

Abstract
The role of autophagy, a catabolic lysosome-dependent pathway, has recently been recognized in a variety of disorders, including Pompe disease, the genetic deficiency of the glycogen-degrading lysosomal enzyme acid-alpha glucosidase. Accumulation of lysosomal glycogen, presumably transported from the cytoplasm by the autophagic pathway, occurs in multiple tissues, but pathology is most severe in skeletal and cardiac muscle. Skeletal muscle pathology also involves massive autophagic buildup in the core of myofibers. To determine if glycogen reaches the lysosome via autophagy and to ascertain whether autophagic buildup in Pompe disease is a consequence of induction of autophagy and/or reduced turnover due to defective fusion with lysosomes, we generated muscle-specific autophagy-deficient Pompe mice. We have demonstrated that autophagy is not required for glycogen transport to lysosomes in skeletal muscle. We have also found that Pompe disease involves induction of autophagy but manifests as a functional deficiency of autophagy because of impaired autophagosomal–lysosomal fusion. As a result, autophagic substrates, including potentially toxic aggregate-prone ubiquitinated proteins, accumulate in Pompe myofibers and may cause profound muscle damage.
INTRODUCTION
Macroautophagy (hereafter called autophagy) is an evolutionarily conserved degradative pathway by which long-lived intracellular proteins and organelles are delivered to the lysosome for destruction and recycling. During this process, a double-membrane vesicle called an autophagosome develops around a portion of the cytoplasm and organelles, such as mitochondria. The outer autophagosomal membrane fuses with the lysosomal membrane resulting in the delivery of the inner vesicle into the lumen of the lysosome. Within the lysosome, the autophagic cargo is broken down by hydrolases, and the resulting molecules are recycled (reviewed in 1–3). Autophagy is involved in the cellular response to starvation, cellular differentiation, cell death, aging, cancer and neurodegenerative diseases. Since the lysosomes are the final destination of the autophagic pathway, it is not surprising that this pathway has also been implicated in several lysosomal storage disorders (4–7), including Pompe disease (glycogen storage disease type II, GSDII), a deficiency of the glycogen-degrading lysosomal enzyme acid-alpha glucosidase (GAA).
The deficiency of this enzyme results in the failure to metabolize lysosomal glycogen to glucose leading to progressive accumulation of glycogen and the enlargement of lysosomes in multiple tissues. Cardiac and skeletal muscles are the major tissues affected by the storage. Both severe cardiomyopathy and skeletal muscle myopathy are observed in patients with complete or near complete enzyme deficiency. Affected infants usually die within the first year of life. In patients with milder late-onset forms, cardiac muscle is spared, but slowly progressive skeletal muscle weakness eventually leads to premature death due to respiratory insufficiency (8).
Pompe disease itself and the basic biochemistry of the genetic defect have been known for decades, but the mechanism of profound muscle damage has remained unclear. The accumulation of glycogen, presumed to be transported to the lysosomes via the autophagic pathway, is not limited to the lysosomes in skeletal muscle, but is also found in autophagic vacuoles containing cytoplasmic degradation products (8,9). We have demonstrated that the great extent of autophagy and its role in muscle damage make the autophagic process a critical player in the pathogenesis of Pompe disease. Our findings in both humans and GAA knockout mice (GAA KO) revealed the presence in the core of muscle fibers of huge non-contractile inclusions containing cellular debris, fragmented mitochondria, remnants of lysosomal membranes and a large number of autophagosomes (10,11). In the GAA KO these inclusions are limited to muscles rich in glycolytic fast fibers and appear to be responsible for the disappointing response to enzyme replacement therapy (ERT) with recombinant human enzyme (12,13). This enzyme (alglucosidase alfa, Myozyme®, Genzyme Corp. Framingham, MA) has recently become available for ERT in Pompe patients.
