

Abstract
Exon/intron architecture varies across the eukaryotic kingdom with large introns and small exons the rule in vertebrates and the opposite in lower eukaryotes. To investigate the relationship between exon and intron size in pre-mRNA processing, internally expanded exons were placed in vertebrate genes with small and large introns. Both exon and intron size influenced splicing phenotype. Intron size dictated if large exons were efficiently recognized. When introns were large, large exons were skipped; when introns were small, the same large exons were included. Thus, large exons were incompatible for splicing if and only if they were flanked by large introns. Both intron and exon size became problematic at ≈500 nt, although both exon and intron sequence influenced the size at which exons and introns failed to be recognized. These results indicate that present-day gene architecture reflects at least in part limitations on exon recognition. Furthermore, these results strengthen models that invoke pairing of splice sites during recognition of pre-mRNAs, and suggest that vertebrate consensus sequences support pairing across either introns or exons.
Vertebrate genes are typically split into numerous small exons (average size, 134 nt) interrupted by much larger introns (1). Large introns present problems for initial splice site recognition via models that postulate interactions between factors that concertedly recognize the 5′ and 3′ splice sites located at intron termini. In vitro, interactions between the ends of introns have been observed during early spliceosome assembly both in yeast and in mammalian systems (reviewed in refs. 2–7). Most experiments with the mammalian system have utilized pre-mRNAs with internally deleted introns. Extrapolation of results from experiments with precursor RNAs containing small introns to more natural pre-mRNAs with large introns is difficult. An alternate spliceosome assembly mode for pre-mRNAs with small exons and large introns (reviewed in refs. 5–7), which we have termed exon definition, suggests that the earliest interactions between 3′ and 5′ splice sites occurs across exons rather than across introns. In vitro expansion of an internal exon to >300 nt severely inhibits the ability to detect ATP-dependent spliceosome formation (8), suggesting an experimental limit on exon size during exon definition that agreed well with known vertebrate exon sizes. Few experiments have addressed the ability of the in vivo vertebrate splicing machinery to recognize large exons.
Large exons are only a rarity in vertebrates. Lower eukaryotes have an inverted exon/intron architecture compared with higher eukaryotes such that many genes have small introns and large exons (1). Thus, many Schizosaccharomyces pombe or Caenorhabditis elegans genes have introns smaller than 100 nt, and ≈50% of the introns in Drosophila melanogaster are <100 nt. Expansion of such small introns in both S. pombe and D. melanogaster causes either a loss of splicing or incorrect splicing via utilization of cryptic splice sites within the expanded intron (refs. 9 and 10; J. Wise, personal communication). These results as well as differences in gene architecture across the eukaryotic kingdom could be interpreted to indicate an optimal intron size in genes with small introns, and an optimal exon size in genes with small exons. Furthermore, it suggests that genes containing both large exons and large introns might be problematic for the splicing machinery.
To test this hypothesis, exon size was altered via expansion with cDNA sequences, and the ability of such expanded exons to be included in spliced RNA was examined when the exons were flanked by small versus large introns with identical splice sites. To circumvent unavoidable problems associated with altering sequence as length is altered, multiple exon expansion sequences were tested for the ability to confer large exon recognition. The results indicate that large exons are effectively included in mRNA when they are flanked by small introns, but that the same large exons are skipped when the flanking introns are also large, irrespective of splice site strength. These results suggest that present day exon/intron architecture is limited by a requirement to keep either exons or introns short.
MATERIALS AND METHODS
Constructs.
Recipient genes to test exon size were the mouse metallothionein II (MT) gene driven by the Rous sarcoma virus enhancer-promoter and the complete, natural hamster adenine phosphoribosyl-transferase (APRT) gene driven by the APRT promoter. Exon expansion cassettes were derived from three different cDNAs: mouse adenosine deaminase (ADA), chicken ovalbumin (OVA), and hydroxy phosphoribosyl-transferase (HPRT). The mouse MT natural second exon was the recipient exon for the expansion cassette at its natural internal BamHI site. Expanded exons were transferred between the MT and APRT genes—the transferred cassette included flanking intron sequences derived from mouse MT II extending from the SmaI site upstream of exon 2 to the SmaI site downstream of exon 2. Thus, the expanded exons were always flanked by the splice sites natural to MT exon 2.
