

Abstract
Peroxynitrite-mediated damage has been linked to numerous neurological and neurodegenerative diseases, including stroke, Alzheimer’s and Parkinson’s Diseases, amyotrophic lateral sclerosis and multiple sclerosis. Studies on the toxic effects of peroxynitrite in neurons have focused primarily on adverse effects resulting from the nitration of cellular proteins as the principal mode of toxicity while the consequences of the modulation of kinase pathways by peroxynitrite have received relatively less attention. Our results show that treatment of primary rat neurons with the peroxynitrite donor, SIN-1, leads to decreases in glutathione (GSH) levels and cell viability via a novel extracellular-signal-related kinase (ERK)/c-Myc phosphorylation pathway and a reduction in the nuclear expression of NF-E2-related factor-2 (Nrf2) that down-regulate the expression of glutamate cysteine ligase, the rate limiting enzyme for GSH synthesis. The flavonoid fisetin protects against the SIN-1-mediated alterations in ERK/c-Myc phosphorylation, nuclear Nrf2 levels, glutamate cysteine ligase levels, GSH concentration and cell viability. We also show that inhibition of mitogen-activated protein kinase kinase or Raf kinase can increase GSH levels in unstressed primary rat neurons through the same ERK/c-Myc phosphorylation pathway. Together, these results demonstrate that distinct signaling pathways modulate GSH metabolism in unstressed and stressed cortical neurons.
Keywords: Fisetin, peroxynitrite, ERK, c-Myc, Nrf2, Raf1
Introduction
Peroxynitrite-mediated toxicity has been implicated in numerous pathological states. Increased generation of nitric oxide (NO) and superoxide (O2-) in the diabetic coronary vascular endothelium leads to production of peroxynitrite (OONO-) with subsequent endothelial dysfunction and heart disease (Hoeldtke et al., 2003). A similar mechanism leads to both kidney (Ishii et al., 2001; Thuraisingham et al., 2000) and retinal (El-Remessy et al., 2003) damage in diabetes. In the brain, focal traumatic brain injury (Singh et al., 2006) , ischemic stroke (Eliasson et al., 1999) and numerous neurodegenerative disorders including Alzheimer’s Disease, Parkinson’s Disease, multiple sclerosis and amyotrophic lateral sclerosis (reviewed in Torreilles et al., 1999) have all been linked to the increased production of peroxynitrite. Although peroxynitrite itself is not a free radical, it is a uniquely damaging molecule because it can initiate strong oxidation reactions through decomposition into a hydroxyl radical and nitrogen dioxide (Beckman et al., 1990). It can also form a highly reactive nitroderivative in the presence of transition metals (Beckman et al., 1992; Ischiropoulos et al., 1992) and interact directly with protein and non-protein thiol groups leading to the depletion of cellular antioxidant defenses (Radi et al., 1991).
Since neurons have a high energy demand and thus produce more superoxide from the electron transport chain (Nakamura et al., 2001), and activated microglia cells have the ability to release both nitric oxide and superoxide into the extracellular space (Dringen, 2005), many studies on peroxynitrite have used neurons and glia as models of cell damage. Much of this work has focused on the direct nitration of proteins and how this nitration modifies normal protein function (Iravani et al., 2006; Manabe et al., 2001; Naoi et al., 2005). However, peroxynitrite also has been shown to alter kinase pathways involved in signal transduction (Bapat et al., 2001; Klotz, 2002; Levrand et al., 2005; Oh-hashi et al., 1999; Schieke et al., 1999; Zouki et al., 2001).
The flavonoid family of dietary polyphenolic compounds has received much interest recently for their ability to protect cells against a wide variety of stressors (reviewed in Heim et al., 2002; Lau et al., 2005; Middleton et al., 2000; Rahman et al., 2006; Ramassamy, 2006) including peroxynitrite (Klotz and Sies, 2003). Individual flavonoids can protect neurons against oxidative stress by causing an increase in glutathione (GSH) levels, a decrease in calcium influx or by acting as a direct antioxidant (Ishige et al., 2001). They can also block the formation of protein nitrotyrosine residues (Pannala et al., 1997), alter kinase activation (Choi et al., 2005; Wu et al., 2005), and increase heme-oxygenase-1 expression (Andreadi et al., 2006; Chen et al., 2006b; Hanneken et al., 2006; Lin et al., 2004; Maher and Hanneken, 2005; Maher, 2006; Wu et al., 2006).
Fisetin is a member of the flavonol subgroup of flavonoids that has a low EC50, enhances memory in rodents (Maher et al., 2006) and has a high protective capacity against oxidative glutamate toxicity (Ishige et al., 2001) and ischemia in vivo (Dajas et al., 2003; Maher et al., 2007). In the current study, we evaluated the toxicity of peroxynitrite in primary rat neurons and the ability of fisetin to protect against peroxynitrite-mediated damage. We show that in these cells, the peroxynitrite donor SIN-1 causes decreased GSH levels and cell death through a novel extracellular-signal-related kinase (ERK)/c-Myc phosphorylation pathway. Fisetin protects against both the GSH decrease and cell death by inhibiting the changes in ERK and c-Myc phosphorylation as well as preventing the decrease in nuclear NF-E2-related factor-2 (Nrf2) localization caused by SIN-1.
Results
To examine the effect of peroxynitrite on neurons and the ability of fisetin to inhibit this toxicity, primary rat cortical neurons were exposed to increasing amounts of the peroxynitrite donor SIN-1 for six hours and cell viability was then determined. Whereas 500 μM of SIN-1 had no effect on primary rat neurons, 1 mM SIN-1 reduced viability to ∼50% of that of control cells (Fig. 1A). A 15 minute preincubation of the cells with 10 μM fisetin, a concentration with maximal efficacy (Sagara et al., 2004), followed by concurrent fisetin/1 mM SIN-1 treatment for six hours increased viability to 92% of that of control cells.
Figure 1.

Effects of SIN-1, peroxynitrite and fisetin on viability and GSH levels in primary cortical neurons. Viability (A) and GSH levels (D) of 7 day rat primary cortical neurons treated with increasing doses of the peroxynitrite donor SIN-1 for 6 hr and/or the neuroprotective flavonoid fisetin (10 μM) and/or the exogenous GSH source glutathione monoethyl ester (GEE) in the absence or presence of the glutathione synthesis inhibitor buthionine sulfoximine (BSO) (1 mM). (B) Viability of 7 day rat primary cortical neurons treated with 1 mM SIN-1 for 6 hr alone or in the presence of 1 mM uric acid or 25 μM FeTPPS. (C) Viability of 7 day rat primary cortical neurons treated with 250 μM peroxynitrite for 24 hr alone or following pretreatment for 30 min or 24 hr with 10 μM fisetin. In the case of the 24 hr pretreatment, fisetin was removed from the cells before the addition of peroxynitrite. Cell viability was measured by the MTT assay. GSH levels were measured using monochlorobimane. Similar results were obtained in three independent experiments. * indicates significantly different (p < 0.05) from control; # indicates significantly different from SIN-1 or peroxynitrite alone; & indicates significantly different from BSO alone; % indicates significantly different from fisetin + SIN-1 treatment.
