

Abstract
Background
Patient prognosis and response to endocrine therapy in breast cancer correlate with protein expression of both estrogen receptor (ER) and progesterone receptor (PR), with poorer outcome in patients with ER+/PR− compared to ER+/PR+ tumors.
Methods
To better understand the underlying biology of ER+/PR− tumors, we examined RNA expression (n > 1000 tumors) and DNA copy number profiles from five previously published studies of human breast cancers with clinically assigned hormone receptor status (ER+/PR+, ER+/PR−, and ER−/PR−).
Results
We identified an expression signature of genes with either elevated or diminished RNA levels specifically in ER+/PR+ compared to ER−/PR− and ER+/PR− tumors. We similarly identified a gene signature specific to ER−/PR− tumors. ER+/PR− tumors, on the other hand, were a mixture of three different subtypes: tumors manifesting the ER+/PR+ signature, tumors manifesting the ER−/PR− signature, and tumors not associating with ER+/PR+ or ER−/PR− tumors (which we considered “true” ER+/PR−). In analyses of both tamoxifen-treated and untreated patients, ER+/PR− breast cancers defined by RNA profiling were associated with poor patient outcome, worse than those with pure ER+/PR+ patterns; these differences were not observed when using clinical assays to assign ER and PR status. ER+/PR− tumors also showed twice as many DNA copy number gains or losses compared to ER+/PR+ and ER−PR− tumors. Targets of transcriptional up-regulation by specific oncogenic pathways, including PI3K/Akt/mTOR, were enriched in both ER+/PR− and ER−/PR− compared to ER+/PR+ tumors.
Conclusion
ER+/PR− tumors as defined by RNA profiling represent a distinct subset of breast cancer with aggressive features and poor outcome, despite being clinically ER+. Multigene assays derived from our gene signatures could conceivably provide an improved clinical assay for inferring PR status for prognostic and therapeutic purposes.
Keywords: breast cancer, estrogen receptor, progesterone receptor, ER+/PR−, gene expression profiling, meta-analysis
Introduction
Estrogen and the estrogen receptor (ER) play key roles in breast cancer development and progression [1, 2]. Approximately 70% of breast cancers express ER (ER+). ER+ tumors are sensitive to endocrine therapy, while ER− tumors are hormone independent [3, 4]. Progesterone receptor (PR) mediates progesterone’s effects in the development of the mammary gland and breast cancer [2, 5]. More than half of ER+ breast cancers express PR [2, 6], and estrogen signaling via ER is necessary to induce PR expression [7, 8]. ER and PR are prognostic factors for patient outcome, though both are considered weak [9]. Although ER is an accepted predictor of response to endocrine therapy [1–4, 10], the predictive ability of PR has been more controversial [2]. PR was found to be a strong predictor of response to tamoxifen in a randomized clinical trial in pre-menopausal women [11]. Additionally, retrospective analysis of adjuvant tamoxifen therapy has shown poorer response in ER+/PR− compared to ER+/PR+ tumors [9, 12, 13]. In metastatic breast cancer, a prospectively designed clinical trial of tamoxifen showed an independent role for PR in predicting response and time to progression [14, 15]. In the adjuvant setting, two large clinical trials of aromatase inhibitor versus tamoxifen showed poorer outcome in PR−negative disease [16, 17]. In contrast, however, the Oxford overview of all trials of tamoxifen therapy in the early breast cancer setting found that PR status did not predict benefit [18].
The inconsistent results of PR status in predicting response to endocrine therapy in the metastatic and adjuvant settings are difficult to explain. Recent clinical correlative studies in large numbers of patients have shown that PR loss is associated with lower levels of ER, more positive nodes, aneuploidy, slightly larger tumor size, higher rates of proliferation, and expression of EGFR and HER2 [12, 19, 20], factors that would be expected to correlate with both a more aggressive tumor phenotype and resistance to endocrine therapy. Recent molecular studies designed to explain loss of PR expression in breast cancer suggests that in most tumors loss is not simply due to low estrogen levels or a malfunctioning ER pathway, but is more likely due to suppression of PR transcription by hyperactive growth factor signaling pathways that can also modify ER functions [2, 21, 22], by hypermethylation of the PR promoter [23], or even by loss-of-heterozygosity at the PGR gene locus [2]. These studies in total suggest that ER+/PR− tumors may be a distinct subset of breast cancer.
Numerous gene expression profiling studies have focused on a molecular classification of breast cancer into different subsets that reflect clinical subtypes [24–27] (e.g. ER+ versus ER−, or high versus low grade) or differences in patient outcome or treatment response [27–34]. Here we examined both gene expression and DNA copy number profiles in human breast cancer, with the goal of defining and characterizing global molecular patterns and potential underlying biology of ER+/PR− tumors. We hypothesized that ER+/PR− tumors represent a specific molecularly-defined subset of human breast cancer completely different from ER+/PR+ tumors and that this subset would demonstrate a unique natural history and response to treatment.
Methods
Breast tumor gene expression profile datasets
The breast tumor gene expression profile datasets used in this analysis were previously processed and publicly available from studies by Wang et al. [30], Miller et al. [35], van de Vijver et al. [28], Sorlie et al. [25], Hoadley et al. [36], Loi et al. [37], and Chin et al. [26]. CGH array data were also obtained from the Chin study. The sources of these datasets are listed in Supplementary Table 1. For the Miller and Loi datasets, only the U133A profiles and not the U133B profiles were considered (for the subset of Loi profiles generated on U133 plus2, the subset of the probes shared by U133A were selected for analysis). For analysis of the Sorlie dataset, gene probes with no data values in over two-thirds of the profiled samples were removed from consideration.
Estimated expression values were log-transformed and centered on the mean centroid (i.e. the mean of the means) of the clinical assay-based ER+/PR+, ER+/PR−, and ER−/PR− groups (Sorlie and Hoadley datasets: mean centroid of the luminal A, luminal B, and basal subtypes). Expression and DNA copy number values were visualized as heat maps using the Cluster and Java TreeView software [38, 39]. Further details on our use of the mean centroid are provided in Supplementary Material.
Determination of ER and PR status
ER and PR protein expression was assessed in most of these studies by either biochemical assay (e.g. Wang, Miller) or immunohistochemistry (e.g. van de Vijver). There were very few clinical assay-based ER−/PR− tumors in the Loi dataset, so these were not considered here. For the Miller, Chin, and Loi datasets, clinical ER and PR status information was provided through the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). For the Miller dataset, four profiles had been designated as PR+, with unknown ER status; on the basis of mRNA expression, these tumors were inferred to be ER+. For the Wang dataset, as indicated [30], ER and PR protein expression was measured mainly by ligand-binding assay (LBA) or enzyme immunoassay (EIA), though a few were by IHC; we called ER values of > 10 fmol/mg protein as ER+ and PR values of > 4 fmole/mg protein as PR+ (thereby being conservative in calling ER+/PR− tumors that truly expressed ER but had little or no PR, though qualitatively we found our overall findings to be essentially the same when considering alternative cut points, e.g. 5 for ER or 10 for PR, see Supplementary Material and Figure S2 in Additional File 1). For the Wang dataset, 22 profiles that had been called as ER−/PR+ were not considered in the analysis (as ER−/PR+ tumors are rare and some are thought to reflect methodological problems resulting in a false-negative ER assay [3, 9]). For the van de Vijver dataset, IHC information for ER and PR was provided in terms of percentage of tumor cells with positive staining for ER and PR expression, respectively; greater than 10% positive cells was called as ER+ or PR+; ten profiles were called as ER−/PR+ and so were removed from the analysis. For the Wang, Miller, and van de Vijver datasets, profiles used in the analysis, along with biochemical assay or IHC values, are available as Additional File 2.
