiophene (2)
This compound was synthesized according to literature procedures.52,53 In brief, under argon 2-bromo-magnesio-3-methylthiophene 1 (745.7, 3.7 mmol) prepared as above was added dropwise to a solution of 5,5'-dibromo-bithiophene (606 mg, 1.85 mmol) and NiCl2(dppp) (16.2 mg; 0.03 mmol) in diethyl ether (52 mL). The reaction mixture was stirred at room temperature for 4-5 h as monitored by thin layer chromatography. The mixture was quenched with water (85 mL) and then extracted with diethyl ether (5 × 100 mL). The ether fraction was dried over anhydrous sodium sulfate, filtered, dried using a rotary evaporator, and then dried under vacuum. The crude product 2 obtained in this way was further purified by column chromatography (dry packed column; silica gel; hexanes eluent), giving a yellow powder after removal of solvent (575 mg, 86.6% yield). 1H NMR (CDCl3) δ 2.42 (6H, s, CH3), 6.88 (2H, d), 7.04 (2H, d), 7.12 (2H, d), 7.14 (2H, d). UV-vis, λmax. (DCM): 381 nm. IR (KBr): ν = 614, 692, 790, 831, 1065, 1447 (alkane C-C), 2852 (alkane C-H), 2918 (alkane C-H), 3073 (aromatic C-H) cm-1.
5,5"'-Diformyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene (3)
Under argon, phosphorous oxychloride (4.4 mL, 48.12 mmol) was slowly added to a solution of dry dimethylformamide (3.73 mL, 48.12 mmol) and DCE (7 mL) pre-cooled to 0°C. This mixture was allowed to stir for 30 min and warm to room temperature after which point it was added dropwise to a solution of 3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene 2 (575.11 mg, 1.604 mmol) in dichloroethane (73 mL). The reaction mixture was then heated to 80°C and allowed to undergo reflux overnight. After cooling, the reaction was quenched via the addition of an aqueous solution of NaOH (2 M; 160 mL) and allowed to stir for 30 min at room temperature. The product was then extracted with dichloromethane (5 × 100 mL), subject to preliminary drying using a rotary evaporator, and then subsequently dried under vacuum. The crude product 3 obtained in this way was further purified using column chromatography (dry packed column; alumina; DCM/hexanes (50:50) eluent) to give an orange powder (501.1 mg, 75% yield). 1H NMR (CD2Cl2) δ 2.49 (6H, s, CH3), 7.29 (4H, dd), 7.58 (2H, s), 9.82 (2H, s). UV-vis, λmax. (DCM): 414 nm. Anal. Calcd for C20H14O2S4: C, 57.94; H, 3.40; O, 7.72; S, 30.94. Found: C, 57.80; H, 3.45; O, 7.89, S, 30.91.
5,5"'-Bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene (bMdMQT) (4)
Under argon, a solution of 5,5"'-diformyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene 3 (364.7 mg, 0.88 mmol) in dry tetrahydrofuran (THF) (750 mL) was dissolved at 50°C and then degassed for 30 min. NaBH4 (83.2 mg, 2.2 mmol) was added to the reaction mixture, which was then stirred at 60°C for 2 h. The solvent was removed on the rotary evaporator and the product was sonicated in water (80 mL) for 30 min, filtered, and dried under vacuum overnight to give a yellow-orange powder in quantitative yield. 1H NMR (CDCl3) δ 2.36 (6H, s, CH3), 4.77 (4H, d), 6.81 (2H, s), 7.03 (2H, d), 7.12 (2H, d). 13C NMR (400 MHz, DMSO-d6) δ (ppm) = 15.44, 58.26, 124.80, 126.06, 128.46, 128.72, 133.63, 135.09, 135.32, 144.66. UV-vis, λmax. (THF): 409 nm. FT-IR (KBr): ν = 760, 852, 1009, 1170, 1455 (alkane C-C), 2930 (alkane C-H), 3382 (O-H) cm-1. Td = 335°C. Tm = 160 - 170°C. Anal. Calcd for C20H18O2S4: C, 57.38; H, 4.33; O, 7.64; S, 30.64. Found: C, 57.54; H, 4.47; O, 7.71, S, 30.33.
5,5"'-Bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene-co-adipic acid polyester) (QAPE) (5)
Under argon, a solution of freshly distilled adipoyl chloride (0.139 mL, 0.955 mmol) in dry THF (1.3 mL) was added to 5,5"'-bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene 4 (400 mg, 0.955 mmol) in dry pyridine (7 mL) at 0°C. The reaction mixture was slowly warmed to room temperature and then heated to 100°C and held at reflux for 6 h or 24 h. Pyridine was removed in vacuo and the product was washed with water (4 × 5 mL). The resulting polymer was purified by dissolution in THF and reprecipitation from methanol to give a brown powder (322.7 mg). 1H NMR (DMSO-d6) δ 1.55 (m, CH2), 2.31 (m, CH3), 2.34 (m, CH2), 4.56 (m, CH2), 5.17 (m, CH2), 6.70 (m, CH=), 7.15 (m, CH=), 7.31 (m, CH=). 13C NMR (DMSO-d6) δ 15.9, 24.5, 24.6, 33.7, 33.8, 34.0, 60.5, 125.6, 125.7, 127.3, 133.6, 134.4, 135.3, 136.2, 136.8, 173.2, 175.0. ESI MS (-): 543.33, 673.34, 1073.23, 1090.89, 1156.07, 1203, 1218.70, 1490.66, 1618.55. High resolution ESI MS (-): 545.0591 ± 0.002 (C26H25O5S4-1. 673.1059 ± 0.002 (C32H33O8S4-1). UV-vis, λmax. (THF): 398 nm. UV-vis, λmax. (acetonitrile/THF/FeCl3): 412 nm and 595 nm. FT-IR (KBr): ν = 1440 (alkane C-C stretch, ring C-C stretch), 1698 (C-O-C=O), 2924 (alkane C-H), 3056 (aromatic C-H), 3401 (O-H) cm-1. Td = 170 - 190°C. Tm = 142°C.
Film Preparation
All films or coatings were applied to glass coverslips of 1.2 cm in diameter, which had been cleaned previously in 7 M nitric acid solution and autoclaved. Solutions of bMdMQT, 4.0 mg/mL (9.56 mM), and QAPE, 3.1 mg/mL (5.93 mM, based on monomer molecular weight), were made in DMSO. Films were solvent cast by evenly applying 20.0, 30.0, or 100.0 μL of bMdMQT or QAPE solution onto coverslips. This generated 0.17, 0.25, or 0.85 μmol/cm2 bMdMQT and 0.11, 0.14, or 0.52 μmol/cm2 QAPE-coated surfaces. Solvent cast films were dried in a vacuum oven for 24 h at 25°C and then washed three times in 1 mL of fresh water with slight agitation for 10 min each. Slides were allowed to dry in a laminar flow hood for at least 2 h. For the CellTiter-Blue® cell viability assay, films were placed in the wells of 24 multiwell plates. For the Live/Dead® viability/cytotoxicity assay, films were sandwiched between a polydimethylsulfoxide well (1 cm in diameter) and a microscope glass slide using silicone grease as an adhesive. In both cases, films were sterilized under UV light for 30 min.