The ADA cDNA sequences represented a nested set of sequences with common 3′ termini and increasingly expanded 5′ termini from a mouse ADA cDNA provided by R. Kellems (Baylor College of Medicine). The largest ADA expansion cassette was from a 721 nt BamHI–BglII fragment; added to the natural MT exon 2, this expansion cassette produced an exon of 787 nt. Smaller inserts were prepared by internal deletion of this construct with BamHI–BspMI, BamHI–PstI, BamHI–BalI, or BamHI–BsmI to create exons of 526, 432, 314, or 222, respectively.
Chicken OVA cDNA and human HPRT cDNA inserts were prepared as nested sets with identical 5′ termini and inserted into mouse MT exon 2 at its BamHI site. OVA inserts were prepared from pOV250 provided by B. O’Malley (Baylor College of Medicine) via EcoRI–AccI, EcoRI–FokI, EcoRI–PstI, or EcoRI–StuI digestion to create exons of 219, 343, 532, or 855 nt, respectively. HPRT inserts were prepared from a cDNA provided by J. Wilson (Baylor College of Medicine) via NaeI–XhoI, NaeI–XhoII, NaeI–HindIII, or NaeI–BsmI digestion to create exons of 232, 354, 566, or 894 nt, respectively.
Intron expansions were performed by inserting increasing larger fragments of the human HPRT gene including exon 2 and its flanking intron sequences into the mouse MT gene such as to replace exon 2 and its intron flanks. The HPRT exon 2 was subsequently replaced with the test exon and its intron flanks. As constructed, the expansions represented internal expansion of the natural MT introns with insertion points distal to all known splicing consensus signals. The final constructs contained three size classes of introns flanking the test exon as denoted on Fig. 2.
Figure 2.
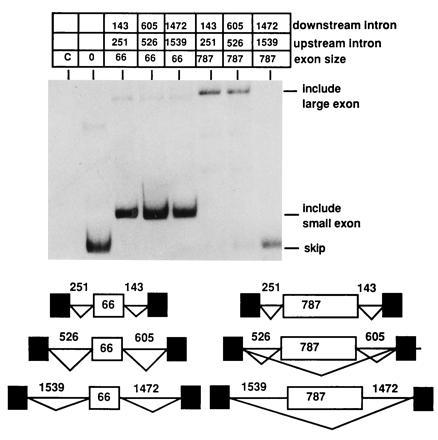
Expanding small introns flanking a large exon causes exon skipping. The MT constructs shown in Fig. 1C containing either the natural 66-nt second exon from MT or the 787-nt internally expanded second exon were altered to increase the lengths of both introns. Intron lengths in the final constructs are indicated. RNA splicing phenotypes were determined by RT-PCR as described in Fig. 1 using radiolabeled PCR primers. Products resulting from skipping of the middle exon or inclusion of either the small or large internal exon are indicated.
Reverse Transcription–PCR (RT-PCR) Analysis of Splicing Phenotypes.
RNA was prepared from transfected NIH 3T3 cells or CHO cells 48 hr after for analysis of the Rous sarcoma virus-driven MT gene or from stable transfections of CHO cells lacking both copies of the endogenous APRT gene (cell line U1S36, provided by J. Wilson, Baylor College of Medicine) for analysis of the APRT gene. For stable transformants, one T-75 flask containing cells at ≈60% confluency was cotransfected with 10 μg of test plasmid DNA and 1 μg of plasmid pSV2neo. Drug-containing medium [400 μg/ml geneticin (GIBCO/BRL) in medium] was added to the cells 48 hr after transfection. Resulting drug-resistant colonies were pooled and maintained in drug-free medium.
Total cell RNA was isolated using RNAzol B for RT-PCR amplification as previously described for other exons tested in the MT or APRT backbone genes (12). RT was performed in actinomycin D using oligonucleotide primers to exon 3 of MT or exon 4 of APRT (11). PCR amplification of produced cDNA was performed using 20–30 cycles. Oligonucleotide primers were specific for exons 2 and 5 of the APRT gene or from exons 1 and 3 of the MT gene (the primer to exon 1 was unique to the minigene to prevent detection of the endogenous gene). Products using unlabeled primers were displayed on 5% acrylamide gels and silver stained; reactions with labeled PCR primers were detected by autoradiography.