Mapping between array datasets
Mapping gene expression patterns between profile datasets was done either using the gene probe identifier, in the case where the datasets were generated on the Affymetrix U133 platform (Wang, Miller, Chin, and Loi datasets), or using the Entrez gene identifier, in the case where the datasets were generated on a different platform (van de Vijver, Sorlie, and Hoadley datasets). Where a gene was represented by more than one probe in the van de Vijver, Sorlie, and Hoadley datasets, the “best” probe was selected in an unbiased manner to represent the gene for each dataset (van de Vijver, the probe with the most variation; Sorlie and Hoadley, the probe with the most unflagged values, followed by the probe with the most variation). For the ER+/PR+ gene signature, 144, 100, and 113 genes of the original 152 could be mapped to the van de Vijver, Sorlie, and Hoadley datasets, respectively; while for the ER−/PR− gene signature, 762, 570, and 538 genes of the original 811 genes could be mapped to the van de Vijver, Sorlie, and Hoadley datasets, respectively.
Definition of ER+/PR+ and ER−/PR− gene signatures
Two-sample t-tests were performed as criteria for determining significant differences in mean gene mRNA levels between groups of samples. When defining gene signatures specific to ER+/PR+, ER+/PR−, or ER−/PR− tumors (Figure 1A), one tumor class was compared separately with each of the other two tumor classes (i.e. ER+/PR+ versus ER+/PR−, and ER+/PR+ versus ER−/PR−, as compared by t-test, for the ER+/PR+ signature), and genes showing concordant high or low expression in both comparisons at a particular nominal p-value (see Supplementary Material) were selected to represent the class-specific signature for a particular dataset. Permutation testing was used to assess for false positives: for each of the two comparisons, the sample labels were permuted 100 times, and the genes intersecting with concordant direction between the two comparisons were tabulated for each permutation test. The same type of permutation testing was carried out for the DNA copy number profile analysis.
Figure 1.

Global gene expression patterns associated with ER+/PR+ (“+/+”), ER+/PR− (“+/−“), and ER−/PR− (“−/−“) clinical assay-assigned human breast tumors. (A) Number of RNA transcripts found over- or under-expressed specifically in one of the three breast tumor subtypes in each of the Wang and Miller expression profile datasets, as well as the number of subtype-specific transcripts common to both datasets (“Wang | Miller”). +/+ gene signature, genes/transcripts expressed specifically in ER+/PR+ compared to both ER+/PR− and ER−/PR− tumors (p < 0.01 each comparison in Wang dataset, p < 0.05 each comparison in Miller dataset). −/− gene signature, genes expressed specifically in ER−/PR− compared to both ER+/PR+ and ER+/PR− tumors. (B) Heat map representation of expression patterns of the ER+/PR+ and ER−/PR− gene signatures in the Wang and Miller profile datasets. Rows of the map represent genes; columns, profiled tumors. Relative expression is represented as colorgram (yellow: high expression). Expression values centered on the mean of the clinical group centroids. RNA expression patterns for genes ER, PR, and HER2 corresponding to the tumors are also indicated, as well as protein expression of ER and PR in the Wang dataset (protein data not available for Miller dataset), and recurrence events (gray: missing data). (C) Heat map representation of ER+/PR+ and ER−/PR− gene signatures in ER+/PR− tumors only (expanded from part B). Profiles were manually sorted to highlight those similar to ER+/PR+ and those similar to ER−/PR− tumors.
Gene classifier
Classification of the breast tumor profiles on the basis of the gene signatures specific to ER+/PR+ or ER−/PR− (Figure 2) was carried out in the following steps: (1) each transcript in a gene signature of interest was represented as 1 or −1 (for “up” or “down,” respectively); (2) after centering the tumor profiles on the mean of the centroids of the clinical assay-defined subgroups for the given dataset, the Pearson’s correlation was computed for each tumor profile between its expression values and the gene signature pattern; (3) tumor profiles with a positive correlation ( > 0) to the ER−/PR− signature were classified as ER−/PR−; the rest of the tumors were classified as ER+/PR+ if they had positive correlation with the ER+/PR+ signature. A “leave one out” approach for determining the mean centroid without the sample profile being classified was found to yield identical results to those using the global mean centroid (Supplementary Material).
Figure 2.
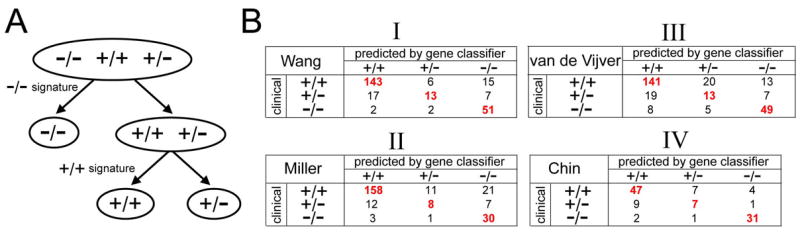
Classification of breast tumors into ER+/PR+, ER+/PR−, and ER−/PR− on the basis of gene expression profiles, as compared to clinical assay-based classification. (A) Decision tree for classifying the subtype of a tumor profile using the ER+/PR+ and ER−/PR− gene signatures (Figure 1). (B) Confusion matrices comparing subtype assignments based on gene classifier with the clinical-based assignments in the Wang and Miller datasets used to originally define the signatures (panels I and II, respectively), as well as in additional datasets from van de Vijver et al. [28] and Chin et al. [26] (panels III and IV, respectively), which were not used to define the signatures. Red bold denotes a significant number of assignments of an actual class within a predicted class (p < = 0.002, one-sided Fisher’s exact).
Results
Gene expression signature patterns of ER+/PR+ and ER−/PR− breast tumors
In studying the gene expression patterns of ER+/PR− tumors, our preliminary analysis (Supplementary Material and Figure S1 in Additional File 1) indicated that we had to consider the patterns of ER+/PR− relative to both ER−/PR− and ER+/PR+ tumors, as many of the genome-wide expression patterns of ER+/PR− tumors were shared by ER−/PR− tumors. We therefore set out to define gene signatures specific to ER+/PR+, ER+/PR−, and ER−/PR− tumors by identifying genes with high or low expression in only one of the three subtypes compared to the other two subtypes (Figure 1A). We relied upon two independent gene expression profile datasets of breast cancer for which ER and PR status was available: one from Wang et al. [30] of 256 tumors—164 ER+/PR+, 37 ER+/PR−, and 55 ER−/PR−; and one from Miller et al. [35] of 251 tumors—190 ER+/PR+, 27 ER+/PR−, and 34 ER−/PR−. For both datasets, ER and PR were determined by biochemical assay as part of the routine clinical evaluation. We did not consider clinical assay-defined ER−/PR+, as these tumors are rare and thought to represent methodological problems with assay measurements [3, 9].
From the two datasets separately, we first obtained sets of genes fitting each of our pre-defined patterns (Figure 1A), and then took the intersection of the Wang and Miller results, thereby defining patterns that were reproducible. An ER+/PR+ signature common to both datasets contained 182 transcripts (Figure 1A-I, 152 unique named genes, 74 high in ER+/PR+). Similarly, an ER−/PR− signature common to both datasets contained 1005 transcripts (Figure 1A-III, 811 genes, 349 high). The expected false positive number of genes in these signatures was very low (close to zero), and the concordance of results between the two datasets was high (Supplementary Material). The numbers of transcripts in the ER+/PR− signatures (Figure 1A-II) were considerably closer to chance expected, and no transcripts were shared by the two datasets, suggesting that few genes are uniformly higher (or lower) in ER+/PR− relative to the ER+/PR+ and ER−/PR− tumors.
Genes down in the ER−/PR− signature shared some overlap with the genes up in the ER+/PR+ signature (17 shared genes), and genes up in the ER−/PR− signature, with genes down in the ER+/PR+ signature (16 genes); such overlap would happen where, for instance, a gene is high in ER+/PR+, intermediate in ER+/PR−, and low in ER−/PR− tumors, and all three groups are significantly different from each other. Additional File 3 includes the complete list of genes in the ER+/PR+ and ER−/PR− signatures. Many estrogen-regulated genes or those previously associated with ER+ tumors (e.g. ESR1, GATA3, IRS1, IGF1R, CCND1, BCL2, NRIP1, GREB1) were among the genes low in the ER−/PR− signature (i.e. these genes were high in both ER+/PR+ and ER+/PR− tumors). PGR (the PR gene) was represented in the ER+/PR+ but not the ER−/PR−signature (because its expression was similar in the ER−/PR− and the ER+/PR− groups).
