

Abstract
An androgen-repressed human prostate cancer cell line, ARCaP, was established and characterized. This cell line was derived from the ascites fluid of a patient with advanced metastatic disease. In contrast to the behavior of androgen-dependent LNCaP and its androgen-independent C4-2 subline, androgen and estrogen suppress the growth of ARCaP cells in a dose-dependent manner in vivo and in vitro. ARCaP is tumorigenic and highly metastatic. It metastasizes to the lymph node, lung, pancreas, liver, kidney, and bone, and forms ascites fluid in athymic hosts. ARCaP cells express low levels of androgen receptor mRNA and prostate-specific antigen mRNA and protein. Immunohistochemical staining shows that ARCaP cells stain intensely for epidermal growth factor receptor, c-erb B2/neu, and c-erb B3. Staining is negative for chromogranin A and positive for bombesin, serotonin, neuron-specific enolase, and the c-met protooncogene (a hepatic growth factor/scatter factor receptor). ARCaP cells also secrete high levels of gelatinase A and B and some stromelysin, which suggests that this cell line may contain markers representing invasive adenocarcinoma with selective neuronendocrine phenotypes. Along with its repression of growth, androgen is also found to repress the expression of prostate-specific antigen in ARCaP cells as detected by a prostate-specific antigen promoter–β-galactosidase reporter assay. Our results suggest that the androgen-repressed state may be central to prostate cancer progression and that advanced prostate cancer can progress from an androgen-independent to an androgen-repressed state.
Keywords: gene expression, androgen receptor, prostate-specific antigen, metalloproteinases, bone metastasis
Progression of human prostate cancer from the androgen-dependent to the androgen-independent state is a well established phenomenon in both the clinic (1, 2) and the laboratory (3, 4). Once prostate cancer becomes androgen-independent, however, the disease is perceived as not progressing further, because patients with androgen refractory disease seldom respond to secondary endocrine therapy (5, 6). Recently, some patients with androgen-independent disease [defined as evidence of disease progression or an increase of serum prostate-specific antigen (PSA) in surgically or hormonally castrated hosts] were shown to respond to secondary hormonal manipulation, such as flutamide withdrawal (7) or intermittent androgen therapy (8). This phenomenon supports the concepts that (i) the secondary hormonal therapy may trigger the tumor cells to redifferentiate, thus allowing them to become apoptotic upon a subsequent androgen withdrawal cycle, or (ii) the circulating serum testosterone may impede the progression of prostate cancer from an androgen-independent to an androgen-repressed state by suppressing the growth of the androgen-repressed clones of prostate cancer cells. The predicted phenotypes of the androgen-repressed cells, therefore, should be such that the growth of these cells in vivo and in vitro could be repressed by testicular or exogenous androgen. If such a phenotype exists, it offers a possible explanation for disease that responds positively to flutamide withdrawal or intermittent androgen therapy. It may be that endogenous adrenal or testicular androgens/growth factors released by flutamide withdrawal and/or intermittent androgen therapy retard the progression of disease. This communication focuses on the establishment and characterization of an androgen-repressed human prostate cancer cell line, ARCaP, from a man with advanced metastatic cancer.
MATERIALS AND METHODS
Cell Culture.
ARCaP cells were derived from the ascites fluid of an 83-year-old Caucasian man diagnosed with metastatic carcinoma of the prostate for more than 1 year before he was transferred to the University of Texas M. D. Anderson Cancer Center. At the time of admission, cancer had metastasized to the liver, spine, and long bones. The patient had extensive accumulation of ascites fluid that contained tumor cells and a protein factor, PSA-stimulating autocrine factor, that is associated with androgen-independent prostate cancer progression (9). The patient had undergone bilateral orchiectomy 11 months before admission to the University of Texas M. D. Anderson Cancer Center, but the disease progressed rapidly after surgery, with a latent period of 12 months between surgery and death. At the time of initial diagnosis, serum PSA was 350 ng/ml. It dropped sharply (25 ng/ml) 6 months after surgery and remained at a low level (14–17 ng/ml) even after the disease became widely disseminated. The original ascites specimen was found to stain negatively for PSA, prostatic acid phosphatase, and chromogranin A, which implied that the cancer was poorly differentiated and was not a typical neuroendocrine tumor. The ARCaP cell line was established after orchiectomy from the patient’s ascites fluid by a standard procedure used by our laboratory for the establishment of LNCaP sublines from parental LNCaP chimeric tumors (4). The LNCaP cell line was provided by Gary J. Miller of the University of Colorado Health Science Center, Denver, CO. Cells were routinely cultured in T medium [80% DMEM (GIBCO)/20% F12K (Irvine Scientific, Santa Ana, CA)/3 g/liter NaHCO3/100 units/ml penicillin G/100 μg/ml streptomycin/5 μg/ml insulin/13.6 pg/ml triiodothyronine/5 μg/ml transferrin/0.25 μg/ml biotin/25 μg/ml adenine) containing 5% fetal bovine serum (FBS). The cells were passaged weekly at a 1:5 to 1:20 dilution following trypsinization. The androgen-independent and metastatic LNCaP subline, C4-2, was derived from LNCaP/bone fibroblast chimeric tumors passaged in castrated hosts as reported (4). To determine the in vitro effects of sex steroids, DHT (5α-dihydrotestosterone or 5α-androstan-17β-ol-3-one), R1881 (a synthetic analog of testosterone, 17β-OH-17-methylestra-4,9,11-triene-3-one), or 17β-estradiol (estra-1,3,5[10]-triene-3,17β-diol) was added to charcoal-stripped serum to determine the effect on the growth or gene expression of the tested cell lines; the specificity of DHT or R1881 on target cells is confirmed by blocking with Casodex (10).