Poly-L-lysine (PLL)-coated coverslips were prepared as standards for cell viability assays. Coverslips were placed in a 24-multiwell plate and 400 - 500 μL PLL (50 μg/mL) was dispensed to cover the 1.2 cm diameter coverslips in the well. These were allowed to sit for 1 h. The PLL solution was then aspirated off and the coverslips were rinsed twice with 1 mL sterile water. PLL-coated coverslips were air dried overnight in a tissue culture hood. Prior to use, the coverslips were sterilized under UV light for 30 min.
Degradation Assay
In vitro biodegradability of the polymer by esterases was evaluated by incubating the polymer films in 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (HEPES) (pH~7) with or without cholesterol esterase (CE) (Lot #077K4005; 1.47 U/mg solid as determined by Sigma-Aldrich) in solution at 37°C under conditions of gentle agitation. QAPE (0.14 μmmol/cm2) films were prepared as indicated above and were first washed in sterile deionized water for 24 h at 37°C. QAPE films were either treated with a solution of CE (25 units) in HEPES (0.5 mL) or simply HEPES (0.5 mL). Both experimental and control samples were prepared in triplicate. In addition, two background controls were established, both with n = 2; the first involved the addition of CE (25 units) in HEPES (0.5 mL), whereas the second involved HEPES (0.5 mL) alone. All solutions contained 1% BSA, as this has been found to enhance CE activity54 and 0.1% sodium azide to maintain sterility over the extended period of the experiment. Samples were gently agitated at 37°C for up to 10 days. At each time point (every 2 days), 200 μL aliquots of the solutions from all wells were placed in a clean multiwell plate and analyzed by fluorescence spectroscopy (ex: 360/20 nm; em: 460/40 nm) using a BioTek plate reader. Solutions were then returned to their appropriate wells to allow continuation of the experiment. Prior to returning the multiwell plate to the incubator, experimental wells and CE background control wells were supplemented with additional CE, 10 units in 37 μL HEPES. HEPES controls were supplemented with 37 μL HEPES alone. Fluorescence at the designated excitation and emission wavelengths was utilized to detect the presence of bMdMQT and other QAPE degradation products containing bMdMQT in solution.
Dopant Optimization
UV-vis spectroscopy was used to follow changes in the absorbance spectrum of QAPE in THF (3.78 × 10-5 mmol/mL) as varying quantities of either FeCl3 or Fe(ClO4)3 solutions were added. The molar ratio of FeCl3 to QAPE was varied by adding 150, 300, 450, 600, 750, 900, 1050, or 1100 μL aliquots of FeCl3 in acetonitrile (1.07 × 10-3 mmol/mL) to 600 μL solutions of QAPE in THF (3.78 × 10-5 mmol/mL). This generated samples with FeCl3:QAPE ratios of 7, 14, 21, 28, 42, 49, or 56, respectively. Similarly, the molar ratio of Fe(ClO4)3 to QAPE was varied by adding 25, 50, 100, 150, 300, 450, or 900 μL aliquots of Fe(ClO4)3 in acetonitrile (1.07 × 10-3 mmol/mL) to 600 μL solutions of QAPE in THF (3.78 × 10-5 mmol/mL). This produced samples with Fe(ClO4)3:QAPE ratios of 1.2, 2.4, 4.7, 7, 14, 21, or 42. Solutions were mixed briefly and analyzed 1.167 min following addition.
X-ray Photoelectron Spectroscopy (XPS)
XPS was carried out on a Kratos AXIS Ultra XPS system employing a monochromatic Al Kα1 source. The binding energy of the instrument was calibrated using Au 4f7/2, Ag3d5/2 and Cu2p3/2 at 84, 368.3 and 932.6 eV, respectively. Typical operating conditions were: 1×10-9 Torr chamber pressure; 15 kV; and 150 W for the Al X-ray source. High-resolution elemental scans were collected with a pass energy of 20 eV using takeoff angles of 90 degrees between the sample and the analyzer.
Cyclic Voltammetry
The electroactivities of QAPE and bMdMQT were characterized by cyclic voltammetry (CV). CV was performed using a CHI627C Electrochemical Workstation (CH Instruments, Inc.) and a standard three-electrode set-up consisting of a platinum mesh counter electrode, indium tin oxide glass slide working electrode, and a silver wire reference electrode. Ferrocene (5 mM) was added as an internal standard. QAPE (0.43 mg/mL) or bMdMQT (0.41 mg/mL) was added to a 50 mM solution of tetrabutylammonium perchlorate in DMSO and the potential was swept from -1.8 V to 1.8 V vs. Ag with a scan rate of 100 mV/s for 5 cycles.
Water Contact Angle
The hydrophobicity of the QAPE films was assessed via water contact angle measurements made using a Dektak 6M goiniometer and a FTA 32 video camera. Measurements were made after a water droplet was applied to the surface for 30 sec. All measurements were performed in triplicate.
Cell Culture
Primary rat Schwann cells (purchased from Sciencell Research Laboratories or isolated from P4 postnatal rat pups using Brocke's method55) were used for in vitro studies. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (HyQ-DMEM-RS, Hyclone), 10% fetal bovine serum (Hyclone), forskolin (2 μM), and bovine pituitary extract (3 μg/mL) on PLL-coated tissue culture polystyrene dishes. Cells were maintained at 37°C in a humid atmosphere of 5% CO2 and were fed every 3-4 days and passaged every 6-7 days. Cells used for experimental work were from passages 4-6 and were grown in medium plus serum and supplements (as indicated above) with the addition of 1% penicillin/streptomycin/amphotericin B solution (Sigma-Aldrich).
Cell viability was qualitatively assessed with the Live/Dead® viability/cytotoxicity assay (Invitrogen) as detailed below. This assay consists of two dyes: calcein AM, which stains live cells fluorescent green (ex/em ~495 nm/~515 nm); and ethidium homodimer-1, which stains the nuclei of dead cells fluorescent red (ex/em ~495 nm/~635 nm). Cells were seeded onto bMdMQT (0.254 μmol/cm2), QAPE (0.135 μmol/cm2), and PLL-coated coverslips at a density of 25,000 cells in 300 μL medium per well. Cells were allowed to adhere and proliferate for 48 h in a tissue culture incubator, after which cells were treated with live/dead stain and imaged using phase contrast and fluorescence microscopy (Olympus IX70 inverted microscope).
Viability was quantified using CellTiter-Blue®, a fluorescence-based cell viability assay (Invitrogen). This assay was performed at 4 h and 48 h on two types of films: 0.847 μmol/cm2 bMdMQT-coated coverslips and 0.524 μmol/cm2 QAPE-coated coverslips. As indicated above, all coverslips were individually placed in the wells of a 24-well culture plate. A standard curve was prepared for both time points by seeding cells on PLL-coated coverslips at 4 different cell densities: 30,000, 15,000, 7500, 3750 cells in 400 μL (n = 3). All experimental films were seeded with 15,000 cells in 400 μL (n = 3). Controls consisted of 400 μL medium only (no cells) with experimental or PLL-coated coverslips or without any coated coverslips (n = 2). Cells incubated for 4 h and 48 h were treated with 80 μL of cell titer blue reagent for 3 h and 30 min, respectively, prior to measuring the fluorescence using a BioTek fluorescence plate reader.