To determine if glycogen reaches the lysosome via autophagy and to establish if autophagic buildup in Pompe disease is a consequence of an induction of autophagy and/or impaired resolution of autophagosomes due to defective fusion with lysosomes, we have generated muscle-specific autophagy-deficient Pompe mice. We have found that the transport of glycogen from the cytoplasm to lysosomes does not require autophagy. We also demonstrated that autophagy is upregulated in both slow (type I) and fast (type II) fibers in the GAA KO, but only in fast fibers is there also inefficient disposal of autophagic cargo and the accumulation of ubiquitin-positive bodies, putative mediators of muscle damage. In addition, suppression of autophagy in the wild-type (WT) control mice allowed us to explore the role of this pathway in normal muscle, of which little is yet known. Here we demonstrate that the consequences of loss of autophagy in normal muscle are dependent on fiber type, indicating that autophagy fulfills different purposes in the diverse fiber types.
RESULTS
Generation of skeletal muscle-specific autophagy-deficient GAA KO mice
We generated muscle-specific autophagy-deficient mice on the GAA KO background (AD-GAA KO); these mice contain a Cre recombinase transgene under the control of the human skeletal actin promoter (HSA-Cre) and an Atg5 gene, in which exon 3 is flanked by loxP sites (Atg5flox/flox). The HSA promoter drives the expression of Cre recombinase in both fast and slow muscle fibers resulting in muscle-specific inactivation of Atg5, a critical gene in autophagosome formation (14). The need for tissue-specific suppression of autophagy is justified by the fact that mice with global deletion of autophagic genes (Atg5 or Atg7) die soon after birth (15,16).
Of note is the fact that in the GAA KO mice, the GAA gene, which is disrupted by a neo cassette in exon 6 (6neo/ 6neo), contains two loxP sites in introns 5 and 6 (17). The expression of Cre recombinase would therefore remove exon 6 (Δ6) and the neo from the GAA gene in skeletal muscle. We have previously demonstrated that in homozygous Δ6/Δ6 mice, pathological and biochemical changes in skeletal muscle are indistinguishable from those in the 6neo/6neo mice (17). Nevertheless, to obtain the most adequate control we have generated GAA KO mice, in which Cre recombinase, driven by the HSA promoter, is expressed in the muscle, resulting in the removal of exon 6 of the GAA gene specifically in the muscle but not in other tissues. WT mice and muscle-specific Atg5-deficient mice on a WT background (AD-WT) served as additional controls; the data concerning the AD-WT mice will be discussed in a separate section.
The suppression of autophagy in the skeletal muscle was evaluated by the level of a specific autophagosomal marker, LC3-II. The microtubule-associated protein 1 light chain 3 (LC3), the mammalian homologue of the yeast autophagosomal marker, Atg8, exists as a soluble form, LC3-I, which is modified into the PE (phosphatidylethanolamine)-conjugated form LC3-II (18). Conversion of the soluble LC3-I to the membrane associated LC3-II form is an Atg5-dependent process (19,20). In AD-GAA KO only the LC3-I form is present, indicating complete suppression of autophagy in the skeletal muscle in these mice (Fig. 1A).
Figure 1.

Generation and characterization of muscle-specific AD-GAA KO mice. (A) Western blotting of protein lysates from gastrocnemius muscle of 3-month-old mice with LC3 antibody. Lane 1: WT; Lane 2: GAA KO; Lane 3: AD-GAA KO. The absence of LC3-II in AD-GAA KO indicates an efficient suppression of autophagy (dozens of mice were analyzed for the suppression of autophagy). (B) PAS-stained sections of gastrocnemius muscle from 5-month-old WT, GAA KO and AD-GAA KO; PAS-positive material (small dots) is seen in both GAA KO and AD-GAA KO. Centrally located ‘holes’ found in the GAA KO, but not in the AD-GAA KO fibers represent areas of autophagic buildup (arrows). Magnification: 335×. A 4.5-month-old AD-GAA KO mouse shows profound muscle wasting.