Northern and Western Blot Analyses.
Northern blot analysis was performed to determine the levels of expression of PSA and androgen receptor (AR) mRNA. A PSA cDNA probe was prepared as a 0.4 kb StyI fragment isolated from a cDNA clone (11). The AR cDNA probe is a 0.5 kb EcoRI fragment from human AR cDNA (12). The cDNA probes were labeled with 32P by the random oligonucleotide method using the Megaprime DNA labeling kit (Amersham). Total RNA was extracted by RNAzol (Tel-Test, Friendswood, TX). For Northern analysis, 10–50 μg of total RNA was denatured in 18% formaldehyde and 50% formamide at 55°C and electrophoresed onto a 0.9% denaturing formaldehyde agarose gel. Samples were transferred onto a Zeta-probe membrane (Bio-Rad) by capillary action. The membrane was crosslinked by a UV Stratalinker, prehybridized in rapid hybridization buffer (Amersham) at 65°C for 3 hr. RNA hybridization was subsequently carried out at 65°C overnight with a 32P-labeled cDNA probe at 1 × 106 cpm/ml. The membranes were washed with 2× standard saline citrate. Autoradiograms were prepared by exposing the membranes to Kodak XAR film at −80°C. Equal loading of RNA samples was verified either by ethidium bromide staining before transfer or by reprobing the filters with GAPDH cDNA probe, or by both methods. To determine the level of expression of PSA protein, Western blot procedures described by Towbin and Gordon (13) were followed with some modification. Cultured cells were harvested at 80% confluence. Cell protein was extracted in a homogenization buffer containing 20 mM NaH2PO4/Na2HPO4, 150 mM NaCl, 1% Triton X-100, 10 μg/ml aprotenin, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin, pH 7.40. Protein concentration was determined by the method of Lowry et al. (14) using bovine serum albumin as the standard. Twenty-five micrograms of LNCaP and 50-μg ARCaP protein samples were separated by SDS/PAGE (10% gel) and transferred to nitrocellulose membrane (NitroPlus, MSSI, Westboro, MA). The membrane was blocked with 5% skim milk in PBS for 1 hr and incubated with 2 μg/ml monoclonal antibody against PSA (BioGenex Laboratories, San Ramon, CA) for an additional 1 hr. The signals were detected by enhanced chemiluminescence–Western blot reagents and exposed to Hyperfilm (Amersham).
Mutational Analysis of AR by Reverse Transcription–PCR and DNA Sequencing.
To determine potential structural mutation at a DNA-binding domain (exons 2 and 3), a hinge region (exon 4), and a steroid-binding domain (exons 4–8) of the AR cDNA, 1 μg of total RNA (extracted by RNAzol, Tel-Test) was used for the first-strand cDNA synthesis. The reaction mixture (20 μl) contained 5 mM MgCl2, 1× PCR buffer II (500 mM KCl/100 mM Tris, pH 8.30), 1 mM 4dNTP, 1 unit/ml RNase inhibitor, 2.5 mM oligo d(T)16, and 2.5 units/ml murine leukemia virus reverse transcriptase (Perkin–Elmer). The reaction mixture was incubated in a Perkin–Elmer DNA Thermal Cycler at 42°C for 15 min and at 99°C for 5 min, and then chilled at 4°C for 5 min.
Primers employed for the PCR and DNA sequencing of the AR DNA and steroid-hormone-binding domains are as follows: forward primers, 5′-AGTCCCACTTGTGTCAAAAGC-3′ (2071–2091), 5′-CAGAAGCTGACAGTGTCACA-3′ (2497–2516), and 5′-GTCATTCAGTACTCCTG-3′ (2734–2750); reverse primers, 5′-ATGGCTTCCAGGACATTCAGA-3′ (2547–2567) and 5′-CAAGGCACTGCAGAGGAGTA-3′ (3364–3383). The numbering system for the AR cDNA sequence is based on the report by Chang et al. (15).
For the PCR, 20 μl of the first-strand synthesis mixture was incubated with 2 mM MgCl2, 1× PCR buffer II, 0.15 mM upstream and 0.15 mM downstream primers (Genosys, The Woodlands, TX) and 2.5 units of AmpliTaq DNA polymerase (Perkin–Elmer) in a final volume of 100 μl. Cycling conditions were denaturation at 95°C, 2 min; 95°C, 1 min; annealing and extension at 60°C for 35 cycles; 60°C for 7 min, then chilling at 4°C. The amplified PCR products were electrophoresed on a 1.2% low electroendosmosis agarose gel containing 0.5 mg/ml ethidium bromide and photographed under UV illumination. The PCR products were cloned into PCRII vector (Invitrogen) by the T-A cloning method (16). They were routinely sequenced by using three independent clones recovered from the PCRII vector by plasmid sequencing (17) using the sequenase V2.0 sequencing system from American Science (Arlington Heights, IL). The DNA sequence was reconfirmed by direct PCR product sequencing.
Immunohistochemical Analyses.