RESULTS
Internal Expansion of a Vertebrate Internal Exon Can Cause Exon Skipping.
To experimentally investigate the in vivo relationship between exon and intron size in mammalian pre-mRNA processing, we placed internally expanded exons into genes with small and large introns and tested the effect of exon expansion on splicing phenotype. The 66-nt middle exon from the mouse MT II gene was expanded with increasingly larger cDNA cassettes derived from the mouse ADA, chicken OVA, or human HPRT genes. The expansion cassettes increased the exon length from a minimum of 70 to a maximum of 894 nt. The expanded exons (including their flanking splice sites) were tested for in vivo exon inclusion within both the homologous MT and heterologous hamster APRT genes. In APRT the flanking introns created by the fusion were 642 and 449 nt and in MT the flanking introns were 251 and 143 nt.
The resulting constructs were transfected into cultured cells and the splicing phenotypes were analyzed by RT-PCR amplification of total cell RNA using primers directed against the exons flanking the inserted expanded exon. Fig. 1 shows the observed splicing phenotypes of exons expanded with ADA cDNA sequences (Fig. 1A) when placed into either the APRT (Fig. 1B) or MT (Fig. 1C) genes. The same exons had strikingly different phenotypes when assayed in the two different genes. When the expanded exon with its splice sites was inserted into APRT, the test exon was included when it was 66, 70, 222, or 314 nt. When the exon was 432 nt, both inclusion and skipping were observed. When the exon was 526 nt, the majority of the observed product resulted from exon skipping. When the exon was 787 nt, the only product observed resulted from exon skipping. Thus, as the size of the exon increased, exon inclusion decreased and exon skipping increased.
Figure 1.
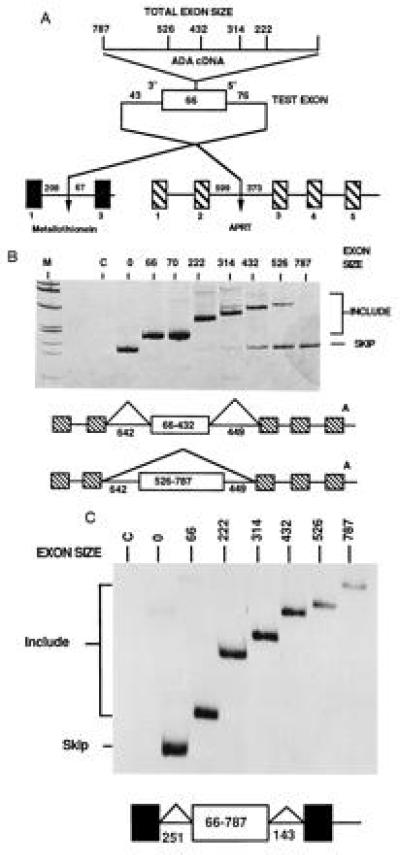
Large internal exons have different splicing phenotypes when placed in different genes. (A) Structure of the genes used to test exon size limits. Exon expansion cassettes derived from cDNAs were used to expand the natural second exon of mouse MT II which was then placed into test genes. (B) In vivo splicing phenotypes of expanded exons containing an expansion cassette from ADA cDNA within the CHO APRT gene. The hatched exons are from the resident APRT gene, the white exon is the expanded exon 2 from MT. Splicing phenotypes were determined by RT-PCR analysis of total cell RNA. To emphasize visualization of RNAs resulting from exon inclusion, the amplification products were detected by silver staining; therefore, an equal intensity of skipping to inclusion products with exons of 400–800 nt represents a strong bias toward skipping. Lanes C and 0 indicate amplification of RNA from nontransfected cells and the parental APRT gene, respectively. (C) In vivo splicing phenotypes of the same expanded exons tested within the MT gene. The gel shown is an autoradiogram of an amplification reaction using labeled PCR primers. The results shown are from transfection of CHO cells; a similar result was observed when the recipient cells were NIH 3T3 cells.
