

Abstract
Replication protein A (RPA) is a highly conserved single-stranded DNA-binding protein, required for cellular DNA replication, repair, and recombination. In human cells, RPA is phosphorylated during the S and G2 phases of the cell cycle and also in response to ionizing or ultraviolet radiation. Saccharomyces cerevisiae exhibits a similar pattern of cell cycle-regulated RPA phosphorylation, and our studies indicate that the radiation-induced reactions occur in yeast as well. We have examined yeast RPA phosphorylation during the normal cell cycle and in response to environmental insult, and have demonstrated that the checkpoint gene MEC1 is required for the reaction under all conditions tested. Through examination of several checkpoint mutants, we have placed RPA phosphorylation in a novel pathway of the DNA damage response. MEC1 is similar in sequence to human ATM, the gene mutated in patients with ataxia-telangiectasia (A-T). A-T cells are deficient in multiple checkpoint pathways and are hypersensitive to killing by ionizing radiation. Because A-T cells exhibit a delay in ionizing radiation-induced RPA phosphorylation, our results indicate a functional similarity between MEC1 and ATM, and suggest that RPA phosphorylation is involved in a conserved eukaryotic DNA damage-response pathway defective in A-T.
Eukaryotic cells contain a heterotrimeric single-stranded DNA-binding protein, replication protein A (RPA), first characterized as a necessary component of the cell-free simian virus 40 (SV40) DNA replication system (1–3). Like its prokaryotic counterparts, RPA functions to destabilize the double helix during DNA replication, thereby permitting the parental DNA strands to serve as templates for DNA synthesis. Despite this functional similarity, RPA is structurally more elaborate than the homomeric prokaryotic single-stranded DNA-binding proteins. While the large subunit of RPA contains the single-stranded DNA-binding activity, the holoprotein is required for the initiation of DNA synthesis in the SV40 system. Antibodies directed against any single RPA subunit inhibit DNA replication in vitro (4–7), and studies in yeast have indicated that the three genes encoding the RPA subunits are essential for viability (8, 9). Therefore, it is likely that all three subunits are involved directly in cellular DNA replication, but the functional organization of RPA remains largely unresolved.
In addition to its complex quaternary structure, RPA exists as a phosphoprotein. In both human and yeast cells, the middle subunit of RPA (RPA2) is phosphorylated during the S and G2 phases of the cell cycle (10). A similar reaction occurs during the course of SV40 DNA replication in human cell extract, and the pattern of this phosphorylation resembles that of the cell cycle-regulated reaction (11). Phosphorylation of RPA2 is also induced upon exposure of human cells to either ionizing or ultraviolet (UV) radiation (12, 13). We report here that both the ionizing and UV radiation-induced reactions occur in yeast, demonstrating that DNA damage-mediated RPA phosphorylation is also conserved among eukaryotes. As yet, the functions of RPA phosphorylation during normal growth and in response to genotoxic stress are unknown.
We previously established that the DNA-activated protein kinase (DNA-PK) is required for RPA phosphorylation during SV40 DNA replication in vitro (14). The catalytic subunit of this enzyme (DNA-PKc) is also required for V(D)J recombination and DNA double-strand break repair, and mutation of the mouse DNA-PKc gene results in the severe combined immune deficiency defect (15–18). Our studies indicated that immunodepletion of DNA-PK from human cell extract abolishes DNA replication-dependent RPA phosphorylation without affecting replication activity. Therefore, it is unlikely that the analogous cell cycle-regulated phosphorylation reaction is involved directly in the mechanics of DNA replication. An alternative possibility is that RPA phosphorylation during the normal cell cycle is involved in a signaling pathway, perhaps coordinating DNA replication with other cell cycle events (14). It is significant that DNA-PKc is homologous to ATM, the gene mutated in patients with ataxia-telangiectasia (A-T) (19, 20). This disease results in a diverse array of characteristic phenotypes, including cerebellar degeneration, immunodeficiency, and increased risk of certain cancers (for review, see ref. 21). These defects are thought to result at least partially from defects in checkpoint signaling pathways that arrest cell cycle progression in response to DNA damage (22, 23).
An interesting correlation was revealed by examination of radiation-induced RPA phosphorylation in A-T cells. While UV radiation-induced RPA2 phosphorylation appears normal in these cells, the ionizing radiation-induced reaction is delayed (12). It is not clear whether this phenomenon relates directly to the A-T disease mechanism, but the specificity of this defect matches the specific sensitivity of A-T cells to ionizing radiation (21, 24). There is also evidence that A-T cells do not efficiently accumulate the tumor suppressor p53 following ionizing radiation exposure during the G1 phase of the cell cycle (25, 26). Thus, it is possible that RPA and p53, which physically interact (27, 28), both have a role in the biochemical pathway defective in patients with A-T.