To confirm that the toxicity of SIN-1 was due to the production of peroxynitrite, we tested the protective effects of two compounds that have been reported to interact with peroxynitrite but not nitric oxide or superoxide. Both uric acid, a peroxynitrite scavenger (Zhang and Rosenberg, 2004), and FeTPPS, a peroxynitrite decomposition catalyst (Misko et al., 1998), protected the primary rat neurons from SIN-1 toxicity (Fig. 1B). We also tested the ability of fisetin to protect the rat primary neurons from authentic peroxynitrite. As shown in Figure 1C, 250 μM peroxynitrite killed ∼75% of the neurons when cell death was determined after 24 hr. 10 μM fisetin significantly increased cell survival whether it was added 30 min or 24 hr before the addition of peroxynitrite. Importantly, in the case of the 24 hr pretreatment, the medium was replaced with fisetin-free medium before the addition of peroxynitrite indicating that the protection was not due to a direct interaction between fisetin and peroxynitrite.
Since both fisetin (Ishige et al., 2001; van Acker et al., 2000) and peroxynitrite (Bolanos et al., 1995; Brito et al., 2006; Ho et al., 2006; Lim et al., 2004; Nowak et al., 2003) have previously been shown to differentially affect the levels of intracellular GSH, the major antioxidant in neurons, we evaluated the possibility that the alterations in neuronal viability resulting from the above treatments correlated with changes in GSH concentrations. 1 mM SIN-1 treatment for six hours significantly decreased GSH levels in the primary rat neurons as compared to control cells, and this decrease was significantly reduced by fisetin treatment (Fig. 1D). The inhibition by fisetin of both the SIN-1-mediated decrease in GSH levels and SIN-1 toxicity was blocked by treatment of the cells with buthionine sulfoximine (BSO), an inhibitor of the rate limiting enzyme in GSH biosynthesis, glutamate cysteine ligase (GCL) (Fig. 1A and 1D). In contrast, the protection against SIN-1 toxicity afforded by treatment with glutathione monoethyl ester (GEE), an exogenous source of GSH, was not affected by BSO (Fig. 1A), consistent with the ability of GEE to maintain GSH levels in the presence of BSO (Fig. 1D). Together these results suggest that the ability of fisetin to maintain GSH levels plays an important role in its neuroprotective activity against SIN-1 toxicity.
Since a number of studies have shown that peroxynitrite (Klotz, 2002; Schieke et al., 1999; Zhang et al., 2000) Pesse, 2005 #1390] as well as some dietary phenolic compounds, including fisetin (Sagara et al., 2004), modify ERK1/2 phosphorylation, and ERK activation can alter GSH metabolism (Benassi et al., 2006), we next evaluated the effect of SIN-1 and fisetin treatment on ERK1/2 phosphorylation, a measure of its activation. While phospho-ERK1/2 was nearly undetectable in control cells, fisetin treatment alone for six hours led to a small increase in phospho-ERK1/2 (Fig. 2A), consistent with previous results (Sagara et al., 2004). 1mM SIN-1 treatment for six hours led to the hyperphosphorylation of ERK1/2 which was significantly decreased by fisetin treatment. A time course of SIN-1 treatment showed that ERK1/2 was not phosphorylated at time points earlier than 4 hours and phosphorylation continued to increase up to 6 hours (Fig. 2B). Since ERK1/2 is downstream of MEK, we used the MEK inhibitor PD98059 to determine if the fisetin- and SIN-1-mediated alterations in ERK1/2 phosphorylation occur via MEK activation. PD98059 blocked the low level of basal ERK1/2 phosphorylation present in control cells and the fisetin-mediated ERK1/2 phosphorylation in control cells as well as a significant amount of the SIN-1-mediated increase in ERK1/2 phosphorylation, indicating that peroxynitrite acts mainly at or upstream of MEK in primary rat neurons to hyperphosphorylate ERK1/2. However, PD98059 treatment did not alter the GSH level or viability of the primary neurons either alone or in combination with SIN-1 (Fig. 1), consistent with previous results that have used MEK inhibitors in similar paradigms (Bapat et al., 2001; Zhang et al., 2004). These results suggest that the relationship between the changes in GSH levels and ERK1/2 activation caused by SIN-1 are complex.
Figure 2.

Analysis of ERK1/2 phosphorylation in primary cortical neurons treated with SIN-1, fisetin and PD98059. (A) 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 6 hr with 1 mM SIN-1, 10 μM fisetin and/or 40 μM PD98059 as indicated in the figure. Cells were pre-treated with fisetin or PD98059 for 15 min before the addition of SIN-1. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho-ERK along with antibodies to total ERK2 demonstrating no changes in overall protein levels. Similar results were obtained in three independent experiments. The average phosphoprotein signal from the blots was quantified by densitometry, normalized to total protein and plotted ± S.D. * indicates significantly different (p < 0.05) from control; # indicates significantly different from SIN-1 alone. (B) 7 day primary cortical neurons in 35 mm dishes were treated for 15 min to 6 hr with 1 mM SIN-1. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho-ERK along with antibodies to total ERK2 demonstrating no changes in overall protein levels. Similar results were obtained in three independent experiments. Fis = Fisetin, PD = PD98059
A recent report showed that the phosphorylation and activation of ERK1/2 is positively correlated with the phosphorylation of the transcription factor c-Myc in human melanoma cells (Benassi et al., 2006) and that the phosphorylation of c-Myc causes its recruitment to the GCL promoter, leading to increased transcription of GCL and thereby increased GSH levels. Therefore, given our results with fisetin, SIN-1 and ERK1/2 phosphorylation, we were interested in the effects of SIN-1 and fisetin treatment on phospho-c-Myc levels in primary rat neurons. Using an antibody specific to the phospho form of c-Myc, we found that treatment with 1 mM SIN-1 led to a significant decrease in phospho-c-Myc levels at the four and six hour time points, but not earlier (Fig. 3A). These are the same time points that showed a significant increase in SIN-1-mediated ERK1/2 phosphorylation (Fig. 2B). The decrease in phospho-c-Myc after 6 hr of SIN-1 treatment was prevented by either fisetin or PD98059 (Fig. 3B), both of which also blocked the SIN-1-mediated increase in ERK1/2 phosphorylation (Fig. 2A). These results suggest that in rat primary cortical neurons, c-Myc phosphorylation is inversely, rather than directly, correlated with ERK1/2 phosphorylation.
Figure 3.