Cells grown in chambered slides were fixed in methanol/acetone (3:1), treated with 3% hydrogen peroxide, blocked with Super Block (Scytek Laboratories, Logan, UT), and incubated with a primary antibody, followed by biotinylated goat anti-immunoglobulins and peroxidase-conjugated streptavidin (ref. 18; BioGenex Laboratories). The substrate used was 3,3′-diaminobenzidine or 3-amino-9-ethylcarbazole. Monoclonal antibodies against PSA, chromogranin A, and neuron-specific enolase, and polyclonal antibodies against serotonin and bombesin, were obtained from BioGenex Laboratories. Monoclonal antibodies against epidermal growth factor receptor (Ab-4) and c-erb B2/neu (Ab-3) were from Oncogene Science. Polyclonal antibodies against c-met and c-erb B3 protooncogenes were from Santa Cruz Biotechnology. Monoclonal antibody against AR was kindly provided by Jan Trapman (Erasmus University, Rotterdam, The Netherlands). Normal rabbit serum, normal mouse serum, and/or a monoclonal antibody against bacterial protein (trpE, Ab-1; Oncogene Science) were used as the negative staining controls.
Chromosome Preparation and Banding.
ARCaP cells at 75–80% confluence in fresh media were exposed to Colcemid (20 ng/ml; Sigma) for 30 min at 37°C, washed two times with Hanks’ balanced salt solution, and exposed to 0.01% trypsin solution for 5–7 min. The dislodged cells were neutralized with RPMI 1640 medium containing 10% FBS. The pelleted cells were exposed to hypotonic solution (0.06 M KCl) for 20 min at room temperature. After centrifugation, the cells were fixed in methanol/acetic acid (3:1) and washed two times in the fixative. Conventional air-drying chromosome preparations were made by routine procedures (19).
Effects of Androgen and Estrogen on the Growth of ARCaP, LNCaP, and C4-2 Cells in Vitro.
To determine the effects of DHT and 17β-estradiol on the growth of LNCaP, C4-2, and ARCaP cells in vitro, 2 × 104 cells per well were grown in T medium with 5% FBS for 24 hr and replaced with fresh T medium containing 5% charcoal-dextran-treated FBS for another 24 hr. Then, DHT (10 pM to 1 μM) or 17β-estradiol (10 nM to 100 μM) was added to the culture medium and incubated for 72 hr. The cells were fixed in 1% glutaraldehyde and stained with 0.5% crystal violet. Plates were rinsed and air-dried, and the dye was eluted with Sorensen’s solution (9 mg of trisodium citrate in 305 ml of H2O, 195 ml of 0.1 M HCl, and 500 ml of 90% ethanol). Absorbance at 590 nm was measured by a precision microplate reader (Molecular Devices). Control experiments demonstrated that absorbance is directly proportional to the number of cells in each well (20).
Tumorigenicity and Metastatic Potentials in Athymic Mice.
Athymic BALB/C nude (nu/nu) mice (6–8 weeks old), congeneically inbred, were obtained from Charles River Breeding Laboratories. Intact or castrated athymic BALB/C nu/nu mice were used as hosts. Cells were injected either subcutaneously (2 × 106 in 100 μl of T medium per site) or orthotopically (1 × 106 in 50 μl of T medium per site) into the dorsolateral lobe of the prostate according to a previously described method (20). Metastases to distant lymph nodes and other organ sites were confirmed by necropsy, followed by histologic analysis. Tumors were measured weekly, and the volumes were calculated as length × width × height × 0.5236 (21).
Matrix Metalloproteinase (MMP) Determination.
Zymogram analyses were performed as described by Sang et al. (22). For detection of 72-kDa type IV collagenase/gelatinase A (MMP-2) and 92-kDa type IV collagenase/gelatinase B (MMP-9), 1 mg/ml gelatin copolymerized into 9% polyacrylamide gel was run under nondenaturing conditions. The gel was incubated in the collagenase assay buffer containing 50 mM Tricine, 0.2 M NaCl, 10 mM CaCl2, 50 mM ZnSO4, 0.05% Brij 35, and 0.02% NaN3 (pH 7.5) at 37°C for 1 day. For detection of stromelysin (MMP-3), 1 mg/ml casein copolymerized into 12% polyacrylamide gel was used. To prepare conditioned media, cells (1 × 106 per 10-cm dish for LNCaP cells; 2 × 106 cells per 10-cm dish for ARCaP cells) were cultured in a T medium with 5% FBS for 24 hr, switched to a T medium with 2% TCM (a serum-free defined medium supplement; Celex Company, Minnetonka, MN) for 24 hr, and then added DHT (10 nM for ARCaP cells; 1 nM for LNCaP cells) for an additional 3 days. The media were collected and filtered through a 0.2-μm filter. Cell extracts and serum-free conditioned media derived from the same cell numbers were analyzed.
Assessment of PSA-Promoter Reporter Activity.
An adenoviral infection system was chosen for the analysis of PSA-promoter reporter activity because of the low efficiency of gene transfer into ARCaP cells by conventional gene transfection techniques (unpublished results). We have constructed a recombinant PSA promoter by deleting a 3.3-kb fragment (−3, 900 to −630) from a 6-kb (−6, 100 to +12) PSA promoter (kindly provided by Jan Trapman, Erasmus University, Rotterdam, The Netherlands); the p64 promoter retained tissue-specific and androgen-responsive activities (23). The p64 promoter β-galactosidase (β-gal) was cloned into a shuttle vector, pΔE1SP1A (24). The replication-defective recombinant adenovirus was produced by cotransfecting the 293 cells with pΔE1SP1A p64-β-gal (shuttle vector) and pBHG-11 (recombinant vector) plasmids as described by Bett et al. (24). The recombinant virus was then cloned from individual plaques, amplified, and purified by the CsCl2 centrifugation method. The virus stock was dialyzed against 10 mM Tris, pH 7.5/1 mM Mg2+ containing 10% glycerol. The plaque-forming unit (pfu) of the viruses was measured by a standard plaque assay (25). ARCaP cells were grown to 50–60% confluence in T medium containing 5% FBS. The adenoviruses were added at 20 pfu per cell and incubated overnight. The adenovirus-containing media were replaced with fresh T media and 5% charcoal-stripped serum containing various concentrations of a synthetic androgen, R1881, and incubated for 36 hr. The level of β-gal activity was determined in the cell lysates (26). For comparison, PSA promoter activity was assayed in LNCaP cells transfected by DOTAP-mediated (Boehringer Mannheim) DNA transfer of 5 μg of p64 PSA promoter–luciferase construct.