Results and Discussion
Design Considerations
The 5,5"'-bishydoxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene-co-adipic acid polyester (QAPE) of this study was designed to incorporate alternating electroactive quaterthiophene subunits and potentially biodegradable ester groups. Conduction is predicted to occur via both intra- and inter-oligomer chain electron hopping.56-58 As previously mentioned, there is some evidence that such conduction can occur through oligothiophenes within a polyester framework.51,59 The choice of quaterthiophenes as the conducting subunits was based on the well-known conductivity and oxidative stability of oligothiophenes.2,42,60-62 Quaterthiophene oligomers were targeted instead of terthiophene subunits or higher order thiophene oligomers so as to balance the need for conductivity with ease of synthesis. Although it was considered likely that longer oligothiophene subunits would be more easily oxidized and doped, thereby improving conductivity, it was also expected that higher order oligothiophenes would prove harder to prepare. Additionally, it was appreciated that such species would display relatively poor solubility, which would impact processibility. Although this latter problem could presumably be overcome by increasing the extent of thiophene alkylation, an overly high degree of alkylation was expected to lead to perturbations in the planar conformation of the conjugated system and therefore decreased conductivity. In the present instance, the use of quaterthiophene-bearing methyl substituents at the 3 and 3"' positions was expected to produce an appropriate balance between ease of synthesis, organic solubility, and undesirable reductions in electroactivity (i.e., increase in the band gap). Such expectations are in agreement with published studies53 and are corroborated by UV-vis spectroscopic studies (see supplementary information). Finally, by functionalizing the quaterthiophene subunit with hydroxyl substituents, ester linkages could be incorporated to form a polyester material, thus introducing key hydrolysable subunits.
Based on previous findings demonstrating the esterase-catalyzed degradation of a number of ester-based unnatural substrates,50,63-66 it was expected that an ester-based polymer would be biodegradable. The biological relevance of esterase, specifically cholesterol esterase (CE), has been reported, as well.67-69 CE and other esterases are naturally occurring in mammals and have been found in most animal tissues, including the pancreas, liver, muscle, and brain.67,70 More significantly, CE is produced by mononuclear phagocytic cells (i.e., macrophages and monocytes), which are up-regulated during the inflammatory response that accompanies invasive procedures, such as scaffold implantation.69,71-74 In addition, the use of ester linkages would allow the degree of quaterthiophene-quaterthiophene separation to be controlled, although this particular modification has not been extensively pursued by the authors.a The choice of adipoyl chloride-derived linkers reflected a need to balance polymer solubility with a quaterthiophene-quaterthiophene separation that was not so large as to preclude conductivity. Ultimately, the synthetic pathway developed permits flexibility in design, facilitating changes in the length of the ester linker and the conductive unit to allow for future optimization of properties, such as conductivity, hydrophobicity, and degradation rate.
Synthesis of QAPE
The synthesis of QAPE 5 was readily accomplished in a small number of high yielding steps, as shown in Scheme 1. The first two steps were performed according to literature and consisted of making the methyl thiophene Grignard reagent 1, which was then coupled to dibromobithiophene in a nickel-catalyzed reaction to form quaterthiophene 2.52,53 Subjecting the alpha-free quaterthiophene 2 to Vilsmeier formylation followed by reduction produced the bismethoxy-dimethyl quarterthiophene (bMdMQT) 4. QAPE 5 was then synthesized, according to a modified literature procedure50, via a polycondensation reaction between bMdMQT 4 and adipoyl chloride in pyridine at reflux. The use of pyridine was found to promote polymerization, presumably by sequestering released HCl. The resulting crude product was purified by reprecipitation to remove unreacted starting material, monomers, and pyridine hydrochloride.
Scheme 1.

5,5"'-bishydroxymethyl-3,3"'-dimethyl-2,2',5',2",5",2"'-quaterthiophene quaterthiophene-co-adipic acid polyester synthesis.
Spectroscopic analysis of QAPE provided support for the expected ester bond formation, as well as for successful polymer formation. For instance, comparison of the 1H NMR spectra of bMdMQT 4 and QAPE 5 (Figure 1) revealed a downfield shift, from δ 4.55 ppm to δ 5.25 ppm, in the allylic proton signal for QAPE, a finding that is consistent with increased deshielding of these protons as a result of ester bond formation. FT-IR analyses also revealed energy absorption at 1730 cm-1 in the FT-IR spectrum of QAPE but not that of bMdMQT. This signal is consistent with an ester carbonyl stretching mode, whereas the smaller IR adsorption between 1650-1700 cm-1 is ascribed to a carboxylic acid carbonyl stretching mode. These findings provide evidence in support of the conclusion that condensation of bMdMQT with adipoyl chloride proceeds as expected and likely gives rise to a species containing terminal carboxylic acid groups (Figure 2).
Figure 1.

1H NMR of A) 5,5"'- bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene (QAPE); B) 5,5"'- bishydroxymethyl-3,3"'-dimethyl-2,2':5',2":5",2"'-quaterthiophene-co-adipic acid polyester. Preservation of the aromatic signals (a, b, and c) is seen between 6.8 and 7.4 ppm and a downfield shift of the allylic protons (e) is observed in the NMR spectrum of QAPE.
Figure 2.

Infrared spectra of QAPE (black) and infrared spectra bMdMQT (grey). Peaks characteristic of bMdMQT are also seen in the IR spectrum of QAPE; however, there is the appearance of a new peak at ~1730 cm-1, indicating the presence of C=O stretch.
While providing support for the presence of ester bonds, neither the 1H NMR nor FT-IR spectroscopic analyses provide direct insights into the nature of the condensation products. In particular, they do not allow assignments of chain lengths; nor do they provide evidence for whether the putative polymeric products are linear or cyclic. 13C NMR spectroscopic studies lead us to suggest that the majority of the polymeric species are linear; this is because such analyses reveal the presence of two different carbonyl carbon signals between δ 173 - 175 ppm, a finding that would correlate well with the presence of ester bonds and terminal carboxylic acid carbons. However, it is still possible that a mixture of linear and cyclic species is present.
Because the QAPE polymer formed after 24 h reaction proved only sparingly soluble in most organic solvents, and to better gage the progression of chain growth, the polymerization reaction was stopped after 6 h and subject to analysis by electrospray mass spectrometry (ESI MS). Mass peaks were observed at approximately 543, 673, 1073, 1091, 1156, 1203, 1218, 1490, and 1618, correlating well with the formation of several singly and doubly charged linear oligomeric species and doubly charged dimer species (as is often the case with ESI, some of these species were accompanied by a sodium ion). High resolution ESI revealed peaks at 545.0591 ± 0.002 and 673.1059 ± 0.002 amu, as would be expected for products based on a 1:1 and 1:2 condensation between bMdMQT and adipoyl chloride. On this basis, we propose that the majority of QAPE produced during the polymerization process is linear and that as the reaction proceeds the length of the oligomeric chains increases.