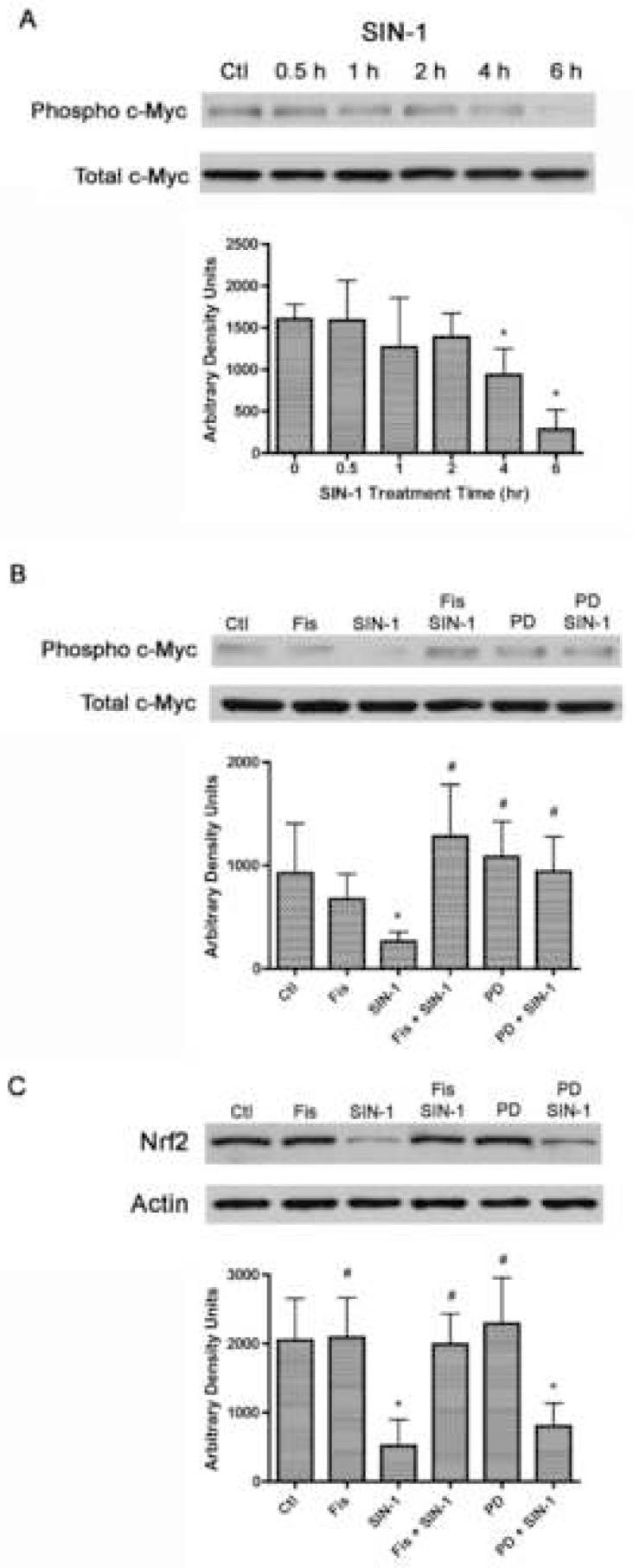
The effects of SIN-1, fisetin and PD98059 on the transcription factors c-Myc and Nrf2. (A) Time course of the effects of SIN-1 on c-Myc phosphorylation. 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 30 min to 6 hr with 1 mM SIN-1. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho-c-Myc along with antibodies to total c-Myc demonstrating no changes in overall protein levels. Similar results were obtained in three independent experiments. The average phosphoprotein signal from the blots was quantified by densitometry, normalized to total protein and plotted ± S.D. (B) Inhibition of c-Myc dephosphorylation by fisetin and PD98059. 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 6 hr with 1 mM SIN-1, 10 μM fisetin and/or 40 μM PD98059 as indicated in the figure. Cells were pre-treated with fisetin or PD98059 for 15 min before the addition of SIN-1. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho-c-Myc along with antibodies to total c-Myc demonstrating no changes in overall protein levels. Similar results were obtained in three independent experiments. The average phosphoprotein signal from the blots was quantified by densitometry, normalized to total protein and plotted ± S.D. (C) The SIN-1-dependent decrease in Nrf2 is prevented by fisetin but not PD98059. 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 6 hr with 1 mM SIN-1, 10 μM fisetin and/or 40 μM PD98059 as indicated in the figure. Cells were pre-treated with fisetin or PD98059 for 15 min before the addition of SIN-1. Nuclei were prepared and equal amounts of nuclear protein were analyzed by SDS-PAGE and immunoblotting with antibodies to Nrf2 along with antibodies to actin as a loading control. The average Nrf2 signal from the blots was quantified by densitometry, normalized to actin and plotted ± S.D. * indicates significantly different (p < 0.05) from control; # indicates significantly different from SIN-1 alone. Fis = Fisetin, PD = PD98059
Since both fisetin and PD98059 decrease SIN-1-mediated ERK1/2 phosphorylation and increase c-Myc phosphorylation but only fisetin protects against the SIN-1-mediated decreases in GSH and cell viability, this suggested that fisetin but not PD98059 could activate additional signaling pathways involved in the maintenance of GSH homeostasis under conditions of stress. Accordingly, we looked at the nuclear expression of the transcription factor Nrf2, which can also regulate GCL expression (Dickinson et al., 2004; Wild and Mulcahy, 2000). SIN-1 treatment lead to a significant decrease in nuclear Nrf2 expression, and this decrease was blocked by fisetin treatment (Fig. 3C). However, unlike the alterations in phosphorylated ERK1/2 or c-Myc, PD98059 did not prevent the SIN-1-mediated decrease in nuclear Nrf2 (Fig. 3C). These results indicate that fisetin may act through Nrf2 rather than c-Myc to increase GSH under conditions of SIN-1-mediated oxidative stress.
To explore this idea further, we looked at the effects of fisetin and SIN-1 on the expression of GCL, the rate limiting enzyme for GSH biosynthesis. GCL consists of two subunits, a catalytic (GCLC) subunit and a modifier (GCLM) subunit. Six hours of fisetin treatment led to an increase in the expression of both subunits, while the same length of SIN-1 treatment led to a decrease in both subunits (Fig. 4). Importantly, the SIN-1-mediated decrease in both GCL subunits was prevented by treatment with fisetin (Fig. 4).
Figure 4.