The results from the MT gene, however, were remarkably different. In this case, all exons were constitutively included, even an exon of 787 nt. Furthermore, RNA levels were approximately equal for all constructs, indicating that large exon sequences presented no problems for RNA production in this minigene. Note that identical internal exons were used in the experiments in Fig. 1 B and C. Therefore, the RNA products resulting from exon inclusion that are being amplified by RT-PCR in the two experiments contain similar internal sequences, minimizing concerns about the inability to detect exon inclusion in the APRT backbone because of difficulties in amplifying large sequences (see Fig. 2). These results suggested that identical large exons were a problem for the mammalian splicing machinery in some, but not all pre-mRNAs.
Inclusion of an Expanded Internal Exon Depends on the Size of the Flanking Introns.
Two obvious differences exist between the two experiments described in Fig. 1 B and C. In the MT gene the expanded exon had splice sites from the recipient gene and the test exon replaced the natural exon. In the APRT backbone the test exon has splice sites from a heterologous gene, thereby presenting possible splice site incompatibility problems. A second difference between the two employed genes was the size of the two introns flanking the inserted expanded internal exon. In MT the flanking introns were noticeably smaller than those in the APRT gene.
To ascertain if either of these differences was responsible for the phenotypic differences between the two genes, the two introns in the MT gene were internally expanded with intron sequences from the human HPRT gene (Fig. 2). In this context, splice site incompatibility can be ruled out and only effects of intron size or sequence should be assayed. Such an experiment can also rule out any concerns that the difference in phenotypes resulted from a differential ability of PCR amplification to represent the larger RNA species because identical primers and exons were utilized. Analysis of the splicing phenotype of these constructs indicated that as intron size increased, the ability to include a small middle exon of 66 nt was unaffected, but the ability to include a large middle exon of 787 nt severely decreased (Fig. 2). Therefore, altering the size of the intron altered the ability to recognize a large middle exon despite the presence of identical splice site sequences. The large exon was completely skipped when the average size of the flanking introns was 1505 nt. This size is slightly larger than the average size of vertebrate introns (1). When the average size of the flanking introns was 566, the majority of the transcripts included the test exon, but even with introns of this size, some exon skipping was observed. When the average size of the flanking introns was 197 nt, only exon inclusion was observed.
We did notice that the MT gene in Fig. 2 with average expanded flanking introns of 566 nt was better able to afford inclusion of the large 787-nt exon than the APRT gene used in Fig. 1 with average flanking intron size 545 nt. We attribute this difference to the presence of stronger and homologous splice sites within the MT construct as compared with the APRT construct. A compensatory relationship between exon size and splice site strength has been previously reported for exons that are restricted for inclusion because they are very short (12, 13).
The Sequence of the Expanded Exon Dictates if Skipping or Activation of Internal Cryptic Sites Occurs.
The results in Figs. 1 and 2 indicate that exons expanded with ADA cDNA sequences are unable to be included if the neighboring introns are large. Any such experiment is limited by the necessity of altering sequence as exon size is altered. To compare exons of different sequence for their ability to be included, we analyzed the phenotypes of exons expanded with cDNAs derived from OVA or HPRT (Fig. 3). The exons expanded with OVA cDNAs acted similarly to those expanded with ADA sequences. The ADA-expanded exons began to be skipped when their length exceeded 500 nt when they resided within the APRT gene (Fig. 3A), but were constitutively included within the MT gene (data not shown). Observation of skipping with long exons containing expansion cassettes from different cDNA sequences suggested that skipping was a property of length rather than internal exon sequence. The largest tested OVA exon was included better than an exon of similar size expanded with ADA sequences suggesting that some exon sequences are better able to support the inclusion of large exons.
Figure 3.
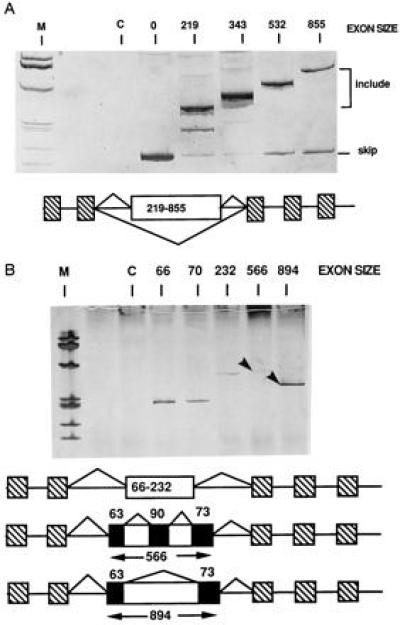
Exon sequence influences splicing phenotype. APRT genes containing an expanded MT exon similar to those described in Fig. 1A were created in which the exon expansion cassette was derived from either chicken OVA (A) or human HPRT (B) cDNAs. RNA splicing phenotypes were determined by RT-PCR as described in Fig. 1. Products resulting from skipping or inclusion of the middle exon are indicated. The unexpected products in B from the constructs with middle exons of 556 or 894 nt are indicated with an arrow and were sequenced. The splicing patterns as determined from sequencing data are depicted below the figure. Both gels were silver-stained to visually emphasize inclusion products. The cryptic spliced transcripts produced in B are denoted with an arrow.