In addition to DNA-PKc, ATMp homologues from several species have been identified, all of which share a putative phosphatidylinositol 3 (PI 3)-kinase domain at the carboxyl terminus (for review, see ref. 29). Mammmalian PI 3-kinase is involved in signal transduction (for review, see ref. 30), and it is possible that ATMp catalyzes lipid phosphorylation to transduce a checkpoint signal in response to genotoxic stress. As yet, enzymatic activities associated with ATMp have not been identified; however, PI 3-kinase activity has not been detected in purified preparations of DNA-PK (19). Mammalian PI 3-kinase also contains a protein kinase activity that requires the consensus PI 3-kinase domain (31), and it has been proposed that members of the “PI 3-kinase” family involved in the DNA damage response, such as ATMp and DNA-PK, are exclusively protein kinases (32). Therefore, it is conceivable that ATMp, like its homologue DNA-PKc, can directly phosphorylate RPA (14), a possibility supported by the defect in RPA phosphorylation characteristic of A-T cells.
To further explore the function of RPA phosphorylation, we chose to investigate the genetically tractable and well-defined yeast, Saccharomyces cerevisiae. Although DNA-PK activity has not been detected in yeast cells, the significant homology of Mec1p with both DNA-PKc and ATMp (19, 20, 33) suggested a potential role for Mec1p in yeast RPA phosphorylation. In addition to its checkpoint functions in response to both DNA damage and the delay of DNA synthesis, MEC1 is essential for viability and is required for DNA repair and recombination (33–35). We report here that phosphorylation of yeast RPA during the normal cell cycle requires MEC1. Furthermore, we show that RPA2 phosphorylation induced by exposure of yeast cells to either ionizing or UV radiation also requires MEC1. Through examination of several checkpoint mutants, we have placed RPA phosphorylation in a novel branch of the checkpoint pathway downstream of MEC1. The results presented support a functional relationship between ATM and MEC1, and suggest that RPA phosphorylation is involved in a DNA damage-response pathway conserved among eukaryotes and defective in patients with A-T.
MATERIALS AND METHODS
Strains and Plasmids.
The strains employed in this study are listed in Table 1. For most experiments, cultures were grown in rich medium (YPD) at 30°C. However, for experiments that included the temperature-sensitive TWY312 (mec2-1), cultures were grown at 25°C. In addition, strains harboring plasmid were grown in a synthetic complete medium lacking uracil.
Table 1.
Strains used in this study
| Strain | Genotype | Ref. |
|---|---|---|
| TWY397 | MATa ura3 his7 leu2 trp1 | 34, 36 |
| TWY308 | MATα mec1-1 ura3 trp1 | 34 |
| DLY285 | MATa mec1-1::HIS3 ura3 his3 leu2 trp1 | 35 |
| TWY312 | MATa mec2-1 ura3 his7 trp1 | 34 |
| TWY316 | MATa mec3-1 ura3 his3 trp1 | 34 |
| TWY398 | MATa rad9Δ::LEU2 ura3 his7 leu2 trp1 | 34, 36 |
| YDM937 | MATa mec1-1 tel1Δ1::HIS3 ura3-52 his3Δ200 leu2Δl lys2-801 trp1Δ1 | 37 |
Plasmids were derived from pRS316 (ARS/CEN, URA3) (38), which also served as a control vector. pDM197, which contains TEL1, was described (37), and the MEC1-containing plasmid pDM207 was constructed in a similar fashion.
Native Extract Preparation and Phosphatase Treatment.
Exponentially growing TWY397 cells (15 ml) were harvested by centrifugation at 4°C. The pellet was washed with 10 ml ice-cold water and the cells were once again collected by centrifugation. The pellet was resuspended in 0.2 ml lysis buffer (50 mM Hepes, pH 7.8/2 mM EDTA/2 mM DTT/20% glycerol/0.2 mM phenylmethylsulfonyl fluoride). An equal volume of acid-washed glass beads (425–600 μm; Sigma) was added, and the cells were disrupted by vortexing as described (10). The supernatant was collected after a 5-min centrifugation at 1900 × g (4°C), frozen with liquid nitrogen, and stored at −80°C.
Native extract was incubated for 30 min at 30°C in 50 mM Tris·HCl, pH 7.8/5 mM DTT/2 mM MnCl2 in the presence or absence of λ protein phosphatase (400 units; New England Biolabs) and the phosphatase inhibitor sodium vanadate (5 μM). RPA2 was analyzed by Western blot analysis as described below.
Cell Synchronization.
Cultures were adjusted to 2.5 × 106 cells per ml and incubated for 2 hr at 30°C. α mating factor (Sigma) was then added to a final concentration of 2.5 μM, and the cultures were incubated for an additional 2 hr at 30°C. The cells were collected by centrifugation at room temperature, washed with water (room temperature, volume commensurate with original culture volume), resuspended in warmed (30°C) YPD, and returned to 30°C.
Hydroxyurea (HU) Treatment.