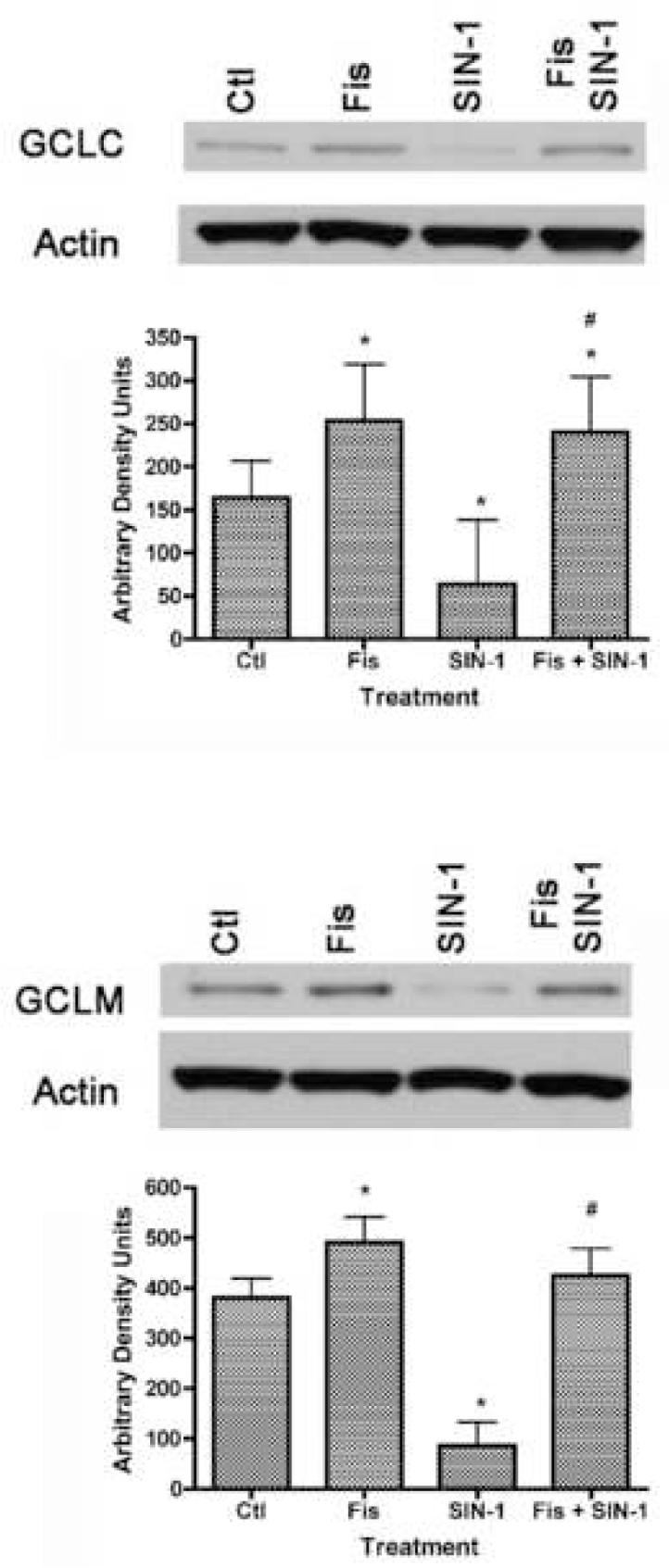
The opposing effects of SIN-1 and fisetin on the levels of the two subunits of glutamate cysteine ligase (GCL), the rate limiting enzyme in GSH biosynthesis. 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 6 hr with 1 mM SIN-1, 10 μM fisetin or 10 μM fisetin + 1 mM SIN-1. Cells were pre-treated with fisetin 15 min before the addition of SIN-1. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to GCLC or GCLM along with antibodies to actin as a loading control. The average GCLC or GCLM signal from the blots was quantified by densitometry, normalized to actin and plotted ± S.D. * indicates significantly different (p < 0.05) from control; # indicates significantly different from SIN-1 alone. Fis = Fisetin
Finally, we wanted to confirm the inverse relationship between the activity of the MEK/ERK cascade and the phosphorylation of c-Myc in rat primary neurons. To do this, we used a different inhibitor of MEK, U0126, as well as an inhibitor of Raf1, the upstream kinase, Raf1 kinase inhibitor 1 (RKI), and looked at the effects of altering ERK1/2 phosphorylation on c-Myc phosphorylation and GCL expression in unstressed primary rat cortical neurons. U0126 was used in this experiment because, unlike PD98059, it is a potent inhibitor of both the MEK1 and MEK 2 isoforms (Alessi et al., 1995; Favata et al., 1998). Six hours of treatment of unstressed neurons with U0126 (20 μM) or RKI (100 nM) significantly reduced the low level of ERK1/2 phosphorylation present in the cells but led to a strong induction of c-Myc phosphorylation, as well as an increase in GCLC and GCLM expression (Fig. 5A). Consistent with the results using PD98059, the nuclear expression of Nrf2 was not affected by either U0126 or RKI. Both U0126 and RKI treatment of the unstressed cells also led to an increase in intracellular GSH levels (Fig. 5B). Taken together, these results provide additional evidence for an inverse relationship between ERK activity and c-Myc phosphorylation and suggest that it plays a role in regulating GSH synthesis in unstressed primary neurons. However, when the cells are stressed, such as following treatment with the peroxynitrite donor, SIN-1, this pathway no longer appears to be able to modulate GSH synthesis. In the presence of stress, fisetin can act through the Nrf2 pathway to increase GSH production and enhance cell viability.
Figure 5.

Inhibitors of the Ras-ERK cascade increase GSH biosynthesis in unstressed primary cortical neurons. (A) 7 day primary cortical neurons in 35 mm dishes were left untreated (Ctl) or were treated for 6 hr with 100 μM of the Raf-1 kinase inhibitor RKI or 10 μM of the MEK inhibitor U0126. Equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho-ERK and ERK2, phospho-c-Myc and c-Myc, Nrf2, GCLC, GCLM and actin. Similar results were obtained in three independent experiments. The average phosphoprotein or Nrf2, GCLC or GCLM signal from the blots was quantified by densitometry, normalized to total protein or actin, as indicated in the figure, and plotted ± S.D. (B) 7 day primary cortical neurons were left untreated (Ctl) or were treated for 6 hr with 100 μM of the Raf-1 kinase inhibitor RKI or 10 μM of the MEK inhibitor U0126. GSH levels were measured using monochlorobimane and plotted relative to untreated, control cells. * indicates significantly different (p < 0.05) from control.
Discussion
Peroxynitrite is one of the most potent oxidative and nitrative molecules present within cells (Klotz, 2002; Torreilles et al., 1999). Numerous studies have focused on its potential to cause damage through oxidative and nitrosative stress, while fewer studies have looked at the roles of the intracellular pathways that are activated or suppressed by peroxynitrite in its toxicity. In the current study, we evaluated the effects of peroxynitrite and the cytoprotective flavonoid fisetin on a kinase cascade and transcription factors previously shown to modulate cellular antioxidant status and viability.