In contrast to the results using expansion cassettes from ADA and OVA, exons expanded with HPRT cDNA sequences showed a prominent sequence effect. In this case, in either the MT (data not shown) or APRT (Fig. 3B) gene backbone, expansion of the exon to lengths greater than 230 nt resulted in aberrant splicing via activation of cryptic splice sites within the cDNA cassette. Sequencing of the RT-PCR products from these transfections indicated that both 3′ and 5′ splice sites were activated within the expanded exons to create new small exons of 63, 73, and 90 nt. Utilization of cryptic splice sites within an expanded exon, like exon skipping, is suggestive of an inability of the mammalian splicing machinery to handle large exons. The observation of cryptic utilization regardless of intron length suggests that cryptic utilization occurs instead of exon skipping merely because of the fortuitous presence of cryptic splice sites within the expansion cassette.
DISCUSSION
Exon/intron architecture varies across the eukaryotic kingdom. Genes with small exons are usual in vertebrates; genes with small introns are normal in invertebrates. To investigate if a compensatory relationship exists between exon and intron sizes, we altered the size of a vertebrate internal exon and tested the ability of this exon to be included in vivo when flanked by introns of different size. Expanded exons were efficiently included if, and only if the flanking introns were modest in size (<500 nt). When expanded exons were surrounded by large introns, the exons were efficiently skipped. This change in splicing phenotype occurred despite the presence of identical splice sites in the precursor RNAs with large and small introns. We conclude from these results that large exons bordered by large introns is an exon/intron architecture that is problematic for splice site recognition. The paucity of genes with such an architecture within existing data bases supports such a restriction on gene structure. Mechanistically, these results support models that invoke initial pairing between 3′ and 5′ splice sites as a necessary step in pre-mRNA recognition, and emphasize the role distance plays in both exon and intron recognition.
The compensatory relationship between exon and intron size observed in this study suggests that pre-mRNAs are recognized via interactions between the factors that bind 3′ and 5′ splice sites in either an intronic or exonic polarity. In vitro splicing experiments have detected interactions occurring between the 5′ and 3′ splice sites that border an intron as well as the 3′ and 5′ splice sites that border an exon (2–7). Thus, interactions have been detected in both polarities. It is unclear if the interactions occurring in an intronic pair are identical to those occurring in an exonic pair of splice sites. Our results suggest that as long as either the intronic or exonic distance between opposite sites is small in a pre-mRNA region, splice site recognition and splicing will occur. Only when both the introns and the exons were large did splicing efficiency decrease. In our study both exons and introns became problematic when they exceeded 500 nt, suggesting that both exonic and intronic pairing of splice sites have a similar size maximum.
One other study has examined the ability of large internal exons to be recognized in vivo (14). This study used an artificial dihydrofolate reductase (DHFR) minigene with two small introns (304 and 275 nt) created using the splice sites from the natural intron 1 of the gene. In agreement with our results, this minigene was able to include middle exons expanded to over 1 kb with bacterial sequences. Thus, two different genes with small introns were able to splice large internal exons. The splice sites used in our gene were from a gene with naturally small introns; the splice sites used in the DHFR study were from a naturally large intron, again suggesting the interconvertability of vertebrate splice site pairing from exonic to intronic pairs and vice versa.