Exponentially growing cells were treated with 0.1 M HU (final concentration) for 2 hr. An equal volume of water was added to control samples. Flow cytometry (see below) was used to verify inhibition of DNA synthesis. For synchronous release into HU, cells were arrested in G1 with α factor as described above, washed with water, and resuspended in warmed (30°C) YPD containing 0.1 M HU.
Ionizing and UV Irradiation.
Cells to be irradiated were first arrested in G1 with α factor (see above). For ionizing irradiation, the arrested cells were chilled to 4°C, harvested by centrifugation, and then resuspended to the original culture volume in fresh ice-cold YPD containing 2.5 μM α factor. The cultures were then irradiated with a Shepard Mark I 137Cs irradiator at 0.67 krad/min, and subsequently incubated at growth temperature (25 or 30°C) for 1 hr. For UV irradiation, the arrested cells were chilled to 4°C, harvested by centrifugation, and resuspended in ≈0.5 ml of the supernatant. The cell suspensions were spread onto 10-ml YPD plates containing α factor (80 μl of a 125-μM solution applied 1–2 hr prior to irradiation), and either mock- or UV-irradiated at 60 J/m2 with a Stratagene UV Stratalinker 2400. The plates were then incubated at 25°C for 1 hr, and cells were harvested from the plates with ice-cold water.
RPA2 Analysis by Western Blotting.
For most of the experiments presented, yeast RPA2 (36 kDa) was analyzed in denatured whole cell extracts. Cells from 0.5–2.0 ml of culture were collected by centrifugation, washed with 1 ml ice-cold water, and stored at −80°C. The cells were resuspended in Laemmli sample loading buffer and broken by three cycles of freezing (liquid nitrogen, 2 min) and heating (≈100°C, 2 min). Cellular debris was removed by a 5-min centrifugation at full speed in a microcentrifuge (room temperature), and aliquots of the denatured extract were analyzed by SDS/PAGE. Proteins were separated on a 12% resolving gel (150:1 acrylamide:bis-acrylamide) and transferred to nitrocellulose (0.2 μm, Schleicher & Schuell) in 10 mM caps-NaOH, pH 11/10% methanol. The nitrocellulose membrane was incubated with rabbit antiserum directed against RPA2 at a 1:5,000 dilution followed by horseradish peroxidase-linked donkey anti-rabbit antibody (Amersham) at 1:10,000. Antibodies were diluted in phosphate-buffered saline containing 0.3% Tween 20 and 5% nonfat dry milk, and incubations were carried out for 1–2 hr at room temperature. Immunoreactive protein was detected using the enhanced chemiluminescence method (Amersham).
Flow Cytometry.
Preparation of cells for flow cytometry was carried out as described (35), with minor modifications. Samples were analyzed for DNA content using a Becton Dickinson FACScan and cellquest software.
RESULTS
MEC1 Is Required for Cell Cycle-Regulated RPA Phosphorylation.
Exponentially dividing wild-type yeast cells contain a significant level of RPA2 that migrates with reduced mobility during denaturing gel electrophoresis (10) (Fig. 1A). Treatment of crude extract with phosphatase converts the two bands into a single species, indicating that the reduced mobility of RPA2 is due to phosphorylation (Fig. 1A). In marked contrast to wild type, the mec1-1 mutant contains little if any phosphorylated RPA2 as measured by this assay (Fig. 1B, lane 1). This difference in RPA2 phosphorylation is also observed when cells are released from G1 arrest and allowed to proceed synchronously through the cell cycle. While RPA2 is phosphorylated in wild-type cells during S and G2, as previously reported (10), RPA2 in the mec1-1 mutant remains unphosphorylated throughout the cell cycle (Fig. 1B). Flow cytometry demonstrates that the two strains progress through S phase with identical kinetics despite the obvious difference in RPA2 phosphorylation (Fig. 1C).
Figure 1.
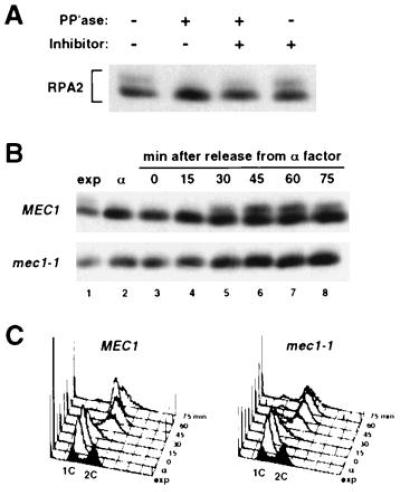
Cell cycle-regulated RPA2 phosphorylation is absent in mec1-1. (A) Western blot analysis of RPA2 from exponentially growing wild-type cells. Crude extract was treated with the indicated combinations of λ protein phosphatase (PP′ase) and the phosphatase inhibitor sodium vanadate. (B) Western blot analysis of RPA2 from synchronized TWY397 (MEC1) and DLY285 (mec1-1) cells. Samples from the exponentially growing cultures prior to α factor treatment (exp) and from the G1-arrested cultures directly prior to release (α) have been included. Equivalent volumes of original cell culture were analyzed. (C) Flow cytometry of synchronized cultures analyzed in B.