ERK1/2 are kinases downstream from Ras, Raf and MEK that are ultimately responsible for the phosphorylation of key activator proteins within the cell (reviewed in Chong et al., 2003, diagrammed in Fig. 6). Our results demonstrate that treatment of rat primary neurons with the peroxynitrite donor SIN-1 leads to the hyperphosphorylation of ERK1/2 and decreases in the phosphorylation of c-Myc, the expression of GCL, the rate limiting enzyme for GSH biosynthesis, and the level of intracellular GSH. All of these alterations were abrogated in the SIN-1-treated neurons by the addition of fisetin. A previous study using melanoma cells also showed that a decrease in phospho-c-Myc leads to a decrease in GCL transcription and GSH levels (Benassi et al., 2006). However, in contrast to the results of our study, Benassi et al (Benassi et al., 2006) demonstrated that in melanoma cells, ERK1/2 and c-Myc phosphorylation are positively correlated. This inconsistency may be due to a fundamental difference in the kinase/phosphatase cascade of the cancer cells used in this earlier study and the primary cortical neurons used in our experiments. Indeed, previous studies using primary neural cultures have found that an increase in ERK phosphorylation can lead to an increase in oxidative stress and death (de Bernardo et al., 2004; Kuperstein and Yavin, 2002; Levinthal and DeFranco, 2004). To our knowledge however, ours is the first study to investigate both ERK1/2 and c-Myc phosphorylation in primary cultures of nerve cells and to ask how the phosphorylation of these proteins relates to cellular antioxidant status and viability.
Figure 6.

Model showing the opposing effects of SIN-1 and fisetin on primary cortical neurons. In stressed cells, treatment with the peroxynitrite donor, SIN-1, leads to the hyperphosphorylation of ERK1/2 by MEK, which results in a decrease in c-Myc phosphorylation, a decrease in GCLC/GCLM levels and a decrease in intracellular GSH. SIN-1 treatment also causes a decrease in Nrf2 levels which contributes to the decrease in GSH levels. Pretreatment of the cells with fisetin prevents these SIN-1-mediated alterations and results in the maintenance of cellular GSH. Large arrows indicate activation, “T” lines indicate inhibition, small arrows indicate direction of change of specific protein phosphorylation events (ERK and c-Myc) or levels of GCLC/GCLM and GSH.
We also demonstrated for the first time that fisetin can prevent SIN-1 toxicity by maintaining the level of GSH in nerve cells. SIN-1 treatment alone leads to a significant decrease in both GSH and viability in these cells and fisetin treatment protects against both of these losses (Fig. 1). To provide additional evidence that the protective effect of fisetin is via the modulation of GSH levels, we treated the nerve cells with the GSH synthesis inhibitor, BSO, in conjunction with fisetin and SIN-1. In the cells treated only with BSO, there was a significant decrease in GSH levels but no significant change in cell viability. Concurrent SIN-1 and BSO treatment led to a further decrease in GSH levels and a significant decrease in viability compared to BSO alone. Under these conditions, fisetin was no longer able to protect against the SIN-1-mediated decreases in GSH levels and viability. In contrast, providing an exogenous source of GSH via the addition of GEE prevented the SIN-1-mediated decreases in both GSH and cell viability. Together, these results strongly suggest that fisetin is neuroprotective via its ability to modulate GSH levels.
Although a previous report indicated that BSO treatment of primary neurons can lead to a decrease in viability (Li et al., 1997), the culture conditions used in that study were markedly different from those used in our experiments. Specifically, the nerve cell medium in our experiments contained a weak antioxidant supplement and only trace levels of copper, while the medium in the Li et al. study (Li et al., 1997) did not contain antioxidants and had a level of copper previously shown to amplify the toxicity of BSO (White and Cappai, 2003). Indeed, when primary nerve cells were cultured in low copper media, BSO was shown to deplete GSH without killing the cells (Klejman et al., 2005). Our results demonstrate that when primary nerve cells are in a non-stressed (control) condition, BSO treatment does not lead to toxicity. However, when the cells are in a stressed condition (e.g. SIN-1 treatment), GSH depletion via BSO treatment does lead to a significant decrease in viability. A similar result was seen in a nerve cell line using exogenous glutamate in conjunction with BSO (Maher, 2006).
SIN-1 treatment induces the hyperphosphorylation of ERK1/2 (Fig. 2) in primary neurons. Since fisetin leads to a small increase in ERK1/2 phosphorylation in primary neurons, but, unlike SIN-1, does not cause a decrease in GSH levels or cell viability (Fig. 1), these data suggest that the hyperphosphorylation of ERK1/2 could be related to the decreases in c-Myc phosphorylation, GCL expression, GSH levels and cell viability. However, while the MEK inhibitor PD98059 did block a significant amount of the SIN-1-mediated hyperphosphorylation of ERK1/2 and restored c-Myc phosphorylation in the primary neurons, it did not alter the SIN-1-mediated decreases in GSH levels or viability. This result suggests that while ERK1/2 phosphorylation may inversely regulate the levels of c-Myc phosphorylation, phosphorylated c-Myc cannot enhance the transcription of GCL in primary neurons under conditions of stress.
Unlike PD98059, fisetin maintains GSH levels and cell viability in the presence of SIN-1. This observation suggested that fisetin activates a pathway distinct from the one involving c-Myc phosphorylation to increase GSH synthesis. We (Hanneken et al., 2006; Maher and Hanneken, 2005; Maher, 2006; Maher et al., 2007) and others (Chen et al., 2006a; Hou et al., 2001; Myhrstad et al., 2002; Valerio et al., 2001) have shown that fisetin can increase the levels of the transcription factor Nrf2, a known regulator of GCL expression (for reviews see Dickinson et al., 2004; Wild and Mulcahy, 2000). Nrf2 acts by binding to the antioxidant response element (ARE; also EpRE,) within the promoter of GCL and various other genes, thereby regulating their inducible production (for reviews see Chen and Kong, 2004; Kensler et al., 2007; Nguyen et al., 2003). Nrf2 normally resides in the cytoplasm bound to a protein called Keap1. In the presence of ARE inducers and/or during times of cellular stress, Nrf2 disassociates from Keap1 and migrates into the nucleus where it initiates the transcription of numerous genes, including GCLC and GCLM. Surprisingly, we found that SIN-1 treatment dramatically reduced the expression of Nrf2 in the primary neurons and this loss was prevented by fisetin but not by PD98059. The effects of SIN-1 and fisetin on Nrf2 expression directly correlated with their effects on GCLC and GCLM expression. Thus, our data strongly suggest that the increase in GCL expression we observed after fisetin plus SIN-1treatment compared to SIN-1 treatment alone is mediated through the Nrf2 pathway. Exactly how fisetin is able to increase Nrf2 expression in the presence of SIN-1 is not clear at this time. ARE inducers can increase Nrf2 levels both by modulating its interaction with Keap1, a substrate adaptor for a Cul3-containing E3 ubiquitin ligase that promotes the ubiquitination and degradation of Nrf2 by the 26S proteasome (for review see Zhang, 2006) and by phosphorylation (for reviews see Chen and Kong, 2004; Nguyen et al., 2003). Studies are underway to identify which mechanism is utilized by fisetin.