RESULTS
ARCaP, a novel androgen-repressed human prostatic cancer cell line, was established from the ascites fluid of a man with a widely disseminated disease. We characterized in detail the biochemical and molecular properties of ARCaP cells and associated the results with the clinical characteristics of the patients. The patient was presumed to have an androgen-independent, hormone-refractory disease, because his disease progressed rapidly despite castration. ARCaP cells stained positively for cytokeratin but negatively for vimentin and desmin (data not shown), thus suggesting that this cell line is a human prostate epithelial cell line. Northern and Western blots showed that ARCaP cells expressed both PSA mRNA and protein (Fig. 1A) and mRNA for AR (Fig. 1B). The levels of PSA and AR are substantially lower than those detected in LNCaP cells. We have sequenced the entire DNA binding, the hinge region, and the steroid-binding domains of ARCaP AR cDNA. We found no mutation in more than 1149 bp sequenced. As a positive control, we confirmed the mutation at codon 877 in LNCaP cells, which resulted in a substitution of threonine by alanine (data not shown).
Figure 1.
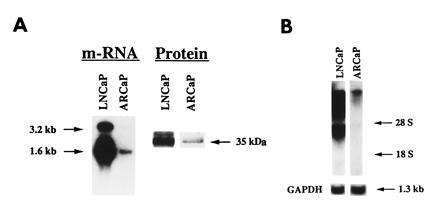
Northern and Western analyses of PSA mRNA and protein and AR mRNA in ARCaP cells. ARCaP cells contain low levels of PSA mRNA and protein (A) and AR mRNA (B) as compared with LNCaP cells. Total cellular RNA used for Northern blots was as follows: PSA, 10 μg for LNCaP cells and 50 μg for ARCaP cells; AR, 20 μg each for LNCaP and ARCaP cells. Western blot analysis of PSA protein was performed by loading 25 and 50 μg of protein derived from LNCaP and ARCaP cells, respectively.
ARCaP cells were cultured in a standard 5% FBS-containing medium. Compared with LNCaP cells, epidermal growth factor receptor and the c-erb B2/neu and c-erb B3 protooncogenes are highly expressed. ARCaP cells stained positively for bombesin, serotonin, neuron-specific enolase, and the c-met protooncogene but stained negatively for chromogranin A. Consistent with the Western blot data, expression of AR and PSA in ARCaP cells was substantially lower than that in LNCaP cells. Other neuronendocrine factors such as neurophysin and substance P were found to stain positively in ARCaP cells. This observation correlates well with the clinical profile of the patient in whom prostate cancer cells from the ascites fluid stained negatively for PSA, prostatic acid phosphatase, and chromogranin A.
Cytogenetic analysis of ARCaP cells (Fig. 2) showed that this cell line is human and consists of at least 11 consistent marker chromosomes (M1–M11) in most of the metaphase spreads: M1 = t(1p;5q); M2 = t(4p;18q); M3 = del(4q); M4 = del(5q); M5 = t(5p;?); M6 = t(7q;8p); M7 = del(8p); M8 = t(12q;?); M9 = inv(16); M10 = del(18q); M11 = t(15q;?). Markers M1, M4, M8, M10, and M11 were present in more than one copy each, whereas M2, M3, M5, M6, M7, and M9 were present in a single copy each. All metaphases showed the presence of X and Y chromosomes in multiple copies. Chromosomes 20 and 22 were disomic, whereas chromosomes 5, 7, and 13 were present in more than 4 copies each. The rest of the autosomes were present in 3 or 4 copies. The presence of two copies of most of the marker chromosomes was an indication that these markers arose before the entire chromosome number had duplicated. After endoreduplication of the entire genome, these markers were present in single copy each. Clonality of all of these markers (M1–M10) was established by their presence in most metaphase spreads. Additional derivatives can be established from parental ARCaP cells by their differences in anchorage-independent growth characteristics. These sublines (fast- and slow-growing clones) also shared some common chromosomal markers with the parental ARCaP cells (unpublished observations).
Figure 2.
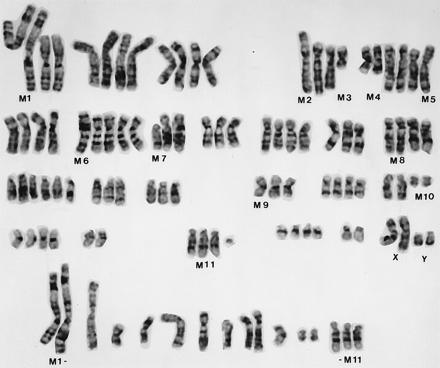
A G-banded karyotype of ARCaP cells. A number of structural markers (M1–M11) and numerical abnormalities were noted. ARCaP cells contain two copies each of the X and Y chromosomes. Marker chromosomes (M1–M11) from additional metaphase spreads are presented at the bottom of the figure.