ER+/PR− tumors share gene expression patterns with both ER+/PR+ and ER−/PR−
Notably, we could not define a gene expression signature specific to ER+/PR− compared to both ER+/PR+ and ER−/PR− tumors (Figure 1A-II), suggesting that ER+/PR− tumors share some expression patterns with ER−/PR− and other patterns with ER+/PR+. We viewed the expression patterns of the ER+/PR+ and ER−/PR− gene signatures as heat maps (Figures 1B and 1C). Almost all of the ER+/PR+ tumors manifested the ER+/PR+ signature patterns (i.e. the tumors had high expression of the “up” genes and low expression of the “down” genes in the signature), and most all of the ER−/PR− tumors manifested the ER−/PR− signature. ER+/PR− tumors, however, were a mixture of three different subtypes (Figure 1C): 1) tumors associating with ER+/PR+ tumors, in terms of manifesting the ER+/PR+ signature; 2) tumors associating with ER−/PR−, in terms of the ER−/PR− signature despite being ER+ clinically; and 3) tumors that could not be aligned entirely with either ER+/PR+ or ER−/PR−. Tumors in this third group shared some similarities with ER−/PR−, in that they did not manifest the gene signature specific to ER+/PR+; this third group also had similarities to ER+/PR+, in that they did not manifest the signature specific to ER−/PR−. The heat maps indicated that some ER+/PR− tumors associating with ER−/PR− had low ER mRNA, and other ER+/PR− associating with ER+/PR+ had high PR mRNA (Figure 1C), suggesting that false positive clinical assay results could be involved; at the same time, however, the correlation of ER/PR mRNA with the gene signature-based associations was not perfect (Figure 1C), indicating that other genes in the signatures were contributing information as well.
To determine how well our gene signatures correlated with clinically measured ER and PR protein status, we developed a gene classifier to stratify breast tumors into ER+/PR+, ER+/PR−, or ER−/PR− on the basis of their gene expression profiles (centered on the mean centroid of the clinical assay-defined groups). The classifier worked in two steps (schematic in Figure 2A). First, profiles were classified as either “ER−/PR−” or “not ER−/PR−,” on the basis of the ER−/PR−signature. Second, the non-ER−/PR− tumors were further classified as either “ER+/PR+” or “not ER+/PR+,” on the basis of the ER+/PR+ signature. Tumors that did not associate with either ER−/PR− or ER+/PR+ were classified as ER+/PR− by default. Importantly, the classifier made use of the known direction of the genes in each signature (by use of the Pearson’s correlation, which indicated whether the “up” genes and the “down” genes in each signature were coordinately up or down, respectively, in the classified sample); in this way, a random selection of genes (with randomly assigned directions) would not have yielded similar results to that of the actual genes used.
We first classified the Wang and Miller tumors using the ER+/PR+ and ER−/PR− signatures. As expected (as the signatures were derived from these datasets), the vast majority of the tumors classified as ER+/PR+ or ER−/PR− by clinical assay were classified as such by gene expression, with 89% ([143+51]/[164+55]) and 84% ([158+30]/[190+34]) accuracies in the Wang and Miller datasets, respectively (Figure 2B, panels I and II). The clinical assay-defined ER+/PR− tumors, however, were only classified as such about 33% of the time are were nearly evenly divided among the three (see Figure 1C). Of the 37 and 27 ER+/PR− tumors defined by clinical assay in the Wang and Miller datasets, respectively, 13 and 8, respectively, were defined as ER+/PR− by our gene classifier; 17 and 12, respectively, were defined as ER+/PR+, and 7 and 7 were defined as ER−/PR−. Despite the high rate of misclassification, the number of clinical ER+/PR− tumors classified as ER+/PR− by profiling was statistically significant (p < 0.001 each dataset, one-sided Fisher’s exact). In addition, some clinical assay-defined ER+/PR+ or ER−/PR− tumors were defined by gene classifier as ER+/PR− (Wang: 6 and 2 tumors, respectively; Miller: 11 and 1, respectively).
The Wang and Miller datasets were originally used to define the ER+/PR+ and ER−/PR− gene signatures on which the classifier was based, and so the concordance between the clinical assay and classifier results were what would be expected, based on the signature heat maps (Figures 1B and 1C). Of note, however, the Wang and Miller results do indicate that, even under the most optimal conditions of using a gene classifier on the same datasets used to develop the classifier, we were unable to classify the ER+/PR− tumors with high accuracy. Rather than this being an indication of poor performance of the classifier, we understood this result to signify that a substantial percentage of ER+/PR− tumors by clinical assay do indeed associate with either ER+/PR+ or ER−/PR− tumors at the global molecular level.
For an independent observation of the Wang and Miller results, we went on to evaluate the classifier on an independent profile dataset of 275 breast tumors from van de Vijver et al.[28], with ER and PR status determined by immunohistochemistry (IHC). The van de Vijver dataset (Figure 2B-III) showed the same patterns as the first two datasets, with most of the IHC-defined ER+/PR+ and ER−/PR− tumors defined as such by gene expression (81% accuracy), and with a much smaller though significant fraction (33%, one-sided Fisher’s exact p < 0.001) of the clinical assay-defined ER+/PR− tumors defined as such by the classifier. While the mean centroid of the IHC-defined ER+/PR+, ER+/PR−, and ER−/PR− groups was used to center the values in the van de Vijver dataset (see Methods), the gene classifier assignments were the same regardless of our using all samples to define the mean centroid, or using a “leave-one-out” approach of redefining the mean centroid without the sample being classified (see Supplementary Material). An additional profile dataset by Chin et al. [26] of 118 tumors showed similar patterns of concordance as for the van de Vijver dataset (Figure 2B-IV).
(Conceivably, in regards to our gene classifier approach outlined in Figure 2A, we could instead have separated out the ER+/PR+ tumors in the first step and then the ER−/PR− tumors in the second step, though in practice the results were essentially the same, with, for example, 98% concordance between the two ways for the van de Vijver tumors. In addition, results using of an alternative “3-way” classification approach by weighted voting were also highly concordant with the results presented here, see Supplementary Material and Figure S3).
ER+/PR− tumors as defined by gene signatures associate with the luminal B breast cancer molecular subtype
Previous gene expression profiling studies of breast cancer have defined six intrinsic molecular subtypes: normal-like, luminal A, luminal B, basal, ERBB2+, and (most-recently) claudin-low [24–26, 36, 40, 41]. The luminal A tumors are thought to mostly represent ER+/HER2− tumors; the luminal B tumors, ER+/HER2+; the basal tumors, ER−/HER2−; and the ERBB2+ tumors, ER−/HER2+. We applied our gene classifier for ER and PR status (Figure 2A) to the dataset from Sorlie et al. [25], which was first used to define the molecular profile subtypes, in order to see which profile subtypes would be associated with tumors defined as ER+/PR− by our classifier. Most (92%) of the luminal A tumors were classified as ER+/PR+ using our ER−/PR− and ER+/PR+ gene signatures, and all of the basal tumors were classified as ER−/PR− (Figure 3A). Most of the ERBB2+ tumors (15/24) were also classified as ER−/PR−. We went on to apply the classifier to a more recent profile dataset from Hoadley et al. [36], where an independent cohort of 248 tumors had previously been classified using the Sorlie-defined intrinsic molecular subtypes. Our classifier again assigned most (91%) of the luminal A tumors to the ER+/PR+ group, all basal tumors to the ER−/PR− group, and most (89%) ERBB2+ tumors to the ER−/PR−group (Figure 3B).