To further explore the relationship between phosphorylated ERK1/2 and c-Myc, GCL expression and GSH levels in unstressed nerve cells, we used the MEK inhibitor U0126 and a Raf-1 kinase inhibitor (RKI) to block upstream kinase activation (Fig. 5). Both U0126 and Raf-1 decrease the basal level of ERK1/2 phosphorylation and increase the phosphorylation of c-Myc, the expression of GCLC and GCLM and the levels of GSH as compared to untreated cells. The greater effect of RKI or U0126 treatment on c-Myc phosphorylation as compared to the effect on the expression of the GCL subunits may be related to the multiple c-Myc binding sites present on each GCL promoter and required for maximal induction of GCL expression (Benassi et al., 2006). We attempted to use ERK and c-Myc RNA interference to further demonstrate the inverse relationship between phosphorylated ERK1/2 and phosphorylated c-Myc, but the loss of ERK or c-Myc protein resulted in significant cell death (data not shown). Nevertheless, our results with the kinase inhibitors in unstressed cells strongly support the inverse relationship between ERK1/2 and c-Myc phosphorylation and demonstrate that phosphorylation of c-Myc is positively correlated with GCL expression as well as GSH levels in unstressed nerve cells. A possible explanation for this novel, inverse relationship between ERK1/2 phosphorylation and c-Myc phosphorylation in primary neurons is that ERK can activate protein phosphatase 2A, a protein known to dephosphorylate c-Myc (Arnold and Sears, 2006). Since the vast majority of the kinase/phosphatase pathways have been worked out in cancer cells, further investigation in primary cells is warranted.
In summary (Fig. 6), our data indicate that the peroxynitrite donor, SIN-1, can lead to GSH loss and neuronal cell death through the down-regulation of both phospho-c-Myc and GCL expression via the hyperphosphorylation of ERK1/2 and a reduction in Nrf2 expression. The flavonoid fisetin prevents these alterations in ERK1/2 and c-Myc phosphorylation, GCL expression and Nrf2 expression as well as the loss of GSH. Our data also suggest that when nerve cells are unstressed, the ERK/c-Myc pathway plays an important role in modulating GSH levels. However, during times of cellular stress, this pathway no longer appears to be able to modulate GSH levels. Instead the positive effects of fisetin on GSH levels and cell viability appear to be mediated by the Nrf2 pathway. Together, these results suggest the presence of distinct, differentially activated pathways for the regulation of GSH levels within primary neurons.
Experimental Procedure
Materials
The mitogen-activated protein kinase kinase (MEK) inhibitors 2′-amino-3′-methoxyflavone (PD98059) and 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126) were from Promega (Madison, WI). The Raf1 kinase inhibitor (RKI) 5-iodo-3-[(3,5-dibromo-4-hydroxyphenyl)methylene]-2-indolinone, glutathione monoethyl ester (GEE) and 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron (III) chloride (FeTPPS) were obtained from Calbiochem (San Diego, CA). Fisetin was from Indofine (Hillsborough, NJ). 3-Morpholinosydnonimime (SIN-1) was obtained from Alexis Biochemicals (San Diego, CA). Peroxynitrite was from Cayman Chemical (Ann Arbor, MI). Buthionine sulfoximine (BSO), uric acid and all other chemicals were from Sigma (St. Louis, MO).
Primary Cortical Neuronal Culture
Primary cortical neurons were prepared from embryonic day 17 Sprague-Dawley rats (Li et al., 1997), but with modifications to the culture media. Briefly, cells were dissociated from the cortex and maintained in serum-free, B27 neurobasal media (Invitrogen, Carlsbad, CA) on polylysine coated dishes. For toxicity studies, cells were plated on polylysine-coated 96-well microtiter dishes at 50,000 cells/100 μl in each well and subjected to the described treatments 7 days after the initial plating.
Cell Viability Assays
Cell viability was determined by the MTT assay (Hansen et al., 1989). MTT reduction was performed in 96-well microtiter plates containing primary rat neuronal cells in 100 μl medium. The reaction was started by adding 10 μl/well of a 2.5 mg/ml MTT stock in Dulbecco’s phosphate-buffered saline (PBS) and terminated after 4 hr by adding 100 μl of a solubilization solution containing 50% N,N’-dimethylformamide and 20% sodium dodecyl sulfate (pH 4.8). Absorbance values at 570 nm were determined on the next day with an automatic microtiter plate reader, using 630 nm as the reference wavelength. In all cases, cell viability was also checked by visual inspection and the possibility of reagent interference with the assay determined by using wells without cells.
Immunoblotting
For Western blot analysis, equal amounts of cell lysate protein were resolved in 10% Bis-Tris gels (Bio-Rad, Hercules, CA) and electrophoretically transferred to polyvinylidene difluoride hybridization membranes (Millipore, Billerica, MA). For immunoblotting of Nrf2, nuclear extracts were prepared as described (Schreiber et al., 1989). The membrane was probed with 1:1000 anti-phospho ERK1/2 (Cell Signaling, Danvers, MA), 1:1000 anti-phospho c-Myc (Cell Signaling), 1:500 anti-Nrf2 (Cell Signaling), 1:10000 anti-glutamate cysteine ligase catalytic subunit, or 1:10000 anti-glutamate cysteine ligase modifier subunit in phosphate buffered saline/0.1% Tween-20 (PBS-T) + 0.05% sodium azide. The anti-glutamate cysteine ligase antibodies were both courtesy of Dr. T. Kavanagh (University of Washington, Seattle). For control blots, the membrane was probed with 1:1000 anti-ERK 2 (BD Biosciences, San Diego, CA), 1:1000 anti-c-Myc (BD Biosciences), or 1:5000 anti-actin (BD Biosciences). After the incubation with the primary antibody, the blots were incubated with horseradish peroxidase—conjugated goat anti-rabbit immunoglobulin G secondary antibody (Bio-Rad) at a dilution of 1:20,000. The antibody conjugates were detected using a chemiluminescence Western blot kit (Amersham, Buckinghamshire, England). For immunoblot analysis, the film was scanned using a Bio-Rad GS-800 densitometer and the bands were quantified with the QuantOne software (Bio-Rad).
Glutathione Measurement
Cellular glutathione concentration was determined as described previously (Sun et al., 2005). Briefly, primary rat neurons in 96 well plates were washed with 100 μl of Hank’s buffered salt solution (HBSS, Invitrogen, Carlsbad, CA) and then incubated with 10 μM monochlorobimane (Sigma) in HBSS. The plate was then immediately read in a Molecular Devices (Sunnyvale, CA) Gemini fluorescent plate reader using the 390 nm excitation / 478 nm emission wavelengths.