Hormonal responsiveness of ARCaP cells was tested in vitro under serum-free conditions. As positive controls, we also examined the growth responsiveness of the androgen-dependent LNCaP cell line and its androgen-independent C4-2 subline in vitro in the presence of 10 pM to 1 μM DHT or 10 nM to 100 μM 17β-estradiol. Fig. 3 shows that while DHT stimulated the growth of LNCaP in a biphasic manner (Fig. 3A), 17β-estradiol stimulated the growth of LNCaP cells in a ligand concentration-dependent manner (Fig. 3B). Both DHT and 17β-estradiol failed to affect the growth of the C4-2 cell line. In sharp contrast, DHT and 17β-estradiol depressed the growth of ARCaP cells in vitro in a concentration-dependent manner (Fig. 3); similarly, R1881 or testosterone also depressed the growth of ARCaP cells, whereas dexamethasone had no effect on the growth of ARCaP cells in vitro (data not shown).
Figure 3.
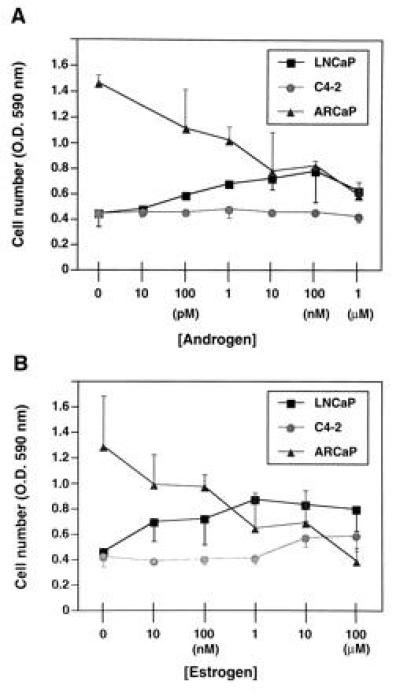
Effects of androgen (5α-dihydrotestosterone; A) and estrogen (17β-estradiol; B) on the growth of LNCaP, C4-2, and ARCaP cells in vitro. Androgen stimulates the growth of LNCaP cells in a biphasic manner but shows no effect on the growth of C4-2 cells. Similarly, estrogen stimulates the growth of LNCaP cells and shows no appreciable effect on the growth of C4-2 cells. In contrast, both androgen and estrogen depress the growth of ARCaP cells in a concentration-dependent manner. Data are expressed as the mean ± SEM of five determinations.
ARCaP cells formed tumors with 100% incidence when administered either subcutaneously or orthotopically to intact or castrated athymic male mice. ARCaP tumors, when maintained as subcutaneous xenografts, grew three times faster in castrated hosts than in intact male hosts (Fig. 4). Tumor growth in castrated hosts was depressed by subcutaneous administration of either testosterone propionate or estradiol dipropionate (Fig. 4). Although metastasis was not noted in animals receiving ARCaP cells subcutaneously, the orthotopically injected ARCaP cells were noted to disseminate to distant organs, such as lymph nodes (18/25, 72%), lung (4/25, 16%), kidney (5/25, 20%), pancreas (3/25, 12%), liver (3/25, 12%), and bone (4/25, 16%), and to produce ascites fluid (6/25, 24%). This pattern of metastasis closely mimicked the clinical presentation of the patient. A typical tumor-bearing animal developed paraplagia (Fig. 5A), with tumor localized on the spine (Fig. 5B) and evidence of osteoblastic and osteolytic responses seen at the histomorphologic level (Fig. 5 C and D, see arrows).
Figure 4.
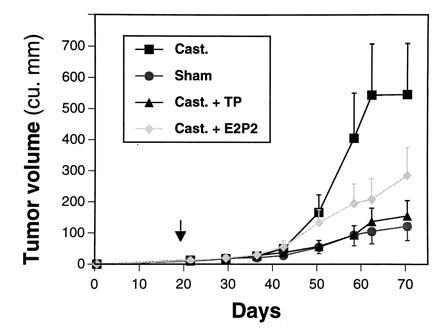
ARCaP tumor growth in sham-operated and castrated male hosts under various hormonal conditions. ARCaP cells were injected subcutaneously into sham-operated or castrated male athymic mice [treated every other day with 50 μl of corn oil, 200 μg of testosterone propionate (TP)/50 μl of corn oil, or 50 μg of estradiol dipropionate (E2P2)/50 μl of corn oil]. Note tumor growth is faster in castrated than in intact hosts, and the growth of ARCaP tumors in castrated hosts is depressed by the administration of either TP or E2P2 alone. Data are expressed as average tumor volume ± SEM of six determinations.
Figure 5.
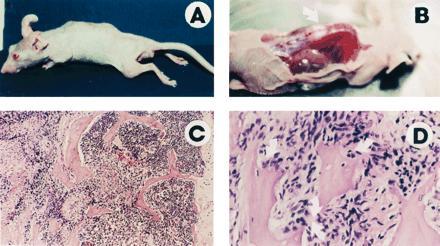
ARCaP cells injected orthotopically into athymic male mice induce paraplegia (A), with visible tumor localized on the spine (B, arrow) and histologically identifiable osteoblastic and osteolytic responses (C and D, arrows). (C, ×75; D, ×150.)
The invasiveness of ARCaP cells in vivo prompted our investigation of the MMP associated with cells as well as secreted in the media. Fig. 6A shows that ARCaP cells secreted a large amount of gelatinase A (72-kDa type IV collagenase, MMP-2) in the media. DHT did not appreciably affect the gelatinase activities associated with LNCaP (Fig. 6A) and ARCaP (Fig. 6B) cells. No stromelysin activity was detected in the cell extract or in the conditioned media of LNCaP cells (data not shown). In contrast, both latent (98 and 52 kDa) and active (42 kDa) forms of stromelysin activity were found to be associated with ARCaP cells. Addition of DHT depressed the basal stromelysin activities (both latent and active forms) associated with ARCaP serum-free medium (Fig. 7A). The level of MMP-3 stromelysin activity associated with ARCaP conditioned medium was five times that of the cell extract (Fig. 7B).