Figure 3.
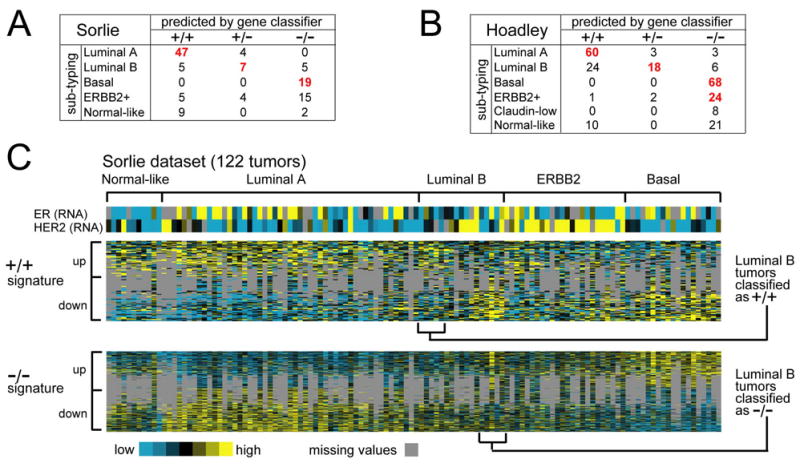
ER+/PR− tumors as defined by gene signatures associate with the luminal B breast cancer molecular subtype. (A) Confusion matrix for the tumor profile dataset from Sorlie et al. [25], comparing subtype assignments using the ER+/PR+ and ER−/PR− gene signatures with the molecular profile subtypes as defined previously by unsupervised analysis. Red bold denotes a significant number of subtype assignments by the gene classifier of Fig. 2a within one of the Sorlie molecular subtypes (P < 0.001, one-sided Fisher’s exact). (B) Confusion matrix for the profile dataset from Hoadley et al. [36]. (C) Heat map of the ER+/PR+ and ER−/PR− gene signatures (100 and 570 genes represented, respectively) in the Sorlie tumor profile dataset (gray: missing data values)
Notably, luminal B tumors in both the Sorlie and Hoadley datasets followed similar patterns of association as observed for the ER+/PR− tumors in the Wang, Miller, and van de Vijver datasets. In particular, the 17 Sorlie luminal B tumors were distributed among ER+/PR+, ER+/PR−, and ER−/PR− groups (5, 7, and 5 tumors, respectively), with a significant number associating with ER+/PR− by gene signatures (p < 0.001, one-sided Fisher’s exact). Of the 48 Hoadley luminal B tumors, 24, 18, and 6 were assigned to ER+/PR+, ER+/PR−, and ER−/PR− groups, respectively, with the ER+/PR− association being significant (p < 1E-09, one-sided Fisher’s exact). When viewing the expression patterns of the ER+/PR+ and ER−/PR− gene signatures in the Sorlie dataset as heat maps (Figure 3C) we observed the same patterns of coordinate expression for the genes as observed in the Wang and Miller datasets: genes previously defined as high in the ER+/PR+ signature tended likewise to be high in luminal A tumors; genes low in the ER+/PR+ signature were low in luminal A tumors; genes high and low in the ER−/PR− signature were likewise high and low, respectively, in the basal tumors. Some of the luminal B tumors associated with ER+/PR+ and some with ER−/PR− in terms of the associated gene signature patterns, with the rest of the tumors not being associated completely with either ER+/PR+ nor ER−/PR−, but sharing features with both (in terms of the ER−/PR− and ER+/PR+ gene signature patterns, respectively).
Patients with tumors designated as ER+/PR− by profiling rather than by clinical assay alone have poorer prognosis
Our gene signature patterns of ER+/PR+ and ER−/PR− cancers were significantly correlated with clinically assigned ER and PR status (Figure 2B), although this correlation was not perfect. Significant differences in patient outcome have often been observed between the three clinical subtypes of breast cancer [2]. Interestingly, when relying on clinical assay assessment of ER and PR status, no significant differences in outcome were observed for ER+/PR− compared to the other two groups for the Wang, Miller, and van de Vijver datasets (Figures 4A–C, left panels). On the basis of clinical receptor assessment, the Miller dataset showed a modest trend for ER+/PR− tumors to have the worst outcome of the three groups; this trend, however, was not evident in the other datasets. When instead the ER and PR status was assigned on the basis of our gene classifier (where, for example, 6+13+2 Wang tumors were considered ER+/PR−, see Figure 2B-I), no differences in time to distant metastases among ER+/PR+, ER+/PR−, and ER−/PR− tumors were observed for the Wang dataset (Figure 4A, right panel), but there were significant differences in disease-specific survival for the Miller dataset and in time to metastases in the van de Vijver dataset (log-rank p = 0.007 and p = 2E-05, respectively, Figures 4B–C, right panels). In both the Miller and van de Vijver datasets, which included patients who received adjuvant systemic therapy, ER+/PR− tumors (defined by gene classifer) showed the shortest time to recurrence, followed by ER−/PR− tumors. However, when comparing the ER+/PR− subset directly with the ER−/PR− tumors a statistically significant difference was not observed, though ER+/PR− tumors differed significantly from ER+/PR+ (log-rank p < 0.002 each dataset).
Figure 4.
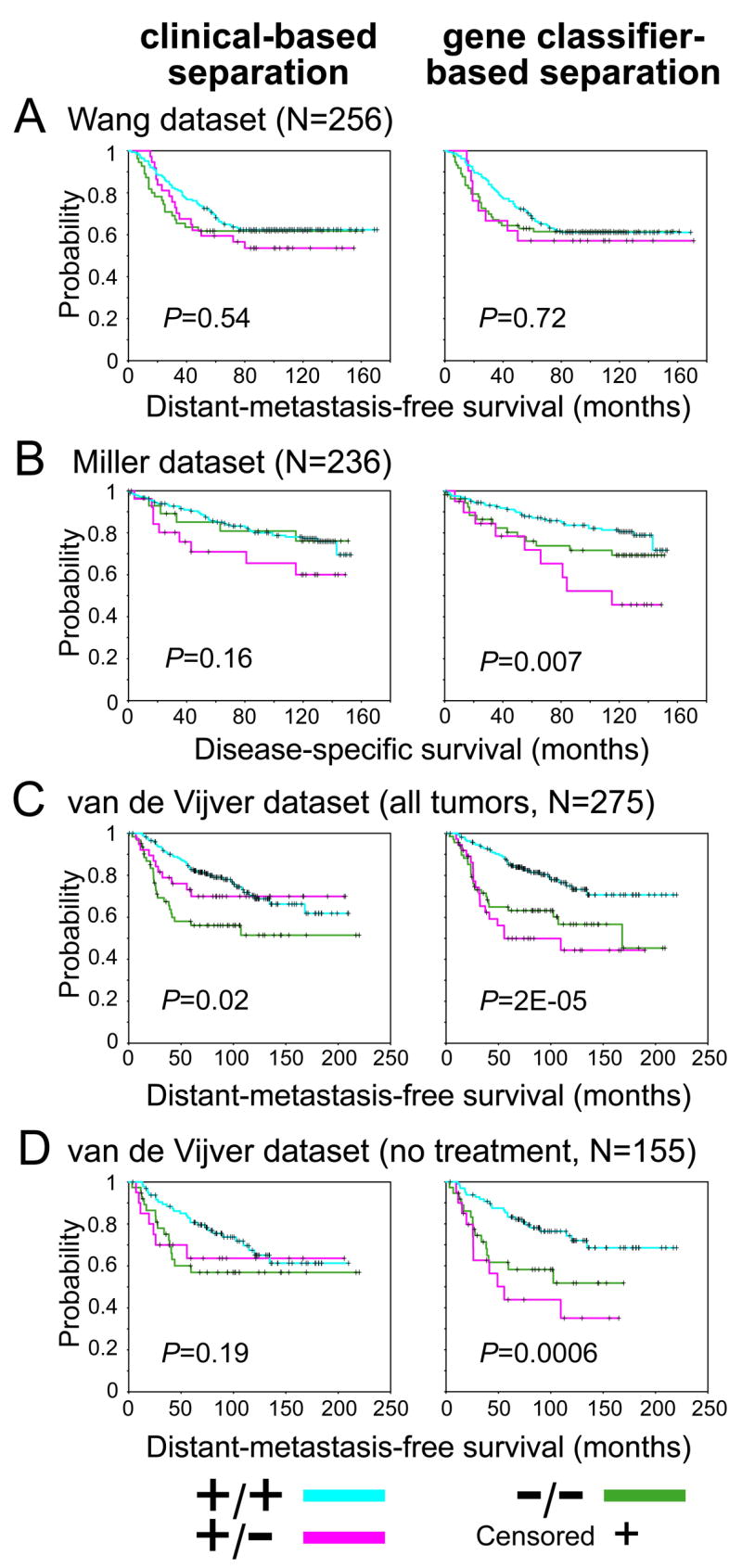
Patients with ER+/PR− breast tumors as defined by gene expression profiling rather than by clinical assay alone tend to have poorer prognosis. Kaplan-Meier analysis of breast cancer patients stratified by ER+/PR+, ER+/PR−, and ER−PR−, where the status was determined by either clinical assay (left panels) or gene classifer (right panels). Profile datasets from Wang (A), Miller (B), and van de Vijver (both all tumors (C), and the subset of tumors that did not receive either hormone therapy or chemotherapy (D), are considered). Log-rank statistic p-values evaluate whether there are significant differences in time to poor outcome event between any of the three groups. For Wang and van de Vijver datasets, measured outcome is distant-metastasis-free survival; for Miller dataset, outcome is disease-specific survival. N, number of patients.