Statistical Analysis
All statistical analyses were performed using the Analyse-It plug in (Leeds, UK) for Microsoft Excel. The density data obtained from the scanned immunoblots was analyzed for statistically significant differences at the p < 0.05 level using the analysis of variance (ANOVA) test and Tukey’s post test for individual group means comparisons. A minimum of three replicates were used for statistical analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- Alessi DR, Cohen P, Dudley DT, Saltiel AR. PD98059 is a specific inhibitor of the mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Andreadi CK, Howells LM, Atherfold PA, Manson MM. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol. Pharmacol. 2006;69:1033–1040. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]
- Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell. Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat S, Verkleij A, Post JA. Peroxynitrite activates mitogen-activated protein kinase (MAPK) via a MEK-independent pathway: role for protein kinase C. FEBS Lett. 2001;499:21–26. doi: 10.1016/s0014-5793(01)02511-x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin JC, Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch. Biochem. Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G, Biroccio A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell. 2006;21:509–519. doi: 10.1016/j.molcel.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Heales SJR, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J. Neurochem. 1995;64:1965–1972. doi: 10.1046/j.1471-4159.1995.64051965.x. [DOI] [PubMed] [Google Scholar]
- Brito PM, Mariano A, Almeida LM, Dinis TCP. Resveratrol affords protection against peroxynitrite-mediated endothelial cell death: A role for intracellular glutathione. Chem. Biol. Int. 2006;164:157–166. doi: 10.1016/j.cbi.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Chen C, Kong A-NT. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Rad. Biol. Med. 2004;36:1505–1516. doi: 10.1016/j.freeradbiomed.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Chen TJ, Jeng JY, Lin CW, Wu CY, Chen YC. Quercetin inhibition of ROS-dependent and -independent apoptosis in rat glioma C6 cells. Toxicol. 2006a;223:113–126. doi: 10.1016/j.tox.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Chen YC, Chow JM, Lin CW, Wu CY, Shen SC. Baicalein inhibition of oxidative stress-induced apoptosis via modulation of ERKs activation and induction of HO-1 gene expression in rat glioma cells C6. Toxicol. Appl. Pharmacol. 2006b;216:263–273. doi: 10.1016/j.taap.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Jeong YJ, Lee YJ, Kwon HM, Kang YH. (-) Epigallocatechin gallate and quercetin enhnace survival signaling in response to oxidant-induced human endothelial apoptosis. J. Nutr. 2005;135:707–713. doi: 10.1093/jn/135.4.707. [DOI] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell. Signal. 2003;15:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Dajas F, Rivera F, Blasina F, Arredondo F, Echeverry C, Lafon L, Morquio A, Heizen H. Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox. Res. 2003;5:425–432. doi: 10.1007/BF03033172. [DOI] [PubMed] [Google Scholar]
- de Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J, Mena MA. Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J. Neurochem. 2004;91:667–682. doi: 10.1111/j.1471-4159.2004.02744.x. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Levonen A-L, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Rad. Biol. Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid. Redox Signal. 2005;7:1223–1233. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- El-Remessy AB, Ali Behzadian M, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB. Experimental diabetes causes a breakdown o fhte blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Amer. J. Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJL, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neuronal damage. J. Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Hanneken A, Lin F-F, Johnson J, Maher P. Flavonoids protect human pigmented retinal epithelial cells from oxidative stress-induced death. Invest. Ophthalmol. Vis. Sci. 2006;47:3164–3177. doi: 10.1167/iovs.04-1369. [DOI] [PubMed] [Google Scholar]
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Heim KE, Tagliaferro AR, Bobilya DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002;13:572–584. doi: 10.1016/s0955-2863(02)00208-5. [DOI] [PubMed] [Google Scholar]
- Ho HY, Wei TT, Cheng ML, Chiu DT. Green tea polyphenol epigallocatechin-3-gallate protects cells against peroxynitrite-induced cytotoxicity: modulatory effect of cellular G6PD status. J. Agric. Food Chem. 2006;54:1638–1645. doi: 10.1021/jf0524372. [DOI] [PubMed] [Google Scholar]
- Hoeldtke RD, Bryner KD, McNeill DR, Hobbs GR, Baylis C. Peroxynitrite versus nitric oxide in early diabetes. Amer. J. Hypertens. 2003;16:761–766. doi: 10.1016/s0895-7061(03)00976-2. [DOI] [PubMed] [Google Scholar]
- Hou D-X, Fukuda M, Johnson JA, Miyamori K, Ushikai M, Fujii M. Fisetin induces transcription of NADPH:quinone oxidoreductase gene through an antioxidant responsive element-involved activation. Int. J. Oncol. 2001;18:1175–1179. doi: 10.3892/ijo.18.6.1175. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Haddon CO, Rose S, Jenner P. 3-Nitrotyrosine-dependent dopaminergic neurotoxicity following direct nigral administration of a peroxynitrite but not a nitric oxide donor. Brain Res. 2006;1067:256–262. doi: 10.1016/j.brainres.2005.10.086. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- Ishii N, Patel KP, Lane PH, Taylor T, Bian K, Murad F, Pollock JS, Carmines PK. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J. Am. Soc. Nephrol. 2001;12:1630–1639. doi: 10.1681/ASN.V1281630. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell Survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Klejman A, Wegrzynowicz M, Szatmari EM, Mioduszewska B, Hetman M, Albrecht J. Mechanisms of ammonia-induced cell death in rat cortical neurons: Roles of NMDA receptors and glutathione. Neurochem. Int. 2005;47:51–57. doi: 10.1016/j.neuint.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Klotz L-O. Oxidant-induced signaling: Effects of peroxynitrite and singlet oxygen. Biol. Chem. 2002;383:443–456. doi: 10.1515/BC.2002.047. [DOI] [PubMed] [Google Scholar]
- Klotz L-O, Sies H. Defenses against peroxynitrite: selenocompounds and flavonoids. Toxicol. Lett. 2003;140-141:125–132. doi: 10.1016/s0378-4274(02)00511-8. [DOI] [PubMed] [Google Scholar]
- Kuperstein F, Yavin E. ERK activation and nuclear translocation in amyloid-beta peptide- and iron-stressed neuronal cell cultures. Eur. J. Neurosci. 2002;16:44–54. doi: 10.1046/j.1460-9568.2002.02056.x. [DOI] [PubMed] [Google Scholar]
- Lau FC, Shukitt-Hale J, Joseph J. The beneficial effects of fruits polyphenols on brain aging. Neurobiol. Aging. 2005;26:128–132. doi: 10.1016/j.neurobiolaging.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Levinthal DJ, DeFranco DB. Transient phosphatidylinositol 3-kinase inhibition protects immature primary cortical neurons from oxidative toxicity via suppression of extracellular signal-regulated kinase activation. J. Biol. Chem. 2004;279:11206–11213. doi: 10.1074/jbc.M314261200. [DOI] [PubMed] [Google Scholar]