Suppression of autophagy in the skeletal muscle does not significantly affect the level of glycogen accumulation, but exacerbates the phenotype of the GAA KO mice
In the WT, a negligible amount of glycogen was detected in skeletal muscle by a biochemical assay, 0.26 ± 0.30% wet weight (n = 17). The levels of accumulated glycogen were comparable in fast (white gastrocnemius) muscle from 4- to 7-month-old AD-GAA KO and GAA KO; 4.0 ± 2.6% wet weight (n = 34) and 5.0 ± 2.8% (n = 36), respectively. The same was true of slow (soleus) muscle; AD-GAA KO 9.7 ± 2.9% wet weight (n = 9), GAA KO 7.8 ± 2.8% (n = 8). Consistent with these data, histological examination of muscle samples from AD-GAA KO mice revealed multiple PAS-positive structures similar to those observed in GAA KO mice, indicating that inactivation of autophagy does not prevent lysosomal accumulation of glycogen (Fig. 1 B, shown for fast muscle).
Despite the similar level of glycogen accumulation in GAA KO and AD-GAA KO, the AD-GAA KO develop a more severe phenotype (Fig. 1B). Like GAA KO, AD-GAA KO are born normal, but at the age of 2–3 months they already show clear signs of lower back muscle weakness and distinct kyphosis. By the age of 4–5 months the AD-GAA KO mice exhibit severe muscle wasting, profound kyphosis, a waddling gait and growth retardation. The symptoms progress rapidly, and by the age of 6–7 months these mice breathe with difficulty, drag their hind limbs in a splayed fashion while walking, cannot lift their bellies off the ground, lose tail strength, and develop a near paralysis of hind limbs. At this stage the AD-GAA KO mice require daily observation and many begin to die. By 8–10 months, the surviving mice are euthanized to prevent suffering. In contrast, the GAA KO mice remained phenotypically normal up to 8–9 months of age, after which point they develop the first clinical signs of muscle wasting consistent with the results reported previously for the GAA 6neo/6neo (17). In the wire-hang task, AD-GAA KO performed less well than GAA KO; AD-GAA KO were able to maintain their hold for an average of 26.5 s, whereas GAA KO held on for an average of 36.1 s (P = 0.035). Both performed significantly less well than WT (average 201.5 s) (P < 0.001).
These changes correlate well with greater muscle atrophy of AD-GAA KO compared with GAA KO, as demonstrated by the significant reduction of the cross-sectional area of myofibers (P < 0.05). GAA KO and AD-GAA KO fibers are both more atrophic compared with the WT (P < 0.001); WT 582 ± 227 µm2 (n = 196); GAA KO 369 ± 116 µm2 (n = 252); AD-GAA KO 286 ± 145 µm2 (n = 273).
The AD-GAA KO mice are significantly smaller than the GAA KO at the age of 5–7 months: 19.9 ± 3.7 g (n = 10) and 25.0 ± 3.6 g (n = 10) (P = 0.03), respectively. In younger, 4–5-month-old mice the difference is not significant: 24.0 ± 3.5 g (n = 7) for the AD-GAA KO and 26.7 ± 3.5 g (n = 6) for the GAA KO. The weights of the GAA KO and AD-GAA KO are significantly lower than the WT at all ages tested (P < 0.05).
Muscle atrophy in either GAA KO or AD-GAA KO is not associated with the common transcriptional program of changes identified in atrophying muscles
It has been shown that muscle loss in different systemic disorders, including cancer, diabetes mellitus, and in multiple models of skeletal muscle atrophy is associated with transcriptional activation of a number of genes termed atrophy-related genes (21,22). In order to determine if these genes are activated in the muscle atrophy in Pompe disease, we selected and monitored by RT-PCR two highly upregulated muscle-specific ubiquitin ligases, atrogin-1/MAFbx and Murf1, as well as the upstream activator FoxO1 and a lysosomal hydrolase Cathepsin L. No activation of any of these genes was detected in GAA KO or the AD-GAA KO. In fact, Atrogin was downregulated up to 3-fold in two of four GAA KO animals, and downregulated up to 2-fold in two of five AD-GAA KO animals. Murf and Cathepsin L were consistently unchanged in both GAA KO and AD-GAA KO animals compared with WT (n = 5 for each group). FoxO1 was downregulated up to 5-fold in two of five GAA KO and in one of seven AD-GAA KO animals. Western analysis revealed no difference in the levels of FoxO3 in GAA KO and AD-GAA KO compared with WT (data not shown). These data indicate that the common program of atrophy is not engaged in Pompe disease and, therefore, there must be a different mechanism of muscle wasting in this disorder.