Figure 6.
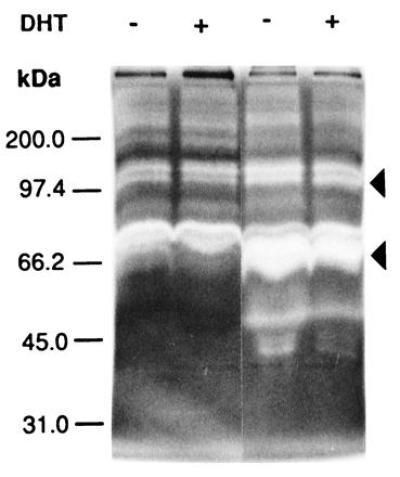
Gelatin zymogram analyses of conditioned media derived from LNCaP (left two lanes) and ARCaP (right two lanes) cells treated either without (−) or with (+) 5α-dihydrotestosterone (DHT). Note that both cell lines secrete more gelatinase A (72-kDa type IV collagenase, or MMP-2) than gelatinase B (92- to 98-kDa type IV collagenase, or MMP-9). ARCaP cells secrete much higher levels of gelatinases A and B, especially the active form of gelatinase A, than do LNCaP cells.
Figure 7.
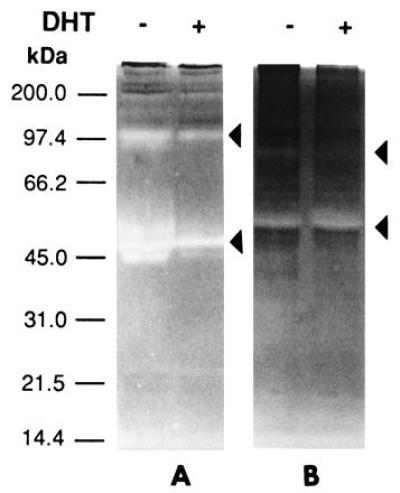
Casein zymogram analyses of conditioned media (A) and cell extracts (B) from ARCaP cells for the detection of stromelysin (MMP-3). Conditioned media (A) contain more stromelysin activity than cell extracts (B). The enzymatic activity in conditioned media was suppressed by DHT by 5- to 10-fold (A).
To determine whether gene expression by ARCaP cells might also be repressed by androgen, we infected these cells with an adenovirus-PSA-β-gal expression vector and exposed the cells to R1881, a synthetic analog, over a 36-hr period. As a positive control, LNCaP cells were transfected with a PSA-promoter luciferase construct, and the reporter activity was determined. Fig. 8 shows that R1881 stimulated PSA-promoter luciferase activity in LNCaP cells but repressed PSA-promoter β-gal activity in ARCaP cells. Both the stimulatory and inhibitory activities of R1881 can be blocked in part (60–80%) by coadministration of Casodex, an antiandrogen (data not shown).
Figure 8.
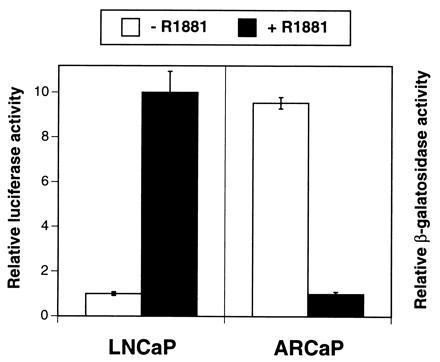
Comparison of PSA-promoter transactivating activity in LNCaP and ARCaP cells. R1881 (10 nM), a synthetic analog of testosterone, stimulates PSA-promoter-driven luciferase activity in LNCaP cells but depresses PSA-promoter β-gal activity in ARCaP cells. Data are expressed as the mean ± SEM of four determinations.
DISCUSSION
The progression of prostate cancer from the androgen-dependent to the androgen-independent state is a critical and well recognized clinical problem. Some cancer cells that acquire the androgen-independent phenotype do respond to secondary hormonal therapy, including flutamide withdrawal (7) and intermittent androgen therapy (8). To understand the molecular basis of these hormonal responses, we began to establish and characterize prostate cancer cell lines from men with androgen-refractory and metastatic disease. In this communication, we present results that document the presence of androgen-repressed phenotypes in a cell clone, ARCaP, derived from the ascites fluid of a man with hormonal-refractory and metastatic disease. Findings that support this conclusion are as follows: (i) the growth of ARCaP cells in vitro is suppressed by DHT, a synthetic androgen, R1881, and 17 β-estradiol; (ii) ARCaP tumor growth in vivo in the castrated hosts is about 3-fold greater than that in intact male hosts; (iii) the growth of ARCaP tumors in vivo in castrated hosts can be suppressed by exogenously administered testosterone propionate or estradiol dipropionate; (iv) consistent with growth inhibition of ARCaP cells by androgen, DHT or R1881 also represses the expression of PSA-promoter reporter activity and stromelysin activities in ARCaP cells. These data demonstrated the first human evidence of a naturally occurring androgen-repressed phenotype of prostate cancer cells.