None of the Wang patients received adjuvant systemic therapy [30]. Many of the Miller and van de Vijver patients did receive hormone therapy or chemotherapy or both, precluding an assessment of prognostic significance. Notably, when removing from consideration the 121 profiles of patients in the van de Vijver dataset that were known to have had adjuvant treatment (treatment information not available for Miller), stratifying the remaining 155 untreated patients on the basis of the gene classifier showed the same recurrence pattern as that for the entire van de Vijver and Miller cohorts (Figure 4D, global log-rank p = 0.0006). Grade or lymph node status alone could not account for the differences in outcome among the three tumor groups (Supplementary Material).
While the gene signatures (Figure 1B) could be used to predict prognosis in untreated patients (Figure 4D), we also tested whether they were prognostic in patients treated with tamoxifen therapy. We used an independent dataset by Loi et al. [37] of ER+ breast tumors treated with only adjuvant tamoxifen. Clinical assay-assigned ER+ tumors were stratified as either ER+/PR+ or ER+/PR− on the basis of the ER+/PR+ signature. The signature was associated with outcome both for the entire set of 349 patients (Figure 5A, log-rank p = 0.008) and for the subset of 263 patients who received tamoxifen monotherapy (Figure 5B, log-rank p = 0.02). For the 86 ER+ tumors from untreated patients, the gene signature showed a trend of being prognostic that was not statistically significant--perhaps due to the small size of this subgroup (Figure 5C, log-rank p = 0.12). For the 202 patients for whom both clinical assay-assigned ER and PR status was available, there was no significant difference between clinical ER+/PR+ and ER+/PR− (Figure 5D, log-rank p = 0.18), though there was a trend for poorer outcome in the ER+/PR− subset. However, for these same 202 patients, the gene signatures showed a difference between ER+/PR+ and ER+/PR− that was of borderline significance (Figure 5E, log-rank p = 0.06).
Figure 5.
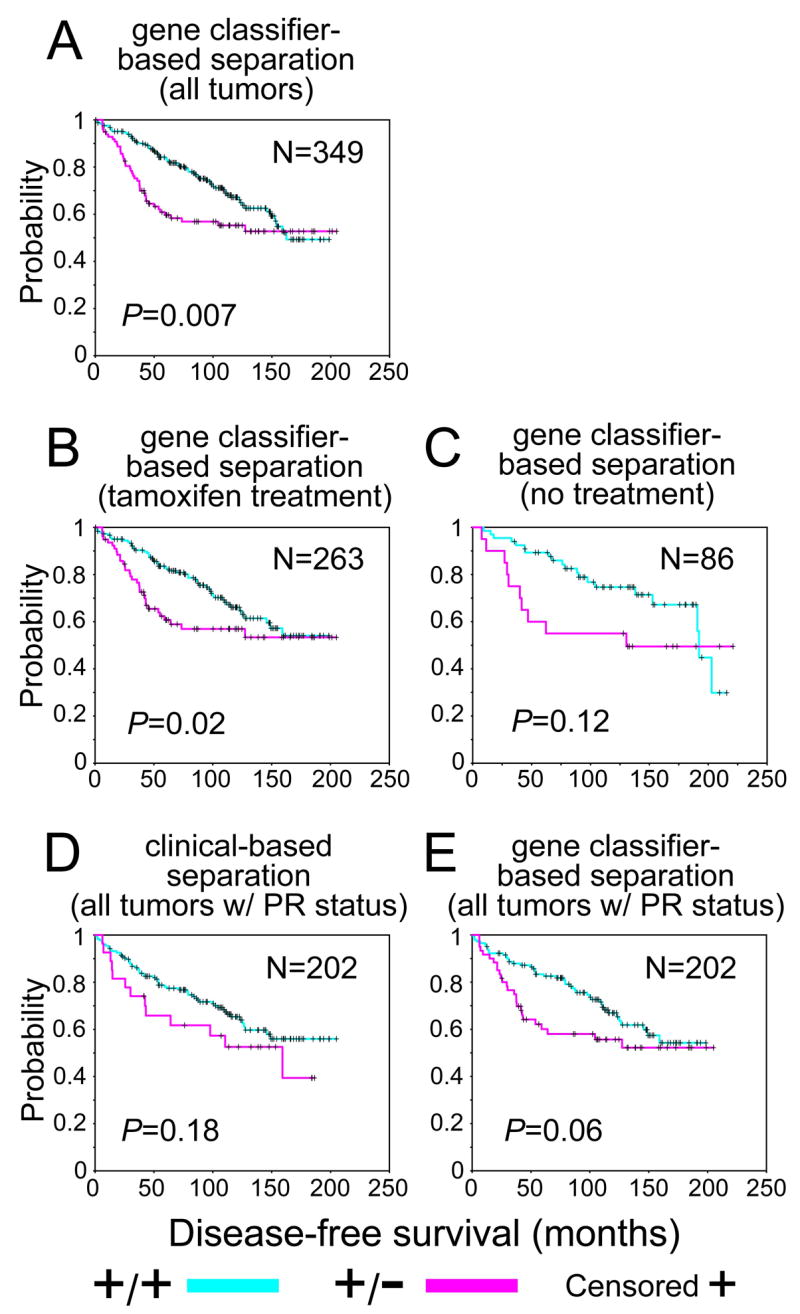
ER+/PR− tumors as defined by gene expression profiling tend to have poorer prognosis in ER+ breast cancer patients receiving tamoxifen hormone monotherapy. Kaplan-Meier analysis of ER+ patients in dataset from Loi et al. [37] stratified by ER+/PR+ and ER+/PR−, where the status was determined by either clinical assay (D) or gene expression profiling (A,B,C,E). For gene classifier-based separation, all patients (A), the subset of patients for which clinical ER and PR status was available (E), and the subset of patients treated with tamoxifen (B) were considered. Outcome is disease-free survival. N, number of patients. P-values by log-rank statistic.
We furthermore considered whether defining the three tumor groups using ER and PR mRNA alone (as measured on the expression array), as opposed to using all of the genes in the signatures, would yield the above findings with respect to patient outcome. As might be expected, we did find a trend for ER and PR alone to be prognostic, though the survival curves did appear better separated when using all of the genes rather than just the two genes (Supplementary Material and Figure S4), which indicated that genes in the signatures in addition to ER and PR were contributing information towards defining the tumor subtype.