It should be noted that exon or intron size alone was not solely determinative for exon recognition. Both intron and exon sequence influenced the ability of large exons to be recognized when flanked by large introns. A similar compensatory relationship between exon and intron sequence and exon size has previously been noticed for other, more normal size exons and for very small exons (5–7, 12, 13). Thus, the ability of an exon to be recognized is the sum of splice site strength, accessory element (exon or intron) recognition, and exon size. The scarcity of natural vertebrate genes with large exons flanked by large introns suggests that exon size is still an important parameter in vertebrate splicing. Perhaps more importantly, organisms with genomes organized into genes with large exons and small introns should not require exonic splicing enhancer sequences.
The observation that large internal exons are problematic for recognition if they are flanked by large introns suggested that naturally occurring large vertebrate exons might be flanked by small introns. Such a restriction on exon and intron length is impossible to examine statistically because there are so few natural large internal exons in vertebrate genes. A few such exons exist, however (1, 15, 16). Interestingly, the two natural, large exons studied to date with respect to splicing (15, 16) are both alternatively utilized, suggesting that nonoptimal size, like suboptimal splice site strength, may be a characteristic of alternative exons. Our data suggest that such exons possess accessory sequences that facilitate their inclusion. We have previously studied a large alternatively recognized exon of over 1 kb that contains such a splicing enhancer sequence (15), and suggest that the presence of splicing enhancer sequences will characterize natural large exons. It should be noted that the restriction on internal exon size discussed here does not apply to 3′-terminal exons. Terminal exons are usually the largest exon in a vertebrate gene and can be quite large (1). Even here, however, large, alternative 3′-terminal exons have been shown to contain enhancer sequences (17, 18).
Supported by PHS GM38526 and the Robert A. Welch Foundation.
Footnotes
Abbreviations: MT, metallothionein II; APRT, adenine phosphoribosyl-transferase; ADA, adenosine deaminase; OVA, ovalbumin; HPRT, hydroxy phosphoribosyl-transferase; RT-PCR, reverse transcription–PCR.
References
- 1.Hawkins J D. Nucleic Acids Res. 1988;16:9893–9908. doi: 10.1093/nar/16.21.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore M J, Query C C, Sharp P A. In: RNA World. Gesteland R F, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 305–358. [Google Scholar]
- 3.Ruby S, Abelson J. Trends Genet. 1991;7:79–85. doi: 10.1016/0168-9525(91)90276-V. [DOI] [PubMed] [Google Scholar]
- 4.Guthrie C. Science. 1991;253:157–163. doi: 10.1126/science.1853200. [DOI] [PubMed] [Google Scholar]
- 5.Berget S M. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 6.Black D L. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 7.Reed R. Curr Opin Genet Dev. 1996;6:215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 8.Robberson B L, Cote G J, Berget S M. Mol Cell Biol. 1990;10:84–94. doi: 10.1128/mcb.10.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo M, Lo P C H, Mount S M. Mol Cell Biol. 1993;13:1104–1118. doi: 10.1128/mcb.13.2.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talerico M, Berget S M. Mol Cell Biol. 1994;14:3434–3445. doi: 10.1128/mcb.14.5.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterner D A, Berget S M. Mol Cell Biol. 1993;13:2677–2687. doi: 10.1128/mcb.13.5.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominski Z, Kole R. Mol Cell Biol. 1991;11:6075–6083. doi: 10.1128/mcb.11.12.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Black D L. Genes Dev. 1991;5:389–402. doi: 10.1101/gad.5.3.389. [DOI] [PubMed] [Google Scholar]
- 14.Chen I-T, Chasin L A. Mol Cell Biol. 1994;14:2140–2146. doi: 10.1128/mcb.14.3.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphrey M B, Bryan J, Cooper T A, Berget S M. Mol Cell Biol. 1995;15:3979–3988. doi: 10.1128/mcb.15.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacke R, Goridis C. Genes Dev. 1991;5:1416–1429. doi: 10.1101/gad.5.8.1416. [DOI] [PubMed] [Google Scholar]
- 17.Tian M, Maniatis T. Cell. 1993;74:105–114. doi: 10.1016/0092-8674(93)90298-5. [DOI] [PubMed] [Google Scholar]
- 18.van Oers C C M, Adema G J, Zandberg H, Moen Y C, Baas P D. Mol Cell Biol. 1994;14:951–960. doi: 10.1128/mcb.14.2.951. [DOI] [PMC free article] [PubMed] [Google Scholar]