ARCaP cells were further characterized biochemically and immunohistochemically. The results showed that ARCaP cells expressed low levels of PSA mRNA and protein, and a low level of wild-type AR (with respect to DNA and steroid-binding domains). Epidermal growth factor receptor and the c-erb B2/neu and c-erb B3 protooncogenes were overexpressed. Some neuroendocrine factors, such as serotonin and bombesin, and the c-met protooncogenes were expressed. These expressed genes [epidermal growth factor receptor (27), c-erb B2/neu (18, 28), and c-met (29)] and neuroendocrine factors [bombesin (30–32) and serotonin (33, 34)] have been associated with rapid androgen-independent tumor progression.
The behaviors of ARCaP cells in athymic mice closely mimicked the clinical disease in the following manner. The disease was characterized by low serum PSA in the patient, which corresponded with low levels of PSA mRNA and protein associated with ARCaP cells in culture. The disease in the patient metastasized to liver and bone and appeared in the ascites fluid. Similar observations were made when ARCaP cells were injected orthotopically into athymic mice, resulting in rapid disease progression and extensive metastasis. These observations suggest that this cell model of prostate cancer does reflect, at least in part, the biological behaviors of clinical prostate cancer. In this context, we speculate that certain types of prostate cancer in men may express low levels of PSA and contain wild-type AR (e.g., no mutation in the steroid-binding domain of the AR) but could pose unexpected risks to patients due to the cancer’s high intrinsic invasive and metastatic potential.
The growth of ARCaP cells in vitro is suppressed by androgen and estrogen. In sharp contrast, the growth of LNCaP cells is stimulated and that of C4-2 is unresponsive to both of these sex steroids. These results are consistent with the contrasting transcriptional activity of the PSA gene in ARCaP and LNCaP cells mediated by AR. These results suggest a possible coupling between growth inhibition and repressed expression of PSA mRNA and protein. It is possible that PSA may be a factor positively associated with growth either through direct stimulation of cell growth via signal-transduction pathways or by indirectly promoting growth factor/growth factor receptor processing, such as proteolytic activation of latent growth factor activities (35) or selective degradation of growth factor binding proteins (36). The possible link between cell growth and PSA expression was proposed previously. However, LNCaP cells, when grown under androgen-deprived conditions for a long period, acquired the androgen-repressed phenotype in which growth was depressed, but PSA expression was stimulated by androgen (37). Overall, because of the relatively low level of PSA expression in ARCaP cells, other regulatory mechanisms might override PSA and contribute to androgen-repressed growth.
The lack of steroid specificity for the induction of growth and PSA expression in LNCaP cells was attributed to a structural mutation found in the steroid-binding domain of the AR (38). In this respect, it is curious that the growth of ARCaP cells containing wild-type AR (with respect to DNA and steroid-binding domains) was inhibited by both androgen and estrogen. It is possible that this inhibition can be explained by one or more of the following possibilities: (i) androgen/estrogen may repress the expression of growth stimulatory autocrine/paracrine factor(s) in ARCaP cells; (ii) androgen/estrogen receptors may form heterodimers with putative cofactors and respond negatively to sex steroid-induced transcriptional activity and the growth of ARCaP cells; (iii) mutations of AR at the transactivation domain (exon 1) could confer the androgen-repressed state; and (iv) ARCaP cells may inherit significant genomic instability and assume an altered signal transduction mechanism that allows androgen/estrogen receptors to negatively regulate its growth and gene expression.
In summary, an androgen-repressed human prostate cancer cell line, ARCaP, was established and characterized. This cell line may represent an advanced form of clinical human prostate cancer. The cell line synthesizes and secretes low levels of PSA and contains a low concentration of AR. This cell line is highly invasive and metastatic. ARCaP is an attractive and useful human prostate cell line for the study of prostate cancer biology and therapy, and the mechanisms of hormone action.
Acknowledgments
Our thanks go to Dr. Christopher J. Logothetis at the University of Texas M. D. Anderson Cancer Center, Houston, Texas, for making the ascites fluid from the patient with prostate cancer available and to Dr. Jonathan Simons from the Johns Hopkins School of Medicine, Baltimore, Maryland, for his clinical as well as scientific input during the process of this study. We thank Ms. Carol Chowdhry for editing the paper. This work was supported by National Institutes of Health Grants CA57361 and CA56307/DK47596 to H.Y.E.Z. and L.W.K.C., respectively.
Footnotes
Abbreviations: PSA, prostate-specific antigen; FBS, fetal bovine serum; DHT, 5α-dihydrotestosterone or 5α-androstan-17β-ol-3-one; AR, androgen receptor; MMP, matrix metalloproteinase; β-gal, β-galactosidase.