ER+/PR− tumors defined by gene expression profiling show increased DNA copy number alteration, including specific regions of gain or loss
In a recent study by Chin et al. [26], profiles of both gene expression and DNA copy number were assessed on the same panel of 89 breast tumors. Analysis of the DNA profiles indicated that there were patterns of copy number alteration (CNA) that were more specific to ER+/PR− tumors [26]. Using expression profile data available for 118 tumors in the Chin cohort (89 of which had DNA profile data), we stratified the tumors into ER+/PR+, ER+/PR−, and ER−/PR− tumors using our gene classifier. As observed above for the other datasets (Figure 2B), most of the clinical assay-defined ER+/PR+ and ER−/PR− tumors in the Chin dataset were defined as such on the basis of the classifier, and the gene classifier called seven of the 17 clinical assay-designated ER+/PR− tumors as ER+/PR−, a significant number (p = 0.002, one-sided Fisher’s exact).
We separated the 89 tumor DNA copy number profiles (consisting of 2149 DNA probes) into ER+/PR+, ER+/PR−, and ER−/PR− groups, as defined by the RNA profiling. We found 46 DNA probes to be higher or lower in ER+/PR− compared to both ER−/PR− and ER+/PR+ tumors (t-test p < 0.01 each comparison, chance mean expected = 5 by permutation testing, SD = 6). We found 56 probes higher or lower in ER−/PR− compared to both ER+/PR+ and ER+/PR− (expected 1), and 8 probes specific to ER+/PR+ tumors. We found the probes specific to ER+/PR− or ER−/PR− tumors to be enriched for probes in chromosomes 11, 12, 17, and 22; CNA patterns for these regions were viewed as a heat map (Figure 6A). ER+/PR− tumors (defined by expression profiling) tended to show DNA copy number gains within regions 11q13 (68–72 Mb), 12q14-q15 (63–68 Mb), and 17q21-q25 (45–73 Mb), and copy number loss within 11q13-q25 (82–134 Mb). Gain of 17q21-q25 in particular appeared almost entirely specific to tumors defined as ER+/PR− by gene expression profiling rather than to clinical assay-defined ER+/PR− tumors. Additionally, we saw evidence for loss of chromosome 22 in many ER+/PR+ and ER+/PR− tumors but not in ER−/PR− tumors.
Figure 6.
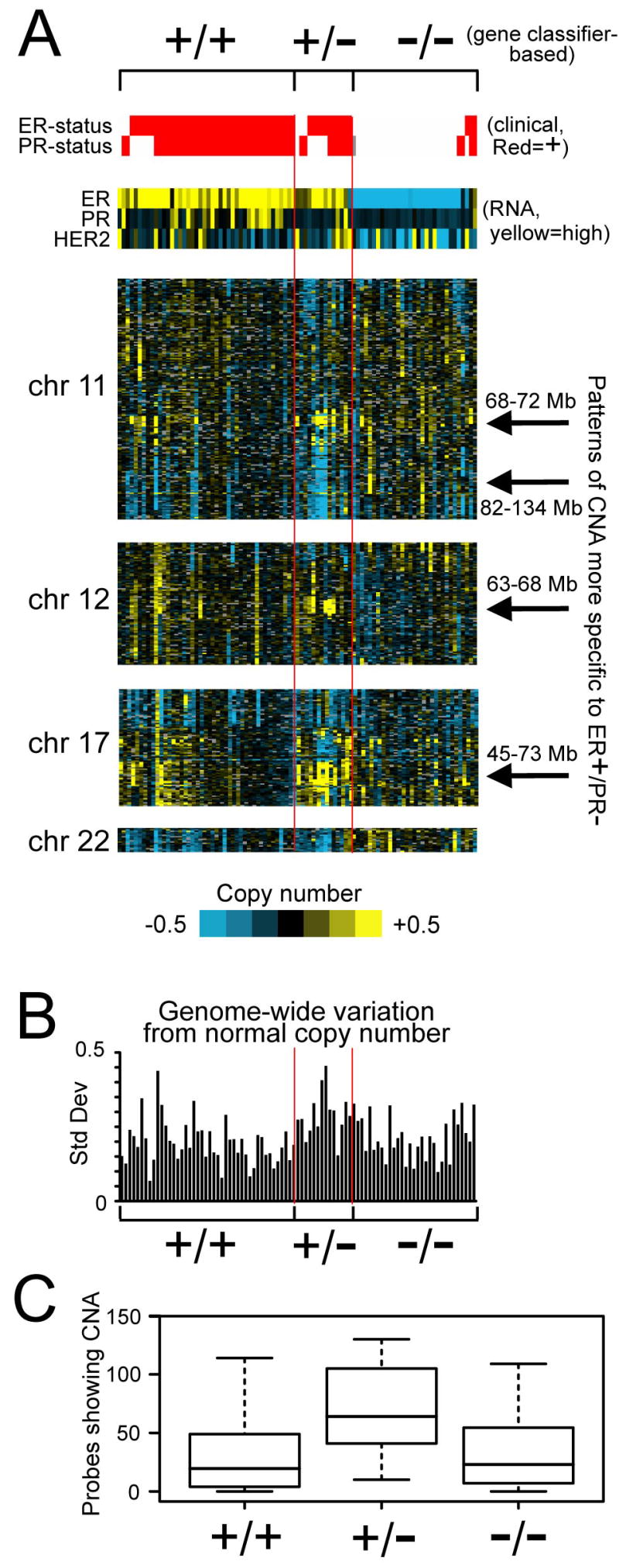
DNA copy number alterations (CNAs) in ER+/PR− breast tumors. (A) Heat map representation of CNA in the Chin tumors within chromosomes 11, 12, 17, and 22. DNA probes are ordered by genome location. Tumors are separated into ER+/PR+, ER+/PR−, and ER−/PR− as defined by gene classifier (see Figure 2A); histology-based ER and PR status are indicated, as well as RNA expression for ER, PR, and HER2. (B) Plot of the standard deviations from normal control across all of the DNA probes for each tumor profile. The ordering of the tumors corresponds to that of part (A). (C) Box plot of the number of DNA probes showing gain or loss of 1.5 times the control among the three tumor groups (as defined by gene classifier), based on 89 DNA copy number profiles from Chin et al. [26].
We also found that ER+/PR− tumors defined by the gene signatures in general showed increased CNA compared to ER+/PR+ and ER−/PR− tumors. When plotting the genome-wide variation (as assessed by standard deviation) from normal copy number in the Chin tumor cohort (Figure 6B), the ER+/PR− tumors tended to show significantly greater variation than either ER+/PR+ or ER−/PR− tumors (p = 0.0001 and p = 0.002, respectively, two-sample t-test). As another assessment of the relative amount of CNA in ER+/PR−, the number of DNA probes showing gain or loss of 1.5 times the control were counted for each tumor profile; ER+/PR− tumors showed the most gains or losses (78 on average compared to 32 for ER+/PR+, p = 0.0004 by t-test, and 38 for ER−/PR−, p = 0.01, Figure 6C). Interestingly, when using the clinical assessment rather than the gene signatures to determine ER and PR status, the patterns of increased CNA in ER+/PR− tumors was not as evident (57 probes with gain or loss of 1.5 in ER+/PR−, compared to 35 in ER−/PR− and 39 in ER+/PR+, p = 0.24 and p = 0.23, respectively, t-test).