It is important to note that all analyses are consistent with the conjugation of the quaterthiophene subunits being retained, something that is considered necessary to impart electroactivity to the polymer. The 1H NMR signals corresponding to aromatic protons of bMdMQT are also observed in the 1H NMR of QAPE (Figure 1), although the peaks are broader for the polymer than for the precursor, as would be expected for a macromolecular species. In addition, IR absorbance at 3065 cm-1 (aromatic C-H stretch) and 2940 and 1440 cm-1 (alkane C-H and C-C stretching, respectively) are seen in QAPE's spectrum, that correlate well with the corresponding signals in the IR spectrum of bMdMQT (Figure 2). Finally, the finding that bMdMQT and QAPE display nearly identical UV-vis spectra, with an absorption maximum at ~387 ± 4 nm in THF, is further support for the retention of thiophene ring integrity following polymerization.
Molecular weight characterization of the QAPE samples produced after a reaction time of 24 h was rendered difficult owing to the poor solubility of the presumably higher molecular weight polymer chains produced under these conditions. However, preliminary analysis of the refractive index of the QAPE formed in this way revealed a dn/dc value of 0.110-0.141 in THF, which was used in conjunction with GPC coupled with light scattering and viscosity measurements to determine MWn, MWw, and PDI (23,482-37,864 Da, 38,108-71,173 Da, and 1.62-1.88, respectively). Analysis of the molecular weights in DMF-LiBr (0.01 M LiBr) using GPC with polystyrene as a reference generated much lower molecular weight values (MWw ~ 6000 Da), as is often found when comparing results from these two GPC systems.75
Biodegradation Properties of QAPE
Although the ester bonds were designed into QAPE to render it biodegradable and, as previously indicated, there is precedence for the biodegradation of polyesters, it was considered important to explore the susceptibility of the final quaterthiophene-based polyester to degradation. Therefore, to investigate QAPE degradation films were incubated in HEPES at 37°C in the presence (experimental) and absence (control) of CE in vitro over a period of 10 days. Degradation products were expected to consist of bMdMQT, adipic acid, and small oligomers containing bMdMQT. Given that the spectral properties of bMdMQT (ex~400 nm; em~470 nm) are distinct from those of the initial buffered solution components (i.e., sodium azide, CE), fluorescence analysis of samples permitted the rate of release of QT-containing species to be monitored readily. (Note: Both the CE and HEPES controls revealed a minimal level of fluorescence background, as would be expected given the composition of these controls.) After the initial 2 days of incubation the fluorescence of experimental solutions was 2.25 times greater than that of the controls and after 10 days experimental solutions were 1.66 times more fluorescent than control solutions. This would indicate an increased release of QT-based species into solution in the presence of CE (Figure 3). The decrease in the rate of release of QT-based species after 2 days may correlate with the fact that enzyme activity is lost over 24 - 48 h and degradation solutions were supplemented every 2 days with 2.5x less enzyme than originally added on day 0. The detection of a fluorescence signal in the absence of enzyme (controls) at all time points indicates that either non-enzymatic hydrolysis and/or material solubilization are possible. Also, only a slightly enhanced fluorescence signal was seen in the experimental solutions compared to the corresponding control solutions; this may indicate that either or both of these decomposition methods could account for the degradation seen in the presence of CE. In other words, it is not clear at present whether enzymatic degradation, hydrolysis, or solubilization, or some combination thereof, is the dominant factor in the biodegradation of QAPE; however, chemical degradation and solubilization would generate the same desired effect, namely the decomposition and clearance of the material in vivo. Thus, these results considered in conjunction with previous studies of CE in biological milieus, lead us to propose that QAPE would prove bioerodible were it administered in vivo.
Figure 3.

UV-vis analysis of the supernatants of control and experimental degradation samples. Absorbance at 385 nm, indicates the presence of bMdMQT and bMdMQT oligomers.
Doping and Electroactiviy of QAPE
Although the presence of ester groups has the likely benefit of imparting bioerodability to QAPE, as noted above, two concerns were: 1) That the presence of ester linkers would preclude effective doping, and 2) the specific choice of adipic acid-derived linkers might increase the inter-quarterthiophene separation and the ratio of insulating to conducting backbone to the point that intra- and inter-chain electron transfer would be reduced to an unacceptably low level. To address the first of these critical concerns, the ability to dope QAPE was explored by subjecting it to ferric chloride (FeCl3) or ferric perchlorate (Fe(ClO4)3). These oxidants/dopants were selected because the counter anions in question, Cl- and ClO -4, are considered biocompatible counteranions,76,77 while any residual iron should result in minimal toxicity.41,78,79 There are several studies that suggest Cl--doped heterocyclic polymers are less stable, compared to polymers doped with less nucleophilic anions;80-83 this motivated us to explore the oxidation and doping of QAPE with Fe(ClO4)3, as well as with FeCl3.
Oxidation and concurrent doping of QAPE in THF in the presence of FeCl3 or Fe(ClO4)3 in acetonitrile was inferred from the observation of 1) a visible color change in the polymer solution from orange/yellow to blue/green, 2) a slight red-shift from ~398 nm to ~412 nm in the absorbance of QAPE and the appearance of new low energy absorbance peaks between 550 nm and 700 nm in the UV-vis spectrum, and 3) an augmented chlorine content relative to that of sulfur, as determined by the XPS analysis of washed doped QAPE samples. The emergence of new low energy bands at ~561 nm (2.2 eV), ~589 nm (2.1 eV), and ~681 nm (1.82 eV) are of particular interest. Not surprisingly, similar low energy band transitions have been observed in both PT and oligothiophene systems following doping.84-87 Specifically, these spectral observations correlate well with the formation of oligothiophene-derived radical cations (i.e., polaron states), which are generally characterized by an absorption signal between 500-600 nm (i.e., in our case ~589 nm) and either the presence of dimers of these radical cations or dicationic species (i.e., bipolarons), both of which are expected to absorb at a smaller and larger wavelength than that characteristic of the radical cation (i.e., in our case at 561 nm and 681 nm).84,85,88
Against this background, efforts were made to determine the effects of dopant concentration on polaron formation within QAPE. More specifically, the dopant concentration necessary to achieve the maximum absorbance of the band assigned to the presence of radical cations was considered to define the potentially optimal doping conditions (i.e., those that were expected to maximize the QAPE conductivity). It was found that as the molar equivalents of FeCl3 or Fe(ClO4)3 were increased with respect to those of QAPE (based on monomer molecular weight) the intensity of the low energy peak at 589 nm increased until what we assumed was the optimal molar ratio of ferric ion oxidant/dopant to QAPE was achieved; after this point, further additions led to a decrease in the absorption intensity of this peak and presumably a loss in conductivity. We hypothesize that as oxidation and doping of the polymer backbone increases, the number of polaron lattice deformations, characterized by absorption at a lower energy wavelength (i.e., ~589 nm), also increases. The corresponding proportional absorption increases of the maximum at ~589 nm, the shoulder at 562 nm, and the secondary maximum at 681 nm lead us to suggest that the radical cations coexist with dimers of these radical species, rather than bipolarons, formed from the radical cations.84 The eventual decrease of the low energy signals is thought to be associated with either the insolubility of aggregated oxidized QAPE or an overoxidation of the quaterthiophene units. Several studies indicate that the process of overoxidation leads to either one or both of the following events in the case of PPy, namely 1) crosslinking between backbone chains (β to β or α to β), and 2) incorporation of hydroxyl groups that subsequently become oxidized to form carbonyl groups at the β-pyrrolic positions.89-92 Both events ultimately, lead to a loss of conjugation and with it conductivity.93 By extension of this analogy, overoxidation of QAPE would lead to a disruption of its polaron bands resulting in a reduction in the absorption intensity at red-shifted (low energy) wavelengths observed in the UV-vis spectrum.