Suppression of autophagy in the skeletal muscle of GAA KO (AD-GAA KO) reveals a buildup of endocytic vesicles in the core of fast muscle fibers
We have previously shown that large areas of non-contractile cellular debris containing numerous LC3-positive autophagosomes (which we refer to as autophagic buildup) accumulate in the core of fast GAA KO fibers. As expected, LC3-positive structures were not seen in fast fibers of the AD-GAA KO. Surprisingly, there was still buildup in the core of the myofibers, now composed primarily of clustered LAMP-1-positive vesicles (Fig. 2A). Double staining of single myofibers with LAMP-1 and cation-independent mannose 6-phosphate receptor (CI-MPR) identified these structures as lysosomes (LAMP-positive/CI-MPR-negative) and late endosomes (LAMP-1/CI-MPR double-positive) (Fig. 2B).
Figure 2.

Buildup of late endocytic vesicles in fast (psoas) fibers from AD-GAA KO. (A) Single fiber stained for LAMP-1 (LP). Arrowhead points to an enlarged lysosome. Bar: 10 µm. (B) Single fiber stained for CI-MPR (MPR) and LAMP-1. Bar: 10 µm. (C and D) Electron microscopy shows intermyofibrillar accumulations of enlarged glycogen-filled lysosomes (arrowhead), clusters of vesicles and inclusions (*). These inclusions likely represent polyubiquitinated protein aggregates (see text and Fig. 4). Some vesicles have the typical morphology of multi-vesicular bodies (right panel in D). Bars: 500 nm.
Consistent with these data, electron microscopy showed accumulation of multiple vesicles surrounding the expanded glycogen-filled lysosomes. Some of these vesicles contained internal vesicles, which identified them as late endosomes, also called multi-vesicular bodies (Fig. 2C and D). The data indicate that a subset of clustered, centrally located, dysfunctional glycogen-filled lysosomes in fast fibers of the AD-GAA KO is unable to fuse with endosomes. It is therefore reasonable to assume that a similar subset of lysosomes in the GAA KO would also be unable to fuse with autophagosomes, leading to the observed autophagic buildup. The consequence of this situation would be a reduction in the turnover of autophagosomes and their substrates. To test this hypothesis we have looked at the levels of autophagic substrates, such as ubiquitinated proteins and P62/SQSTM1 (23–25).
Accumulation of ubiquitinated (Ub) proteins and P62/SQSTM1 in GAA KO and AD-GAA KO
Immunoblotting showed increased levels of high molecular mass Ub-proteins in fast gastrocnemius muscle of the GAA KO, indicating that there is a failure of autophagosomal–lysosomal fusion and a block in autophagosomal turnover—in other words, a functional deficiency of autophagy (Fig. 3A). In the AD-GAA KO, in which there is no autophagy, even more Ub-proteins were detected in both soluble and non-soluble fractions (Fig. 3A). Both FK2 (recognizes both mono- and poly-ubiquitinated proteins) and FK1 (recognizes only polyubiquitinated proteins) antibodies showed a similar pattern on western blots, indicating that at least some (or possibly all) proteins are polyubiquitinated (shown for FK2 in Fig. 3). The accumulation of Ub-proteins in the GAA KO preceded the development of clinical symptoms and increased with age; this increase was most dramatic for non-soluble proteins, suggesting that the formation of Ub-positive protein aggregates parallels the progression of the disease (Fig. 3B).
Figure 3.