Gene signature of oncogenic pathway PI3K/Akt/mTOR is manifested in ER+/PR− tumors
For clues to specific signaling pathways activated in ER+/PR− tumors, targets of transcriptional up-regulation by various pathways as measured in public profile datasets were examined. These pathways included the cell cycle, Myc, c-Src, beta-catenin, E2F3, H-Ras, Akt (both mTOR and non-mTOR branches), cyclin D1, Her2, EGFR, MEK, Raf, MAPK, and PI3K. Using stringent statistical approaches, we found that both the PI3K and Akt/mTOR pathway signatures were significantly enriched in both ER+/PR− and ER−/PR− tumors for each of four tumor profile datasets (complete details in Supplementary Material and Figure S5).
The enrichment patterns for genes up-regulated in the PI3K/Akt/mTOR pathway in ER+/PR− compared to ER+/PR+ breast cancer were evident when viewing the associated expression patterns as a heat map (Figure 7). We selected 670 transcripts more highly expressed in ER+/PR− compared to ER+/PR+ tumors in both the Wang and Miller datasets (p < 0.01 each comparison, using the gene signature-based rather than the clinical assay-based assignments) and viewed the corresponding expression patterns for these genes in the “CMaP” dataset [42] of gene expression profiles of cultured cells (mostly MCF-7 cells) under treatment with 164 different small molecule inhibitors. The PI3K inhibitors, LY-294002 and wortmannin, and the mTOR inhibitor, rapamyacin, all repressed a significant number of the genes that were high in ER+/PR− tumors. Of the 670 transcripts (534 unique named genes) selected on the basis of the breast tumor profile data, 143 (123 genes) were down-regulated (p < 0.01, t-test) in the CMaP LY-294002 and wortmannin profiles (p < 1E-20, one-sided Fisher’s exact test, heat map in Figure 7). Genes with high expression in ER+/PR− compared to ER+/PR+ tumors were similarly expressed in ER−/PR− compared to ER+/PR+ tumors (Figure 7 and Supplementary Figure S1), which accounts for enrichment of the PI3K/Akt/mTOR-associated gene sets in ER−/PR− tumors as well as ER+/PR− tumors.
Figure 7.
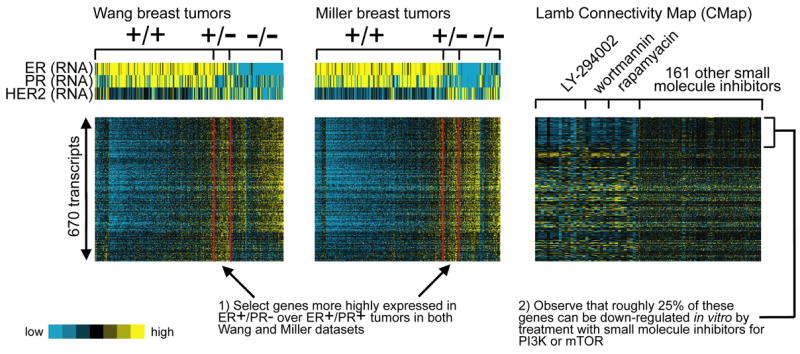
A gene signature of oncogenic pathway PI3K/Akt/mTOR is manifested in ER+/PR− tumors. Heat map of 670 transcripts more highly expressed in ER+/PR− over ER+/PR+ tumors in both the Wang and Miller datasets (p < 0.01 in each, using the gene signature-based rather than the clinically-based assignments). Alongside the patterns of the clinical breast tumor data are the corresponding expression patterns in a compendium of expression profiles from cell lines treated with 164 different small molecule inhibitors (the “Connectivity Map,” or CMap, from Lamb et al. [42]). Profiles in CMap of cells treated with PI3K inhibitors LY-294002 and wortmannin and the mTOR inhibitor rapamyacin are highlighted.
Discussion
We found that ER+/PR− breast tumors as defined by gene expression profiling are a small but distinct molecular subtype from ER+/PR+ or ER−/PR−. ER+/PR− tumors as diagnosed in the clinic were a mixture of three different groups: tumors that appeared ER+/PR+ at the level of genome-wide expression patterns, tumors that appeared ER−/PR−, and tumors exhibiting an expression profile composed of a mixture of the other two groups. Tumors in this latter group we define as “true” ER+/PR− tumors since they are distinct from either the ER+/PR+ or the ER−/PR− subsets. While we did not find a gene expression pattern manifested only in the ER+/PR−group, we did find evidence for DNA copy number alterations that were more specific to tumors of this group. We found no significant overlap between genes within these genomic regions and genes in our ER+/PR+ or ER−/PR− gene signatures (data not shown). The molecularly-defined ER+/PR− subset was also characterized by a gene expression profile indicative of active growth factor signaling via the PI3K/Akt/mTOR pathway.
In most datasets, univariate analysis indicated that the classification of hormone receptor status using molecular profiling was more strongly associated with outcome than clinical assessment by IHC or biochemical assay. The poor outcome of the molecularly-defined ER+/PR− subset was at least as bad as the ER−/PR− group, a subset recognized for its aggressive behavior [4]. This adverse outcome was not observed in the clinical assay-defined ER+/PR− subset in these cohorts (though the trend was evident, just not to statistical significance). In contrast to a number of previous expression profiling studies [27–34, 43], we did not set out to identify gene signatures of poor patient outcome or treatment response; rather, our aim was to further characterize tumors associated with the clinical ER+/PR− designation. We believe our gene signature-defined ER+/PR− subset to bear resemblance to the luminal B subset initially defined by Perou et al. [44], the amplifier phenotype subgroup defined by Chin et al. [26], and the ER+, high grade tumors defined by the gene expression grade index of Loi et al. [37].
This study considered various datasets from different laboratories, in which ER and PR status was defined by different measurement techniques (e.g. IHC, LBA) or different cutoffs. This variability in methodology reflects the current state of how ER and PR is currently assessed in the clinic [45]. We identified robust, reproducible gene sets and gene expression patterns underlying ER+/PR+, ER+/PR−, and ER−/PR− tumors, although the correlation between the clinically-assessed ER and PR and the multigene expression pattern was not perfect. While disparities may be due to technical issues relating to IHC and biochemical assay measurements [45] and/or the choice of cut-off values, it is tempting to speculate that the molecular profile identifies tumors that may behave differently than thought on the basis of ER/PR status alone as determined clinically. This idea is best illustrated in the expression heat maps where, for example, there is a proportion of clinically ER+/PR− tumors that have a profile of ER+/PR+ gene expression (perhaps due to independent loss of PR via LOH, or perhaps due to low estrogen levels in some postmenopausal women insufficient to induce PR), and a proportion of ER+/PR− tumors that show a pattern of ER−/PR−. Studies of endocrine therapy in metastatic breast cancer report that the response rate in ER+/PR− is half that in ER+/PR+ tumors [14, 15], data consistent with the public molecular data showing that this group is likely a mixture of tumor types. Additional study is needed to determine whether those patients with ER+/PR− tumors who do respond to hormone therapy are those whose tumors manifest the ER+/PR+ gene signature. Similarly, not all ER+/PR+ tumors respond and ~5–10% of ER−/PR− tumors do respond to therapy, and these groups could potentially be identified on the basis of molecular profiling.
The fact that the gene expression profiles but not the clinical assay-assigned ER/PR status were able to distinguish high and low risk patient groups raises the intriguing possibility of developing an improved clinical assay for ER/PR status, based on genes selected from our gene classifier. However, Affymetrix expression arrays are not ideal as a clinical diagnostic tool, since they do not quantitate absolute RNA levels; an expression profile from any one individual patient would therefore need to be compared with a comparable population of profiles generated from the same laboratory. On the other hand, RT-PCR assays using RNA from paraffin-embedded tissues have recently been developed [43, 46], and so it would be technically feasible to develop an RT-PCR-based, multi-gene assay for ER and PR status and function, using genes selected from our classifier. Such an assay would naturally include ESR1 and PGR, though our study—along with others [37, 43]—indicates that other genes besides these two would add more information as to the tumor’s true molecular subtype. Finally, the poor prognosis of patients with ER+/PR− tumors suggests that testing of new biological treatment regimens or the addition of aggressive adjuvant chemotherapy for these tumors is warranted.