Based on the above criterion, it was found that it took approximately 7 molar equivalents of Fe(ClO4)3 (relative to moles of QAPE monomer) but ca. 42 molar equivalents of FeCl3 to achieve a level of maximal doping. In addition, at the molar ratios deemed optimal the absorption intensity at 589 nm was ~1.5 times greater, in the case of Fe(ClO4)3, than when FeCl3 was used (Figure 4). This correlates well with our earlier prediction that ClO -4 doping would be more effective and result in higher conductivities. XPS analysis of Fe(ClO4)3 doped QAPE under optimal conditions indicated a doping level of 8.1% as determined by the ratio of chlorine to sulfur (a value that correlates directly with the number of perchlorate anions present per thiophene ring).
Figure 4.

A) The effect of the ratio of molar equivalents of FeCl3 (grey) or Fe(ClO4)3 (black) to (monomer) molar equivalents of QAPE is inferred from the presence of absobance signals at 589 nm. Optimal molar ratios are indicated by an asterisk (*). B) UV-vis spectra of QAPE undoped (dashed), QAPE with optimal molar equivalents of FeCl3 (grey), and QAPE with optimal molar equivalents of Fe(ClO4)3 (black).
The electroactivity (inferred by redox activity) of QAPE was evaluated using cyclic voltammetry (CV) in the presence of ClO -4. CVs of QAPE and bMdMQT in DMSO using tetrabutylammonium perchlorate as the electrolyte indicate that both systems are endowed with similar redox properties and therefore similar electroactivity. There appear to be three redox events. An oxidation event appears to occur at ~1.4 for both bMdMQT and QAPE and two reduction events are also observed. The first reduction peak (at ~ -0.9 V) is essentially the same for both bMdMQT and QAPE; however, the second reduction event is slightly shifted from ~ -1.5 V for bMdMQT to ~ -1.9 V for QAPE, a finding that can likely be attributed to the presence of the insulating aliphatic ester groups. These events do not appear to be particularly reversible since corresponding redox potentials are barely visible in the case of bMdMQT (~ -1.03 and ~ -1.46 V) and are not observable for QAPE (Figure 5). The E1/2 of ferrocene, used as an internal standard in this system, was measured at ~ 770 mV. Initial attempts to quantify the conductivity of the doped QAPE samples using a 4-point probe proved unsuccessful. This disappointing finding is believed to reflect a combination of low doping levels, heterogeneous solvent-cast samples, and poor sample electrode contact. Although the conductivity of QAPE films still needs to be quantified, CV and UV-Vis spectroscopy results indicate that QAPE can undergo redox chemistry and energy bands associated with doped polaron states are observable. Given this form of electroactivity, QAPE may have the ability to pass a current, although further studies are necessary to confirm this. It is hoped that even if conductivity were low the material would still be able to pass low current, which at the nanoscale is potentially relevant for nerve regeneration because it may be sufficient to affect cell behavior in a salutary manner.
Figure 5.

Cyclic voltammagram of bMdMQT and QAPE (~0.4 mg/1 mL DMSO) in 50 mM tetrabutylammonium perchlorate vs. Ag. The counter electrode was platinum and the working electrode was a cleaned indium tin-oxide coated glass slide. Potentials -2.0 V to 1.9 V were scanned at 100 mV/s.
Cytocompatibility of QAPE
The biocompatibility of materials to be used in biomedical applications is critical; hence, in the first steps towards assessing biocompatibility the cytocompatability of QAPE and one of its putative degradation products, bMdMQT, was investigated in the presence of Schwann cells. The selection of this in vitro model reflects our long-term interest in developing biodegradable electroactive scaffolds for peripheral nerve regeneration. Cell viability and adhesion on QAPE and bMdMQT films were evaluated and compared to that on PLL control surfaces. It was observed that cells adhered more slowly on QAPE and bMdMQT films. This was possibly a result of the fairly hydrophobic nature of these films (QAPE water contact angle ≈114°). However, it is possible that the rough topography of bMdMQT and QAPE films (as a result of solvent-casting) and protein adsorption to the surface may have countered the initial hydrophobic effect of these materials since after 48 h cells were observed to adhere and spread just as well on these two quaterthiophene-containing surfaces as on PLL surfaces. In addition, following an incubation time of 48 h, a live/dead stain provided a qualitative indication that the viability of cells grown on the bMdMQT and QAPE films was similar to that seen for the PLL controls (Figure 6). As a result of the cytocompatibility observed qualitatively in the Live/Dead assay, bMdMQT and QAPE films made from higher concentration solutions than those used in the Live/Dead assay were used to assess cytocompatibility quantitatively using the CellTiter-Blue® assay. Results from this assay corroborate the results from the Live/Dead assay and permit quantification of cell viability. According to this latter analysis, the Schwann cell viability after 4 h was nearly 100% on both bMdMQT and QAPE films relative to that on the PLL controls. After 48 h, cell viability was maintained at 75-100% of that of the controls (Figure 7).
Figure 6.