HU-Induced RPA Phosphorylation Is Compromised in the mec1-1 Mutant.
Because RPA2 becomes phosphorylated during S phase, we were interested in monitoring RPA2 phosphorylation in response to HU, which inhibits DNA replication by inactivating ribonucleotide reductase. Phosphorylated RPA2 accumulates in wild-type cells when a G1-arrested population is released into medium containing HU (Fig. 2A), consistent with a previous study employing exponentially growing cells (10). It is likely that HU induces this phosphorylation, because untreated wild-type cells at the same G1/S transition of the cell cycle (Fig. 1C Left, 15 min; flow cytometry in presence of HU not shown) contain little if any phosphorylated RPA2 (Fig. 1B Upper, lane 4). In mec1-1 cells exposed to HU, an aberrant form of phosphorylated RPA2 is generated, which migrates more diffusely during denaturing gel electrophoresis than the phosphorylated RPA2 from wild-type cells (Fig. 2A). This abnormal HU-induced RPA2 phosphorylation in mec1-1 correlates with the extreme sensitivity of this mutant to HU (34).
Figure 2.
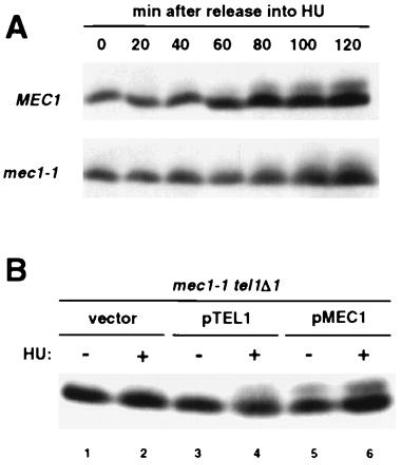
RPA2 phosphorylation in the presence of HU is abnormal in mec1-1. (A) Western blot analysis of RPA2 from synchronized TWY397 (MEC1) and DLY285 (mec1-1) cells released into medium containing HU. Equivalent volumes of original cell culture were analyzed. (B) Western blot analysis of RPA2 from YDM937 (mec1-1 tel1Δ1) harboring pRS316 (vector), the TEL1-containing plasmid pDM197 (pTEL1), or the MEC1-containing plasmid pDM207 (pMEC1) in the absence (−) or presence (+) of HU. Equal numbers of cells were analyzed.
S. cerevisiae TEL1, which is required for telomere maintenance, encodes the yeast protein most similar to ATMp (20, 39). Although deletion of TEL1 has no apparent effect on the yeast DNA damage response, increased dosage of this gene can partially rescue the defects of mec1 mutants, and mec1 tel1 double mutants are synergistically sensitive to genotoxic agents (37). In addition, damage-induced phosphorylation of Mec2p/Rad53p requires either MEC1 or TEL1 (40). Because of the apparent functional homology between MEC1 and TEL1, we investigated RPA2 phosphorylation in a mec1-1 tel1Δ strain. During exponential growth, or in response to HU, the double mutant is completely devoid of phosphorylated RPA2 (Fig. 2B, lanes 1 and 2). When TEL1 is introduced into this strain on an ARS/CEN plasmid, there is still no detectable RPA2 phosphorylation in cycling cells, but the abnormal RPA2 phosphorylation characteristic of the mec1-1 single mutant is observed in the presence of HU (Fig. 2B, lanes 3 and 4). When the double mutant harbors MEC1 on an ARS/CEN plasmid, RPA2 phosphorylation is reconstituted under either condition (Fig. 2B, lanes 5 and 6), and the HU sensitivity is greatly decreased (data not shown). Although the episomal MEC1 and TEL1 genes are under the control of their endogenous promoters in this experiment, the levels of protein expression are unknown. Nonetheless, our data suggest that MEC1, but not TEL1, is required for RPA2 phosphorylation during the normal cell cycle. When DNA synthesis is blocked with HU, MEC1 is required to achieve wild-type levels of RPA2 phosphorylation. However, it is possible that TEL1 contributes to this activity in wild-type cells, since partial TEL1-dependent RPA2 phosphorylation is observed in the absence of functional MEC1.
Radiation Induces MEC1-Dependent RPA Phosphorylation.