Additional Data Files
Additional File 1. Supplementary Material (Methods, Results, and Figures)
Additional File 2. For the Wang, Miller, and van de Vijver datasets, profiles used in the analysis, along with biochemical assay or IHC values
Additional File 3. The complete list of genes in the ER+/PR+ and ER−/PR− signatures
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants P30 CA125123, P50 CA58183, and 5P01 CA30195-25, and the Dan L. Duncan Cancer Center at Baylor College of Medicine
Abbreviations
- ER
estrogen receptor alpha
- PR
progesterone receptor
- IHC
immunohistochemistry
- CNA
copy number alteration
References
- 1.Elledge RM, Fuqua SA. Estrogen and progesterone receptors. In: Harris J, Lippman ME, Morrow M, Osborne C, editors. Diseases of the Breast. Philadelphia: Lippincott, Williams and Wilkins; 2000. pp. 471–488. [Google Scholar]
- 2.Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005;23 (30):7721–7735. doi: 10.1200/JCO.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Osborne C. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998;51(3):227–238. doi: 10.1023/a:1006132427948. [DOI] [PubMed] [Google Scholar]
- 4.Allred DC, Brown P, Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004;6:240–245. doi: 10.1186/bcr938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poole AJ, Li Y, Kim Y, Lin S-CJ, Lee W-H, Lee EYHP. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 6.McGuire W. Hormone receptors: their role in predicting prognosis and response to endocrine therapy. Semin Oncol. 1978;5 (4):428–433. [PubMed] [Google Scholar]
- 7.Horwitz K, McGuire W. Estrogen control of progesterone receptor in human breast cancer. Correlation with nuclear processing of estrogen receptor. J Biol Chem. 1978;253(7):2223–2228. [PubMed] [Google Scholar]
- 8.Horwitz K, Koseki Y, McGuire W. Estrogen control of progesterone receptor in human breast cancer: role of estradiol and antiestrogen. Endocrinology. 1978;103(5):1742–1751. doi: 10.1210/endo-103-5-1742. [DOI] [PubMed] [Google Scholar]
- 9.Bardou V-J, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol. 2003;21(10):1973–1979. doi: 10.1200/JCO.2003.09.099. [DOI] [PubMed] [Google Scholar]
- 10.Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339(22):1609–1618. doi: 10.1056/NEJM199811263392207. [DOI] [PubMed] [Google Scholar]
- 11.Stendahl M, Ryden L, Nordenskjold B, Jonsson PE, Landberg G, Jirstrom K. High progesterone receptor expression correlates to the effect of adjuvant tamoxifen in premenopausal breast cancer patients. Clin Cancer Res. 2006;12(15):4614–4618. doi: 10.1158/1078-0432.CCR-06-0248. [DOI] [PubMed] [Google Scholar]
- 12.Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, Elledge RM. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005;97(17):1254–1261. doi: 10.1093/jnci/dji249. [DOI] [PubMed] [Google Scholar]
- 13.Tovey S, Dunne B, Witton CJ, Forsyth A, Cooke TG, Bartlett JMS. Can molecular markers predict when to implement treatment with aromatase inhibitors in invasive breast cancer? Clin Cancer Res. 2005;11(13):4835–4842. doi: 10.1158/1078-0432.CCR-05-0196. [DOI] [PubMed] [Google Scholar]
- 14.Ravdin PM, Green S, Dorr TM, McGuire WL, Fabian C, Pugh RP, Carter RD, Rivkin SE, Borst JR, Belt RJ. Prognostic significance of progesterone receptor levels in estrogen receptor-positive patients with metastatic breast cancer treated with tamoxifen: results of a prospective Southwest Oncology Group study. J Clin Oncol. 1992;10(8):1284–1291. doi: 10.1200/JCO.1992.10.8.1284. [DOI] [PubMed] [Google Scholar]
- 15.Osborne CK, Yochmowitz MG, Knight WA, McGuire WL. The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer. 1980;46(12 Suppl):2884–2888. doi: 10.1002/1097-0142(19801215)46:12+<2884::aid-cncr2820461429>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 16.Dowsett M, Allred C, Knox J, Quinn E, Salter J, Wale C, Cuzick J, Houghton J, Williams N, Mallon E, et al. Relationship Between Quantitative Estrogen and Progesterone Receptor Expression and Human Epidermal Growth Factor Receptor 2 (HER-2) Status With Recurrence in the Arimidex, Tamoxifen, Alone or in Combination Trial. J Clin Oncol. 2008;26(7):1059–1065. doi: 10.1200/JCO.2007.12.9437. [DOI] [PubMed] [Google Scholar]
- 17.Viale G, Regan M, Maiorano E, Mastropasqua M, Dell’Orto P, Rasmussen B, Raffoul J, Neven P, Orosz Z, Braye S, et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1–98. J Clin Oncol. 2007;25(25):3846–3852. doi: 10.1200/JCO.2007.11.9453. [DOI] [PubMed] [Google Scholar]
- 18.Early_Breast_Cancer_Trialists’_Collaborative_Group. Systemic treatment of early breast cancer by hormonal, cytotoxic, or immune therapy. 133 randomised trials involving 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet. 1992;339(8785):71–85. [PubMed] [Google Scholar]
- 19.Balleine RL, Earl MJ, Greenberg ML, Clarke CL. Absence of progesterone receptor associated with secondary breast cancer in postmenopausal women. Br J Cancer. 1999;79(9–10):1564–1571. doi: 10.1038/sj.bjc.6690249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bamberger AM, Milde-Langosch K, Schulte HM, Loning T. Progesterone receptor isoforms, PR−B and PR−A, in breast cancer: correlations with clinicopathologic tumor parameters and expression of AP-1 factors. Horm Res. 2000;54(1):32–37. doi: 10.1159/000063434. [DOI] [PubMed] [Google Scholar]
- 21.Dowsett M, Harper-Wynne C, Boeddinghaus I, Salter J, Hills M, Dixon M, Ebbs S, Gui G, Sacks N, Smith I. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res. 2001;61(23):8452–8458. [PubMed] [Google Scholar]
- 22.Cui X, Zhang P, Deng W, Oesterreich S, Lu Y, Mills GB, Lee AV. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol Endocrinol. 2003;17(4):575–588. doi: 10.1210/me.2002-0318. [DOI] [PubMed] [Google Scholar]
- 23.Lapidus RG, Nass SJ, Davidson NE. The loss of estrogen and progesterone receptor gene expression in human breast cancer. J Mammary Gland Biol Neoplasia. 1998;3:85–94. doi: 10.1023/a:1018778403001. [DOI] [PubMed] [Google Scholar]
- 24.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100(14):8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo W-L, Lapuk A, Neve RM, Qian Z, Ryder T, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10(6):529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe J-P, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 29.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 31.Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, Cragun J, Cottrill H, Kelley MJ, Petersen R, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12(11):1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- 32.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi M-B, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 33.Ma X-J, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5(6):607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362(9381):362–369. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- 35.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102(38):13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoadley K, Weigman V, Fan C, Sawyer L, He X, Troester M, Sartor C, Rieger-House T, Bernard P, Carey L, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8(258) doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25(10):1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 38.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 40.Herschkowitz J, Simin K, Weigman V, Mikaelian I, Usary J, Hu Z, Rasmussen K, Jones L, Assefnia S, Chandrasekharan S, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007:R76.16. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sotiriou C, Neo S-Y, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100(18):10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 43.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MGDW, Park T, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351 (27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 44.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 45.Gross GE, Clark GM, Chamness GC, McGuire WL. Multiple progesterone receptor assays in human breast cancer. Cancer Res. 1984;44(2):836–840. [PubMed] [Google Scholar]
- 46.Lehmann U, Kreipe H. Real-Time PCR Analysis of DNA and RNA Extracted from Formalin-Fixed and Paraffin-Embedded Biopsies. Methods. 2001;25(4):409–418. doi: 10.1006/meth.2001.1263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.