- Levrand S, Pesse B, Feihl F, Waeber B, Pacher P, Rolli J, Schaller M-D, Liaudet L. Peroxynitrite is a potent inhibitor of NF-kB activation triggered by inflammatory stimuli in cardiac and endothelial cell lines. J. Biol. Chem. 2005;280:34878–34887. doi: 10.1074/jbc.M501977200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997a;19:453–63. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Lim SY, Jang JH, Na HK, Lu SC, Rahman I, Surh YJ. 15-Deoxy-delta 12,14-prostaglandin J(2) protects against nitrosative PC12 cell death through up-regulation of intracellular glutathione synthesis. J. Biol. Chem. 2004;279:46263–46270. doi: 10.1074/jbc.M406555200. [DOI] [PubMed] [Google Scholar]
- Lin HC, Cheng TH, Chen YC, Juan SH. Mechanism of heme oxygenase-1 gene induction by quercetin in rat aortic smooth muscle cells. Pharmacol. 2004;71:107–112. doi: 10.1159/000076947. [DOI] [PubMed] [Google Scholar]
- Maher P, Hanneken A. Flavonoids protect retinal ganglion cells from oxidative stress-induced death. Invest. Ophthalmol. Vis. Sci. 2005;46:4796–4803. doi: 10.1167/iovs.05-0397. [DOI] [PubMed] [Google Scholar]
- Maher P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Rad. Res. 2006;40:1105–1111. doi: 10.1080/10715760600672509. [DOI] [PubMed] [Google Scholar]
- Maher P, Akaishi T, Abe K. The flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. USA. 2006;103:16568–16573. doi: 10.1073/pnas.0607822103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, Salgado KF, Zivin JA, Lapchak PA.A novel approach to screening for new neuroprotective compounds for the treatment of stroke Brain Res 2007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Wang JM, Murakami T, Warita H, Hayashi T, Shoji M, Abe K. Expressions of nitrotyrosine and TUNEL immunoreactivities in cultured rat spinal cord neurons after exposure to glutamate, nitric oxide, or peroxynitrite. J. Neurosci. Res. 2001;65:371–377. doi: 10.1002/jnr.1163. [DOI] [PubMed] [Google Scholar]
- Middleton E, Kandaswami C, Theoharides TC. The effects of flavonoids on mammalian cells: implications for inflammation, heart disease and cancer. Pharmacol. Rev. 2000;52:673–751. [PubMed] [Google Scholar]
- Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J. Biol. Chem. 1998;273:15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- Myhrstad MCW, Carlsen H, Nordstrom O, Blomhoff R, Moskaug JO. Flavonoids increase the intracellular glutathione level by transactivation of the α-glutamylcysteine synthetase catalytical subunit promoter. Free Rad. Biol. Med. 2002;32:386–393. doi: 10.1016/s0891-5849(01)00812-7. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Bindokas VP, Kowlessur D, Elas M, Milstein S, Marks JD, Halpern HJ, Kang UJ. Tetrahydrobiopterin scavenges superoxide in dopaminergic neurons. J. Biol. Chem. 2001;276:34402–34407. doi: 10.1074/jbc.M103766200. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, Shamoto-Nagai M, Yi H, Akao Y, Tanaka M. Oxidative stress in mitochondria. Mol. Neurobiol. 2005;31:81–93. doi: 10.1385/MN:31:1-3:081. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nowak P, Olas B, Bald E, Glowacki R, Wachowicz B. Peroxynitrite-induced changes of thiol groups in human blood platelets. Platelets. 2003;14:375–379. doi: 10.1080/0953710032000141400. [DOI] [PubMed] [Google Scholar]
- Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K-I. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 1999;263:504–509. doi: 10.1006/bbrc.1999.1237. [DOI] [PubMed] [Google Scholar]
- Pannala A, Rice-Evans CA, Halliwell B, Singh S. Inhibition of peroxynitrite-mediated tyrosine nitration by catechin polyphenols. Biochem. Biophys. Res. Commun. 1997;232:164–168. doi: 10.1006/bbrc.1997.6254. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991a;266:4244–50. [PubMed] [Google Scholar]
- Rahman I, Biswas SK, Kirkham PA. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem. Pharmacol. 2006;72:1439–1452. doi: 10.1016/j.bcp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Ramassamy C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur. J. Pharmacol. 2006;545:51–64. doi: 10.1016/j.ejphar.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Vahnnasy J, Maher P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J. Neurochem. 2004a;90:1144–1155. doi: 10.1111/j.1471-4159.2004.02563.x. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Briviba K, Klotz L-O, Sies H. Activation pattern of mitogen-activated protein kinases elicited by peroxynitrite: attentuation by selenite supplementation. FEBS Lett. 1999;448:301–303. doi: 10.1016/s0014-5793(99)00372-5. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nuc. Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006a doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Sun X, Erb H, Murphy TH. Coordinate regulation of glutathione metabolism in actrocytes by Nrf2. Biochem. Biophys. Res. Commun. 2005;326:371–377. doi: 10.1016/j.bbrc.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Thuraisingham RC, Nott CA, Dodd SM, Yaqoob MM. Increased nitrotyrosine staining in kidneys from patients with diabetic nephropathy. Kidney Int. 2000;57:1968–1972. doi: 10.1046/j.1523-1755.2000.00046.x. [DOI] [PubMed] [Google Scholar]
- Torreilles F, Salman-Tabcheh S, Guerin M, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res. Brain Res. Rev. 1999a;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- Valerio LG, Kepa JK, Pickwell GV, Quattrochi LC. Induction of human NAD(P)H:quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin. Toxicol. Lett. 2001;119:49–57. doi: 10.1016/s0378-4274(00)00302-7. [DOI] [PubMed] [Google Scholar]
- van Acker FA, Schouten O, Haenen GR, van der Vijgh WJ, Bast A. Flavonoids can replace alpha-tocopherol as an antioxidant. FEBS Lett. 2000;473:145–148. doi: 10.1016/s0014-5793(00)01517-9. [DOI] [PubMed] [Google Scholar]
- White AR, Cappai R. Neurotoxicity from glutathione depletion is dependent on extracellular trace copper. J. Neurosci. Res. 2003;71:889–897. doi: 10.1002/jnr.10537. [DOI] [PubMed] [Google Scholar]
- Wild AC, Mulcahy RT. Regulation of α-glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Rad. Res. 2000;32:281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- Wu CC, Hsu MC, Hsieh CW, Lin JB, Lai PH, Wung BS. Upregulation of heme oxygenase-1 by epigallocatechin-3-gallter via the phosphatidylinositol 3-kinase/Akt and ERK pathways. Life Sci. 2006;78:2889–2897. doi: 10.1016/j.lfs.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Wu Z, Wu L, Tashiro S, Onodera S, Ikejima T. Phosphorylated extracellular signal-regulated kinase up-regulated p53 expression in shikonon-induced HeLa cell apoptosis. Chin. Med. J. (Engl) 2005;118:671–677. [PubMed] [Google Scholar]
- Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug. Metabol. Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wang Y-Z, Kagan E, Bonner JC. Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J. Biol. Chem. 2000;275:22479–22486. doi: 10.1074/jbc.M910425199. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rosenberg PA. Caspase-1 and poly (ADP-ribose) polymerase inhibitors may protect against peroxynitrite-induced neurotoxicity independent of their enzyme inhibitor activity. Eur. J. Neurosci. 2004;20:1727–1736. doi: 10.1111/j.1460-9568.2004.03651.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apopotosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J. Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouki C, Jozsef L, Ouellet S, Paquette Y, Filep JG. Peroxynitrite mediates cytokine-induced IL-8 gene expression and production by human leukocytes. J. Leukoc. Biol. 2001;69:815–824. [PubMed] [Google Scholar]