In addition to the HU checkpoint defect, mec1-1 cells are unable to delay cell cycle progression in response to ionizing radiation (34). Therefore, we investigated RPA2 phosphorylation in wild-type and mec1-1 cells after ionizing radiation exposure. Cells were blocked in G1 with α factor to distinguish radiation-induced RPA2 phosphorylation from the cell cycle-regulated reaction. Flow cytometry confirmed that the vast majority of cells in each sample were maintained in G1 during the course of the experiment (data not shown), and unirradiated cells contained little if any phosphorylated RPA2 (Fig. 3, 0 krad). Wild-type cells exhibited a dose-dependent induction of phosphorylated RPA2 (Fig. 3), similar to the ionizing radiation response that occurs in human cells (12). In contrast, mec1-1 cells were extremely deficient in the reaction, with only a faint band of phosphorylated RPA2 detected at the higher doses administered. We also analyzed the response of G1-arrested cells to UV radiation, and once again observed an induction of RPA2 phosphorylation activity in wild-type cells that was absent in the mec1-1 strain (Fig. 4C, lanes 1–4). Therefore, the mec1-1 defect in RPA2 phosphorylation correlates with the hypersensitivity of this mutant to both forms of radiation (34). In addition to dependence of both cell cycle-regulated and radiation-induced RPA2 phosphorylation on the same checkpoint gene, the phosphorylated species generated during normal growth and in response to DNA damage comigrate during denaturing gel electrophoresis (Fig. 3).
Figure 3.
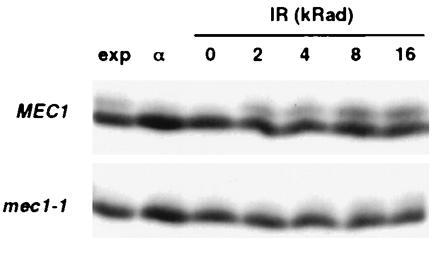
Ionizing radiation induces RPA2 phosphorylation dependent on MEC1. Western blot analysis of RPA2 from TWY397 (MEC1) and DLY285 (mec1-1) cells exposed to increasing doses of ionizing radiation (IR). Samples from the exponentially growing cultures prior to α factor treatment (exp) and from the G1-arrested cultures directly prior to release (α) have been included. Equivalent volumes of original cell culture were analyzed.
Figure 4.
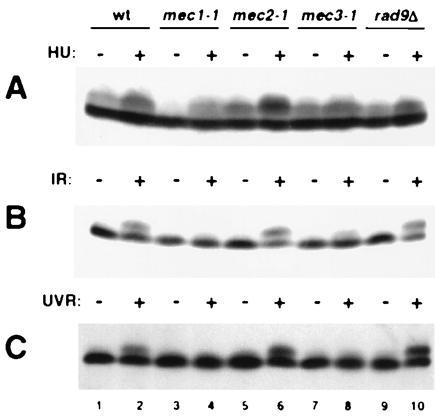
RPA phosphorylation in a collection of checkpoint mutants. Western blot analysis of RPA2 from wild-type and checkpoint mutants (A) in exponentially growing (−) or HU-arrested (+) cells, (B) after mock (−) or ionizing radiation (+, 8 krad) exposure (IR), and (C) after mock (−) or UV radiation (+, 60 J/m2) exposure (UVR). TWY397 was the wild-type control in all three experiments. For the mec1-1 samples, TWY308 was used in A and DLY285 was used in B and C. Within each experiment, equal numbers of cells were analyzed.
Examination of RPA Phosphorylation in Other Checkpoint Mutants Reveals a Novel Checkpoint Pathway.
Several additional genes have been identified in yeast that are required for the HU and/or radiation-induced checkpoint response. We examined RPA2 phosphorylation in a set of these mutants during exponential growth or upon environmental insult. With the exception of mec1-1, the tested mutants exhibited significant levels of RPA2 phosphorylation, both during the normal cell cycle and after exposure to HU (Fig. 4A). In response to ionizing radiation, RPA2 phosphorylation was present in mec2-1 and rad9Δ, but absent in mec1-1 and greatly reduced in mec3-1 (Fig. 4B). Similar results were observed upon exposure to UV radiation, except that mec1-1 and mec3-1 were virtually indistinguishable (Fig. 4C). The specific defect in radiation- but not HU-induced RPA2 phosphorylation in mec3-1 correlates with the sensitivity of this mutant to radiation but not HU (34). Identical MEC1 and MEC3 dependencies have previously been described for phosphorylation of Mec2p/Rad53p (40, 41), which like Mec1p is involved in both the HU and radiation checkpoints (34, 42).
Based on these phosphorylation data, we have placed RPA in a branch of the DNA damage-response pathway downstream of Mec1p but independent of Mec2p/Rad53p (Fig. 5). DNA polymerase ɛ (pol ɛ) is reported to have a role in the HU checkpoint response (43), as shown. RAD9, which is involved in the radiation but not the HU checkpoint response (34, 44), is not included in this diagram because epistasis studies have suggested that this gene lies in a pathway distinct from that of MEC3 (45). The normal RPA2 phosphorylation observed in the rad9Δ mutant is consistent with this hypothesis (Fig. 4).
Figure 5.
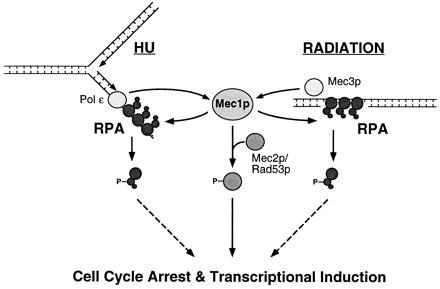
Model of MEC1-dependent RPA phosphorylation. Schematic diagram of the pathways leading from DNA replication inhibition or DNA damage to RPA phosphorylation. In this representation, the DNA-associated RPA becomes phosphorylated, as has been suggested for DNA-PK-catalyzed phosphorylation of human RPA (11, 14).