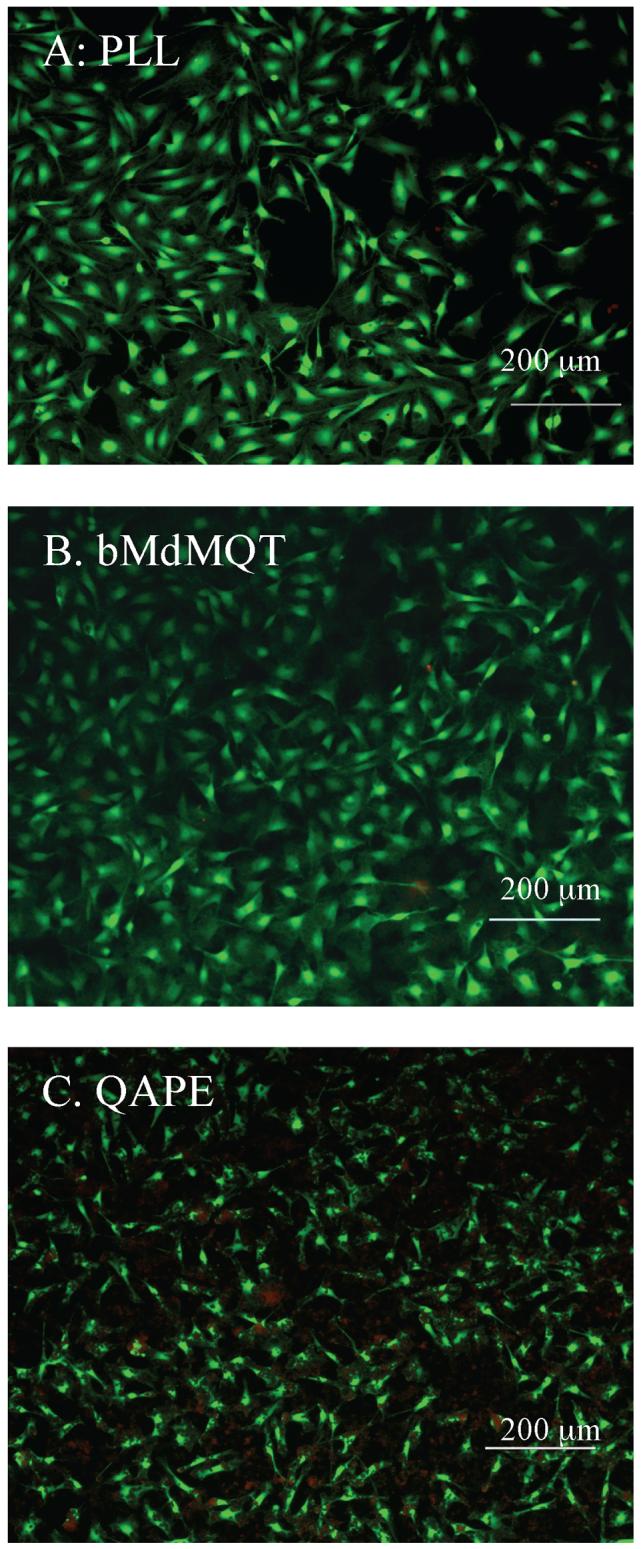
Live/Dead stain of Schwann cells after 48 h of growth on A) PLL, B) bMdMQT, and C) QAPE-coated glass coverslips. Living cells fluoresce green and dead cells fluoresce red. Images were taken individually, with appropriate bandpass filters and subsequently merged. Scale bar 200 μm.
Figure 7.

Cell titer blue cell viability assay after 4 h and 48 h of Schwann cell growth on bMdMQT and QAPE solvent-cast coverslips. After 4 h, cell viability was close to 100% of that for the PLL controls and after 48 h cell viability decreases only slightly.
In vitro compatibility studies have demonstrated the short-term cytocompatibility of QAPE, leading us to suggest that explorations of its long-term compatibility in vitro and in vivo are now warranted to fully assess biocompatibility. In addition, these preliminary results have motivated our efforts to further optimize and characterize QAPE and to develop new QAPE analogues that could result in the creation of a degradable electroactive polymer suitable for use in biomedical applications, including those associated with nerve regeneration.
Conclusion
A novel cytocompatible, degradable, moderately electroactive polymer, QAPE, has been successfully synthesized. Erosion at the surface of QAPE films begins within one to two weeks of exposure to CE. Doping may be effected by treatment with ferric perchlorate, as inferred from XPS analyses, while electroactivity was confirmed using both UV-vis analysis and CV in the presence of perchlorate. Based on these findings, QAPE appears to show promise as a scaffold material for tissue engineering applications, which require biodegradable, biocompatible scaffolds which are inherently conducting (electrically active) when doped with nontoxic anions. However, in spite of the promise of the present QAPE polymer, we feel it is appropriate to develop new QAPE analogues that might be more readily doped, prove equally or more stable in the doped state, display higher conductivity or demonstrate improved degradation properties. Towards this end, we are exploring the use of oligothiophene subunits of greater size (sexithiophene) and diacids other than adipic acid.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Christopher W. Bielawski and his group for the use of their polymer characterization equipment and Christine Smid for her assistance in up-scaling the synthesis of the polymer intermediates. Financial support from National Institute of Health (EB004529 to C.E.S. and CA68682 to J.L.S.) is gratefully acknowledged. We would also like to acknowledge the National Science Foundation, grant number 0618242, for funding the installation of the X-ray photoelectron spectroscopy facility.
Footnotes
Preliminary efforts to synthesize the malonic acid analogue of QAPE are underway. ESI MS characterization of the oligomer species generated in analogy to those reported here is consistent with the coupling between malonic acid and the bMdMQT being successful, leading us to propose that other dicarboxylic acid ester linkers can be incorporated into QAPE in the place of adipic acid.
References
- (1).Heegar AJ. In: Handbook of Conducting Polymers. Skotheim TA, editor. Vol. 2. Marcel Dekker; New York: 1986. pp. 729–756. [Google Scholar]
- (2).Heegar AJ. Angew. Chem. Int. Ed. 2001;40:2591–2611. [PubMed] [Google Scholar]
- (3).Heeger AJ. Synth. Met. 2002;125:23–42. [Google Scholar]
- (4).Shirakawa H, Louis EJ, MacDiarmid AG, Chiang CK, Heeger AJ. J. Chem. Soc. Chem. Commun. 1977:578–580. [Google Scholar]
- (5).Street GB, Clarke TC. IBM J. Res. Develop. 1981;25:51–57. [Google Scholar]
- (6).Gurunathan K, Murugan AV, Marimuthu R, Mulik UP, Amalnerkar DP. Mat. Chem. Phys. 1999;61:173–191. [Google Scholar]
- (7).Abidian MR, Kim D-H, Martin DC. Adv. Mater. 2006;18:405–409. doi: 10.1002/adma.200501726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Abidian MR, Martin DC. Biomaterials. 2008;29:1273–1283. doi: 10.1016/j.biomaterials.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Collier JH, Camp JP, Hudson TW, Schmidt CE. Journal of biomedical materials research. 2000;50:574–584. doi: 10.1002/(sici)1097-4636(20000615)50:4<574::aid-jbm13>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- (10).Cui X, Martin DC. Sens. Actuators, B. 2003;89:92–102. [Google Scholar]
- (11).Cui X, Wiler J, Dzaman M, Altschuler RA, Martin DC. Biomaterials. 2003;24:777–787. doi: 10.1016/s0142-9612(02)00415-5. [DOI] [PubMed] [Google Scholar]
- (12).Guimard NK, Gomez N, Schmidt CE. Prog. Polym. Sci. 2007;32:876–921. [Google Scholar]
- (13).Williams RL, Doherty PJ. J. Mater. Sci: Mater. Md. 1994;5:429–433. [Google Scholar]
- (14).Yang J, Martin DC. J. Mater. Res. 2006;21:1124–1132. [Google Scholar]
- (15).Xiao Y, Martin DC, Cui X, Shenai M. Appl. Biochem. Biotechnol. 2006;128:117–129. doi: 10.1385/abab:128:2:117. [DOI] [PubMed] [Google Scholar]