References
- 1.Whitmore W F. Cancer. 1973;32:1104–1112. doi: 10.1002/1097-0142(197311)32:5<1104::aid-cncr2820320515>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 2.Huggins C, Hodges C V. Cancer Res. 1941;1:293–297. [Google Scholar]
- 3.Isaacs J T. Prog Clin Biol Res. 1987;239:513–576. [PubMed] [Google Scholar]
- 4.Thalmann G, Anezinis P E, Chang S M, Zhau H Y E, Hall C, Pathak S, von Eschenbach A C, Chung L W K. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- 5.Scher H I, Zhang Z F, Cohen L, Kelly W K. Adv Urol. 1995;8:61–95. [Google Scholar]
- 6.Catalona W J. Prostate Cancer. Orlando, FL: Grune & Stratton; 1984. pp. 145–171. [Google Scholar]
- 7.Scher H I, Kelly W K. J Clin Oncol. 1993;11:1566–1572. doi: 10.1200/JCO.1993.11.8.1566. [DOI] [PubMed] [Google Scholar]
- 8.Akakura K, Bruchovsky N, Goldenberg S L, Rennie P S, Buckley A R, Sullivan L D. Cancer. 1993;71:2782–2790. doi: 10.1002/1097-0142(19930501)71:9<2782::aid-cncr2820710916>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh J T, Wu H C, Gleave M E, von Eschenbach A C, Chung L W K. Cancer Res. 1993;53:2852–2857. [PubMed] [Google Scholar]
- 10.Culig Z, Hobisch A, Cronauer M V, Radmay C, Trapman J, Hittmair A, Bartsch G, Klocker H. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 11.Lundwall A, Lilja H. FEBS Lett. 1987;214:317–322. doi: 10.1016/0014-5793(87)80078-9. [DOI] [PubMed] [Google Scholar]
- 12.Chang C, Kokontis J, Liao S. Science. 1988;240:324–326. doi: 10.1126/science.3353726. [DOI] [PubMed] [Google Scholar]
- 13.Towbin H, Gordon J. J Immunol Methods. 1984;72:313–340. doi: 10.1016/0022-1759(84)90001-2. [DOI] [PubMed] [Google Scholar]
- 14.Lowry O H, Rosenbrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 15.Chang C, Kokontis J, Liao S. Proc Natl Acad Sci USA. 1988;85:7211–7215. doi: 10.1073/pnas.85.19.7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichihara Y, Kusosawa Y. Gene. 1993;130:153–156. doi: 10.1016/0378-1119(93)90361-6. [DOI] [PubMed] [Google Scholar]
- 17.Chen E Y, Seeburg P H. DNA. 1985;4:165–170. doi: 10.1089/dna.1985.4.165. [DOI] [PubMed] [Google Scholar]
- 18.Zhau H Y E, Wan D S, Zhou J X, Miller G J, von Eschenbach A C. Mol Carcinog. 1992;5:320–327. doi: 10.1002/mc.2940050413. [DOI] [PubMed] [Google Scholar]
- 19.Pathak S J. Reprod Med. 1976;17:25–28. [PubMed] [Google Scholar]
- 20.Gleave M E, Hsieh J T, von Eschenbach A C, Chung L W K. J Urol. 1992;147:1151–1159. doi: 10.1016/s0022-5347(17)37506-7. [DOI] [PubMed] [Google Scholar]
- 21.Janik P, Briand P, Hartman N R. Cancer Res. 1975;35:3698–3704. [PubMed] [Google Scholar]
- 22.Sang Q-X, Stetler-Stevenson W G, Liotta L A, Byers S W. Biol Reprod. 1990;43:956–964. doi: 10.1095/biolreprod43.6.956. [DOI] [PubMed] [Google Scholar]
- 23.Ko S-C, Gotoh A, Kao C, Chung L W K. J Urol. 1996;155:623A. (abstr.). [Google Scholar]
- 24.Bett A J, Haddara W, Prevec L, Graham F L. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham F L, Prevec L. Mol Biotechnol. 1995;3:207–220. doi: 10.1007/BF02789331. [DOI] [PubMed] [Google Scholar]
- 26.Ko S-C, Gotoh A, Thalmann G N, Zhau H Y E Z, Johnston D A, Zhang W W, Kao C, Chung L W K. Hum Gene Ther. 1996;7:1683–1691. doi: 10.1089/hum.1996.7.14-1683. [DOI] [PubMed] [Google Scholar]
- 27.Habib F K. Prog Clin Biol Res. 1990;357:107–115. [PubMed] [Google Scholar]
- 28.Ware J, Maygarden S J, Kooniz W W, Strom S C. Hum Pathol. 1991;22:254–258. doi: 10.1016/0046-8177(91)90159-m. [DOI] [PubMed] [Google Scholar]
- 29.Pisters L L, Troncoso P, Pollack A, Zhau H Y E, Li W, von Eschenbach A C, Chung L W K. J Urol. 1995;154:293–298. [PubMed] [Google Scholar]
- 30.Bologna M, Festuccia C, Muzi P, Biodri L, Clomei M. Cancer. 1989;63:1714–1720. doi: 10.1002/1097-0142(19900501)63:9<1714::aid-cncr2820630912>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 31.Milovanovic S R, Radulovic S, Grook K, Schally A V. Prostate. 1992;20:269–280. doi: 10.1002/pros.2990200403. [DOI] [PubMed] [Google Scholar]
- 32.Pinski J, Halmos G, Schally A V. Cancer Lett. 1993;71:189–196. doi: 10.1016/0304-3835(93)90115-p. [DOI] [PubMed] [Google Scholar]
- 33.Seuwen K, Magnaldo I, Pouyssegur J. Nature (London) 1988;335:254–256. doi: 10.1038/335254a0. [DOI] [PubMed] [Google Scholar]
- 34.Abdul M, Anezinis P E, Logothetis C J, Hoosein N M. Anticancer Res. 1994;14:1215–1220. [PubMed] [Google Scholar]
- 35.Roberts A B, Sporn M B. In: Peptide Growth Factors. Sporn M B, Roberts A B, editors. Heidelberg, Germany: Springer; 1993. pp. 421–427. [Google Scholar]
- 36.Koutsilieris M. Anticancer Res. 1993;13:443–450. [PubMed] [Google Scholar]
- 37.Kokontis J, Takakura K, Hay N, Liao S. Cancer Res. 1994;54:1566–1573. [PubMed] [Google Scholar]
- 38.Brinkmann A O, Trapman T. Cancer Surv. 1992;14:95–111. [PubMed] [Google Scholar]