DISCUSSION
Cell survival depends on the accurate and complete duplication of the genetic information prior to cell division. The fidelity of this process is maintained through several mechanisms, including the recognition and repair of damaged DNA. To avoid replication or segregation of unrepaired DNA, a “checkpoint” function has evolved that arrests the progression of the cell cycle until the lesion has been encountered and mended (for reviews, see refs. 46 and 47). This delay provides time for restoration of the original DNA content before subsequent cellular processes cause irreversible damage. A checkpoint process also monitors the kinetics of DNA replication itself, inhibiting cell cycle progression when DNA synthesis is delayed to ensure that mitosis does not occur prematurely.
Recent studies have indicated that several proteins required for S phase also function in the checkpoint response. In Schizosaccharomyces pombe, cdc18p (48), cut5p (49), cdt1p (50), and pol-α primase (51) have such dual functionality. Each of these proteins is involved in the initiation of DNA replication, a process that is tightly regulated in eukaryotes. In S. cerevisiae, there is evidence that pol ɛ, a protein involved in the elongation phase of DNA replication, is required for the HU checkpoint in S. cerevisiae (43). Through its association with the replication machinery, pol ɛ is thought to monitor progression of the replication fork and possibly initiate the signal to delay the cell cycle when replication is inhibited (43). Our data show that the phosphorylation of a protein involved in both the initiation and elongation stages of DNA replication is directed by a checkpoint gene, suggesting that yet another protein required for S-phase progression is involved in the S. cerevisiae checkpoint response.
It is possible that MEC1-dependent RPA phosphorylation is involved in the regulation of cell cycle progression or in the transcriptional response to DNA damage (see Fig. 5). This hypothesis is based on the function of Mec2p/Rad53p, which is required for cell cycle arrest and transcriptional induction of certain genes following HU or radiation exposure (34, 42), and is also phosphorylated in a MEC1-dependent manner (40, 41). There are two lines of evidence suggesting that RPA could be involved directly in regulating transcription: (i) RPA physically interacts directly with several transcription factors (27, 28, 52) and (ii) yeast RPA can bind to duplex DNA upstream of many DNA repair and DNA metabolism genes (53). It will be interesting to determine whether transcription of these genes is affected by MEC1-dependent RPA phosphorylation. A recent study has provided evidence for DNA damage-induced transcriptional induction that is MEC1-dependent but MEC2/RAD53-independent (54), and it is conceivable that RPA could be involved in this pathway. However, it should be noted that Mec1p might control many effectors in addition to Mec2p/Rad53p and RPA, each with a specific role in the DNA damage response. Such a mechanism could account for the multifaceted disease state that results from mutation of the homologous ATM gene in humans.
Because RPA is required for DNA replication, repair, and recombination in vivo (55), there remains the possibility that phosphorylation of RPA directly affects the activity of the protein in one or more of these related processes. RPA interacts with certain replication (56, 57), repair (58–60), and recombination (61) proteins, but there has been no direct demonstration that RPA phosphorylation influences any of these associations. While DNA-PK-catalyzed phosphorylation of human RPA has no effect on DNA replication or DNA repair in vitro (14, 62), there is evidence that the phosphorylated RPA induced by UV radiation may have reduced activity in the cell-free SV40 DNA replication system (13). Thus, it is possible that phosphorylation of RPA by various kinases could give rise to forms of phosphorylated RPA that act preferentially in repair or recombination (13). ATMp might be such a kinase, responding specifically in the event of ionizing radiation-induced DNA damage. The delay in ionizing radiation-induced RPA phosphorylation characteristic of A-T cells may be due to a deficiency in ATMp activity that is inefficiently replaced by a homologous protein, similar to the defective HU-induced reaction in mec1-1 cells that requires TEL1 (Fig. 2B).
RPA phosphorylation during normal cell cycle progression, like that observed following DNA damage, also requires the checkpoint gene MEC1 (Fig. 1). This dependence is consistent with the possibility that RPA phosphorylation is involved in a checkpoint process during mitotic growth. Such a pathway could help to ensure the proper order of events in the cell cycle. Alternatively, RPA phosphorylation might “prime” the cell for repair during S phase, for example by inducing production of certain critical repair proteins. From a kinetic viewpoint, the consequences of DNA damage are likely to be most severe during S phase, because ongoing DNA replication could rapidly render mutations permanent. In addition, unwound replication fork structures might increase DNA damage susceptibility. Interestingly, MEC1 is required for a checkpoint response that delays the cell cycle during S phase (35). Further studies will be required to determine whether MEC1 encodes an RPA kinase that directly associates with the replication fork, as has been suggested for DNA-PK in human cells (14).