- (16).Gomez N, Schmidt CE. J. Biomed. Mater. Res., Part A. 2007;81A:135–149. doi: 10.1002/jbm.a.31047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).George PM, LaVan DA, Burdick JA, Chen CY, Liang E, Langer R. Adv. Mater. 2006;18:577–581. [Google Scholar]
- (18).George PM, Lyckmann AW, LaVan DA, Hegde A, Leung Y, Rupali A. Biomater. 2005;26 [Google Scholar]
- (19).Spinks GM, Xi B, Truong V-T, Wallace GG. Synth. Met. 2005;151:85–91. [Google Scholar]
- (20).Smela E. Adv. Mater. 2003;15:481–494. [Google Scholar]
- (21).Deshpande MV, Hall EA. Biosensors Bioelectron. 1990;5:431–448. [Google Scholar]
- (22).Dupont-Filliard A, Billon M, Livache T, Guillerez S. Anal. Chim. Acta. 2004;515:271–277. [Google Scholar]
- (23).Ramanavicius A, Kausaite A, Ramanaviciene A. Sensors Actuators B. 2005;B111-B112:532–539. [Google Scholar]
- (24).Richardson-Burns SM, Hendricks JL, Foster B, Povlich LK, Kim D-H, Martin DC. Biomaterials. 2007;28:1539–1552. doi: 10.1016/j.biomaterials.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Singh S, Chaubey A, Malhotra BD. J. Appl. Polym. Sci. 2004;91:3769–3773. [Google Scholar]
- (26).Singh S, Solanki PR, Pandey MK, Malhotra BD. Sensors Actuators B. 2006;B115:534–541. [Google Scholar]
- (27).Umana M, J W. Anal. Chem. 1986;58:2979–2983. [Google Scholar]
- (28).Chaubey A, Pande KK, Singh VS, Malhotra BD. Anal. Chim. Acta. 2000;407:97–103. [Google Scholar]
- (29).Li M, Guo Y, Wei Y, MacDiarmid AG, Lelkes PI. Biomater. 2006;27:2705–2715. doi: 10.1016/j.biomaterials.2005.11.037. [DOI] [PubMed] [Google Scholar]
- (30).Ateh DD, Vadgama P, Navsaria HA. Tissue Eng. 2006;12:645–655. doi: 10.1089/ten.2006.12.645. [DOI] [PubMed] [Google Scholar]
- (31).Garner B, Georgevich A, Hodgson AJ, Liu L, Wallace GG. J. Biomed. Mater. Res. 1999;44:121–129. doi: 10.1002/(sici)1097-4636(199902)44:2<121::aid-jbm1>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- (32).De Giglio E, Sabbatini L, Colucci S, Zambonin G. J. Biomater. Sci. Polym. Ed. 2000;11:1073–1083. doi: 10.1163/156856200743580. [DOI] [PubMed] [Google Scholar]
- (33).Schmidt CE, Shastri VR, Vacanti JP, Langer R. Proc. Natl. Acad. Sci. USA. 1997;94:8948–8953. doi: 10.1073/pnas.94.17.8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wong JY, Langer R, Ingber DE. Proc. Natl. Acad. Sci. USA. 1994;91:3201–3204. doi: 10.1073/pnas.91.8.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Giaever I, Keese CR. Proc. Natl. Acad. Sci. U S A. 1984;81:3761–3764. doi: 10.1073/pnas.81.12.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kerns JM, Pavkovic IM, Fakhouri AJ, Wickersham KL, Freeman JA. J. Neurosci. Meth. 1987;19:217–223. doi: 10.1016/s0165-0270(87)80005-5. [DOI] [PubMed] [Google Scholar]
- (37).Jaffe LF, Poo MM. J. Exp. Zool. 1979;209:115–128. doi: 10.1002/jez.1402090114. [DOI] [PubMed] [Google Scholar]
- (38).Bassett CA, Pawluk RJ, Becker RO. Nature. 1964;204:652–654. doi: 10.1038/204652a0. [DOI] [PubMed] [Google Scholar]
- (39).Bassett CA, Mitchell SN, Gaston SR. J Bone Joint Surg Am. 1981;63:511–523. [PubMed] [Google Scholar]
- (40).Chen SJ, Wang DY, Yuan CW, Wang XD, Zhang P, Gu XS. J. Mater. Sci. Lett. 2000;19:2157–2159. [Google Scholar]
- (41).Shi G, Rouabhia M, Wang Z, Dao LH, Zhang Z. Biomater. 2004;25:2477–2488. doi: 10.1016/j.biomaterials.2003.09.032. [DOI] [PubMed] [Google Scholar]
- (42).Tourillon G. In: Handbook of conducting polymers. Skotheim TA, editor. Vol. 1. Marcel Dekker; New York: 1986. pp. 293–305. [Google Scholar]
- (43).Waugaman M, Sannigrahi B, McGeady P, Khan IM. Eur. Polym. J. 2003;39:1405–1412. [Google Scholar]
- (44).Yamato H, Ohwa M, Wernet W. J. Electroanal. Chem. 1995;397:163–170. [Google Scholar]
- (45).Welzel HP, Kossmehl G, Engelmann G, Neumann B, Wollenberer U, Scheller F, Schroeder W. Macromol. Chem. Phys. 1996;197:3355–3363. [Google Scholar]
- (46).Gautier C, Cougnon C, Pilard J-F, Casse N. J. Electroanal. Chem. 2006;587:276–283. [Google Scholar]
- (47).Bidez PR, Li S, MacDiarmid AG, Venancio EC, Wei Y, Lelkes PI. J. Biomater. Sci. Polym. 2006;17:199–212. doi: 10.1163/156856206774879180. [DOI] [PubMed] [Google Scholar]
- (48).Wang CH, Dong YQ, Sengothi K, Tan KL, Kang ET. Synth. Met. 1999;102:1313–1314. [Google Scholar]
- (49).Zelikin AN, Lynn DM, Farhadi J, Martin I, Shastri V, Langer R. Angew. Chem,. Int. Ed. 2002;41:141–145. doi: 10.1002/1521-3773(20020104)41:1<141::aid-anie141>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- (50).Rivers TJ, Hudson TW, Schmidt CE. Adv. Funct. Mater. 2002;12:33–37. [Google Scholar]
- (51).Hong Y, Miller LL. Chem. Mater. 1995;7:1999–2000. [Google Scholar]
- (52).Chaloner PA, Gunatunga SR, Hitchcock PB. J. Chem. Soc. Perkin Trans. 1997;2:1597–1604. [Google Scholar]
- (53).Pham CV, Burkhardt A, Shabana R, Cunningham DD, Mark HB, Zimmer H. Phosphorus, Sulfur, and Silicon. 1989;46:153–168. [Google Scholar]
- (54).Nishide T, Sasaki N, Shirai K, Saito Y, Yoshida S. Tohoku J. Exp. Med. 1985;146:123–130. doi: 10.1620/tjem.146.123. [DOI] [PubMed] [Google Scholar]