RPA phosphorylation provides a biochemical link between yeast and human members of the ATM family. The cross-species similarities in the cell cycle-regulated and DNA damage-induced phosphorylation reactions support a conserved role for phosphorylated RPA in eukaryotes. The yeast system will be a valuable tool for defining the role of RPA phosphorylation in the pathway defective in patients with A-T.
Acknowledgments
We thank members of the Kelly laboratory for helpful discussions, Ted Weinert for yeast strains, Steven Brill for anti-yeast RPA2 antiserum, and Jef Boeke and Stephen Desiderio for critically reading the manuscript. This work was supported by grants from the National Institutes of Health and the A-T Children’s Project.
Footnotes
Abbreviations: RPA, replication protein A; RPA2, middle subunit of RPA; A-T, ataxia-telangiectasia; DNA-PK, DNA-activated protein kinase; DNA-PKc, catalytic subunit of DNA-PK; HU, hydroxyurea; SV40, simian virus 40; PI 3, phosphatidylinositol 3.
References
- 1.Wobbe C R, Weissbach L, Borowiec J A, Dean F B, Murakami Y, Bullock P, Hurwitz J. Proc Natl Acad Sci USA. 1987;84:1834–1838. doi: 10.1073/pnas.84.7.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wold M S, Kelly T. Proc Natl Acad Sci USA. 1988;85:2523–2527. doi: 10.1073/pnas.85.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fairman M P, Stillman B. EMBO J. 1988;7:1211–1218. doi: 10.1002/j.1460-2075.1988.tb02933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenny M K, Schlegel U, Furneaux H, Hurwitz J. J Biol Chem. 1990;265:7693–7700. [PubMed] [Google Scholar]
- 5.Erdile L F, Wold M S, Kelly T J. J Biol Chem. 1990;265:3177–3182. [PubMed] [Google Scholar]
- 6.Erdile L F, Heyer W-D, Kolodner R, Kelly T J. J Biol Chem. 1991;266:12090–12098. [PubMed] [Google Scholar]
- 7.Umbricht C B, Erdile L F, Jabs E W, Kelly T J. J Biol Chem. 1993;268:6131–6138. [PubMed] [Google Scholar]
- 8.Heyer W-D, Rao M R S, Erdile L F, Kelly T J, Kolodner R D. EMBO J. 1990;9:2321–2329. doi: 10.1002/j.1460-2075.1990.tb07404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brill S J, Stillman B. Genes Dev. 1991;5:1589–1600. doi: 10.1101/gad.5.9.1589. [DOI] [PubMed] [Google Scholar]
- 10.Din S, Brill S J, Fairman M P, Stillman B. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- 11.Fotedar R, Roberts J M. EMBO J. 1992;11:2177–2187. doi: 10.1002/j.1460-2075.1992.tb05277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu V F, Weaver D T. Mol Cell Biol. 1993;13:7222–7231. doi: 10.1128/mcb.13.12.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carty M P, Zernik-Kobak M, McGrath S, Dixon K. EMBO J. 1994;13:2114–2123. doi: 10.1002/j.1460-2075.1994.tb06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brush G S, Anderson C W, Kelly T J. Proc Natl Acad Sci USA. 1994;91:12520–12524. doi: 10.1073/pnas.91.26.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchgessner C U, Patil C K, Evans J W, Cuomo C A, Fried L M, Carter T, Oettinger M A, Brown J M. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 16.Lees-Miller S P, Godbout R, Chan D W, Weinfeld M, Day R S, III, Barron G M, Allalunis-Turner J. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 17.Blunt T, Finnie N J, Taccioli G E, Smith G C M, Demengeot J, Gottlieb T M, Mizuta R, Varghese A J, Alt F W, Jeggo P A, Jackson S P. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 18.Peterson S R, Kurimasa A, Oshimura M, Dynan W S, Bradbury E M, Chen D J. Proc Natl Acad Sci USA. 1995;92:3171–3174. doi: 10.1073/pnas.92.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartley K O, Gell D, Smith G C M, Zhang H, Divecha N, Connelly M A, Admon A, Lees-Miller S P, Anderson C W, Jackson S P. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 20.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 21.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 662–668. [Google Scholar]
- 22.Painter R B, Young B R. Proc Natl Acad Sci USA. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudolph N S, Latt S A. Mutat Res. 1989;211:31–41. doi: 10.1016/0027-5107(89)90104-8. [DOI] [PubMed] [Google Scholar]
- 24.Taylor A M R, Harnden D G, Arlett C F, Harcourt S A, Lehmann A R, Stevens S, Bridges B A. Nature (London) 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 25.Kastan M B, Zhan Q, El-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J., Jr Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 26.Lu X, Lane D P. Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 27.Li R, Botchan M R. Cell. 1993;73:1207–1221. doi: 10.1016/0092-8674(93)90649-b. [DOI] [PubMed] [Google Scholar]
- 28.Dutta A, Ruppert J M, Aster J C, Winchester E. Nature (London) 1993;365:79–82. doi: 10.1038/365079a0. [DOI] [PubMed] [Google Scholar]
- 29.Zakian V A. Cell. 1995;82:685–687. doi: 10.1016/0092-8674(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 30.Kapeller R, Cantley L C. BioEssays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- 31.Dhand R, Hiles I, Panayotou G, Roche S, Fry M J, Gout I, Totty N F, Truong O, Vicendo P, Yonezawa K, Kasuga M, Coutneidge S A, Waterfield M D. EMBO J. 1994;13:522–533. doi: 10.1002/j.1460-2075.1994.tb06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunter T. Cell. 1995;83:1–4. doi: 10.1016/0092-8674(95)90225-2. [DOI] [PubMed] [Google Scholar]
- 33.Kato R, Ogawa H. Nucleic Acids Res. 1994;22:3104–3112. doi: 10.1093/nar/22.15.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinert T A, Kiser G L, Hartwell L H. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 35.Paulovich A G, Hartwell L H. Cell. 1995;82:841–847. doi: 10.1016/0092-8674(95)90481-6. [DOI] [PubMed] [Google Scholar]
- 36.Weinert T A, Hartwell L H. Mol Cell Biol. 1990;10:6554–6564. doi: 10.1128/mcb.10.12.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrow D M, Tagle D A, Shiloh Y, Collins F S, Hieter P. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- 38.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greenwell P W, Kronmal S L, Porter S E, Gassenhuber J, Obermaier B, Petes T D. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez Y, Desany B A, Jones W J, Liu Q, Wang B, Elledge S J. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 41.Sun Z, Fay D S, Marini F, Foiani M, Stern D F. Genes Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- 42.Allen J B, Zhou Z, Siede W, Friedberg E C, Elledge S J. Genes Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- 43.Navas T A, Zhou Z, Elledge S J. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- 44.Weinert T A, Hartwell L H. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 45.Lydall D, Weinert T. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 46.Hartwell L H, Weinert T A. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 47.Murray A W. Nature (London) 1992;359:599–604. doi: 10.1038/359599a0. [DOI] [PubMed] [Google Scholar]
- 48.Kelly T J, Martin G S, Forsburg S L, Stephen R J, Russo A, Nurse P. Cell. 1993;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- 49.Saka Y, Yanagida M. Cell. 1993;74:383–393. doi: 10.1016/0092-8674(93)90428-s. [DOI] [PubMed] [Google Scholar]
- 50.Hofmann J F X, Beach D. EMBO J. 1994;13:425–434. doi: 10.1002/j.1460-2075.1994.tb06277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Urso G, Grallert B, Nurse P. J Cell Sci. 1995;108:3109–3118. doi: 10.1242/jcs.108.9.3109. [DOI] [PubMed] [Google Scholar]
- 52.He Z, Brinton B T, Greenblatt J, Hassell J A, Ingles C J. Cell. 1993;73:1223–1232. doi: 10.1016/0092-8674(93)90650-f. [DOI] [PubMed] [Google Scholar]
- 53.Singh K K, Samson L. Proc Natl Acad Sci USA. 1995;92:4907–4911. doi: 10.1073/pnas.92.11.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiser G L, Weinert T A. Mol Biol Cell. 1996;7:703–718. doi: 10.1091/mbc.7.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longhese M P, Plevani P, Lucchini G. Mol Cell Biol. 1994;14:7884–7890. doi: 10.1128/mcb.14.12.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dornreiter I, Erdile L F, Gilbert I U, von Winkler P, Kelly T J, Fanning E. EMBO J. 1992;11:769–776. doi: 10.1002/j.1460-2075.1992.tb05110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melendy T, Stillman B. J Biol Chem. 1993;268:3389–3395. [PubMed] [Google Scholar]
- 58.Matsuda T, Saijo M, Kuraoka I, Kobayashi T, Nakatsu Y, Nagai A, Enjoji T, Masutani C, Sugasawa K, Hanaoka F, Yasui A, Tanaka K. J Biol Chem. 1995;270:4152–4157. doi: 10.1074/jbc.270.8.4152. [DOI] [PubMed] [Google Scholar]
- 59.He Z, Henricksen L A, Wold M S, Ingles C J. Nature (London) 1995;374:566–569. doi: 10.1038/374566a0. [DOI] [PubMed] [Google Scholar]
- 60.Li L, Lu X, Peterson C A, Legerski R J. Mol Cell Biol. 1995;15:5396–5402. doi: 10.1128/mcb.15.10.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park M S, Ludwig D L, Stigger E, Lee S-H. J Biol Chem. 1996;271:18996–19000. doi: 10.1074/jbc.271.31.18996. [DOI] [PubMed] [Google Scholar]
- 62.Pan Z-Q, Park C-H, Amin A A, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 1995;92:4636–4640. doi: 10.1073/pnas.92.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]