- (55).Assouline JG, Bosch EP, Lim R. In: A Dissection and Tissue Culture Manual of the Nervous System. Shahar A, de Vellis J, Vernadakis A, Haber B, editors. Alan R. Liss, Inc.; New York: 1989. pp. 247–250. [Google Scholar]
- (56).Chance RR, Bredas JL, Silbey R. Phys. Rev. B: Condens. Matter. 1984;29:4491–4495. doi: 10.1103/physrevb.31.652. [DOI] [PubMed] [Google Scholar]
- (57).Kivelson S. Phys. Rev. B: Condens. Matter. 1982;25:3798–3721. [Google Scholar]
- (58).Kuivalainen P, Stubb H, Isotalo H, Yli-Lahti P, Holmstroem C. Phys. Rev. B: Condens. Matter. 1985;31:7900–7909. doi: 10.1103/physrevb.31.7900. [DOI] [PubMed] [Google Scholar]
- (59).Kunugi Y, Miller LL. Chem. Mater. 1997;9:1061–1062. [Google Scholar]
- (60).Kaneto K, Yoshino K, Inuishi Y. Solid State Commun. 1983;46:389–391. [Google Scholar]
- (61).Chung T-C, Kaufman JH, Heegar AJ, Wudl F. Phys. Rev. B. 1984;30:702–710. [Google Scholar]
- (62).Bredas JL. In: Handbook of conducting polymers. Skotheim TA, editor. Vol. 2. Marcel Dekker; New York: 1986. pp. 859–913. [Google Scholar]
- (63).Wang GB, Labow RS, Santerre JP. J. Biomed. Mater. Res. 1997;36:407–417. doi: 10.1002/(sici)1097-4636(19970905)36:3<407::aid-jbm16>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- (64).Labow RS, Duguay DG, Santerre JP. J. Biomater. Sci. Polym. Edn. 1994;6:169–179. doi: 10.1163/156856294x00293. [DOI] [PubMed] [Google Scholar]
- (65).Santerre JP, Shajii L, Tsang H. J. Dent. Res. 1999;78:1459–1468. doi: 10.1177/00220345990780081201. [DOI] [PubMed] [Google Scholar]
- (66).Woo GL, Mittelman MW, Santerre JP. Biomater. 2001;21:1235–1246. doi: 10.1016/s0142-9612(00)00003-x. [DOI] [PubMed] [Google Scholar]
- (67).Shope RE. J. Biol. Chem. 1928;LXXX:127–132. [Google Scholar]
- (68).Tomita T, Miura S, Chiba T, Mochizuik N, Nemoto K, Tomita I. Annals N.Y. Acad. Sci. 1997;811:471–479. doi: 10.1111/j.1749-6632.1997.tb52028.x. [DOI] [PubMed] [Google Scholar]
- (69).Salthouse TN. J. Biomed. Mater. Res. 1976;10:97–229. doi: 10.1002/jbm.820100204. [DOI] [PubMed] [Google Scholar]
- (70).Brockerhoff, Jensen RG. Lipolytic Enzymes. Academic Press; New York: 1974. [Google Scholar]
- (71).Labow RS, Meek E, Santerre JP. J. Biomed. Mater. Res. 1998;39:469–477. doi: 10.1002/(sici)1097-4636(19980305)39:3<469::aid-jbm18>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- (72).Labow RS, Meek E, Santerre JP. J. Biomed. Mater. Res. 2001;54:189–197. doi: 10.1002/1097-4636(200102)54:2<189::aid-jbm5>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- (73).Li F, Hui DY. J. Biol. Chem. 1997;272:28666–28671. doi: 10.1074/jbc.272.45.28666. [DOI] [PubMed] [Google Scholar]
- (74).Lindhorst E, Young D, Bagshaw W, Hyland M, Kisilevsky R. Biochim. Biophys. Acta. 1997;1339 doi: 10.1016/s0167-4838(96)00227-0. [DOI] [PubMed] [Google Scholar]
- (75).De Boer B, van Hutten PF, Ouali L, Grayer V, Hadziioannou G. Macromolecules. 2002;35:6883–6892. [Google Scholar]
- (76).Li Y, Neoh KG, Cen L, Kang ET. Langmuir. 2005;21:10702–10709. doi: 10.1021/la0514314. [DOI] [PubMed] [Google Scholar]
- (77).Wang X, Gu X, Yang C, Chen S, Zhang P, Zhang T, Yao J, Chen F, Chen G. J. Biomed. Mater. Res. 2004;68A:411–422. doi: 10.1002/jbm.a.20065. [DOI] [PubMed] [Google Scholar]
- (78).Zhang Z, Rouabhia M, Wang Z, Roberge C, Shi G, Roche P, Li J, Dao LH. Artificial Organs. 2007;31:13–22. doi: 10.1111/j.1525-1594.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- (79).Jiang X, Marois Y, Traoré A, Tessier D, Dao LH, Guidoin R. Tissue Eng. 2002;8:635–647. doi: 10.1089/107632702760240553. [DOI] [PubMed] [Google Scholar]
- (80).Chen CC, Rajeshwar K. J. Electrochem. Soc. 1994;141:2942–2946. [Google Scholar]
- (81).Alumaa A, Hallik A, Sammelselg V, Tamm J. Synth. Met. 2007;157:458–491. [Google Scholar]
- (82).Qi Z, Rees NG, Pickup PG. Chem. Mater. 1996;8:701–707. [Google Scholar]
- (83).Masuda H, Asano DK, Kaeriyama K. Synth. Met. 1997;84:209–210. [Google Scholar]
- (84).Hill MG, Mann KR, Miller LL, Penneau J-F. JACS. 1992;114:2728–2730. [Google Scholar]
- (85).Kaufman JH, Colaneri N, Scott JC, Street GB. Phys. Rev. Lett. 1984;53:105–108. [Google Scholar]
- (86).Sato M.-a., Hiroi M. Polymer. 1996;37:1685–1689. [Google Scholar]
- (87).Caspar JV, Ramamurthy V, Corbin DR. JACS. 1991;113:600–610. [Google Scholar]
- (88).Colaneri N, Nowak M, Spiegel D, Hotta S, Heegar AJ. Phys. Rev. B. 1987;36:7964–7968. doi: 10.1103/physrevb.36.7964. [DOI] [PubMed] [Google Scholar]
- (89).Ge H, Qi G, Kang ET, Neoh KG. Polymer. 1994;35:504–508. [Google Scholar]
- (90).Debiemme-Chouvy C, Tran TTM. Electrochem. Commun. 2008;10:947–950. [Google Scholar]
- (91).Palmisano F, Malitesta C, Centonze D, Zambonin PG. Anal. Chem. 1995;67:2207–2211. [Google Scholar]
- (92).Ghosh S, Bowmaker GA, Cooney RP, Seakins JM. Synth. Met. 1998;95:63–67. [Google Scholar]
- (93).Otero TF, Marquez M, Suarez IJ. J. Phys. Chem. B. 2004;108:15429–15433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.