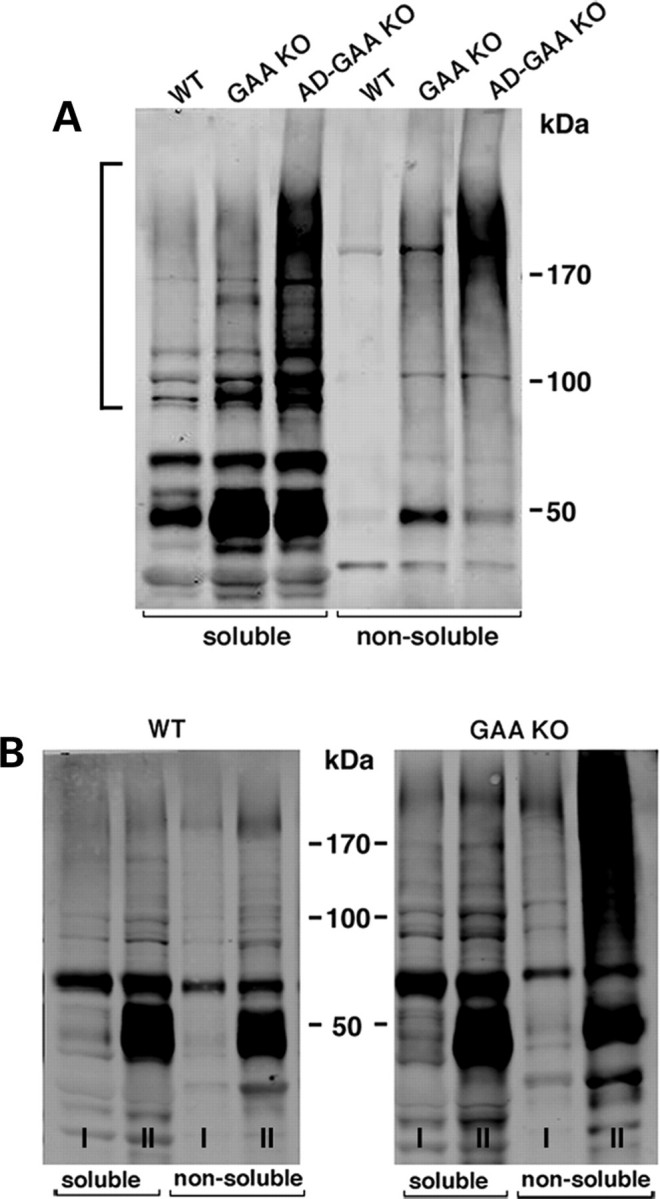
Accumulation of Ub-proteins in fast (gastrocnemius) muscle of GAA KO and AD-GAA KO. Muscle lysates, prepared as detergent (Triton X-100)-soluble and -non-soluble fractions, were analyzed by immunoblotting with anti-ubiquitin (FK2) antibody. (A) Muscle samples were taken from ∼4-month-old WT, GAA KO and AD-GAA KO mice. High molecular mass Ub-proteins are marked by a bracket. Data shown are representative of at least four independent experiments. (B) Age-dependent accumulation of soluble and non-soluble Ub-proteins in muscle from young (6-week-old; I) and old (10-month-old; II) GAA KO. For each time point and each genotype, three mice were used.
Immunostaining of isolated fibers for ubiquitin revealed a dramatic difference in the distribution of Ub-positive structures in the GAA KO and AD-GAA KO. In the GAA KO, inclusions of Ub-proteins were limited to the regions of the autophagic buildup and often located within the LC3-positive autophagosomes (Fig. 4, top panel). In contrast, in the AD-GAA KO, these inclusions were dispersed throughout the fibers, often located next to the lysosomes (Fig. 4 AD-GAA KO, immunostaining), and were identifiable by EM as large irregular non-membrane bound filamentous structures (Fig. 4 AD-GAA KO, EM). The confinement of the inclusions to the regions of autophagic buildup in fast GAA KO muscle fibers indicates that the functional deficiency of autophagy is local.
Figure 4.
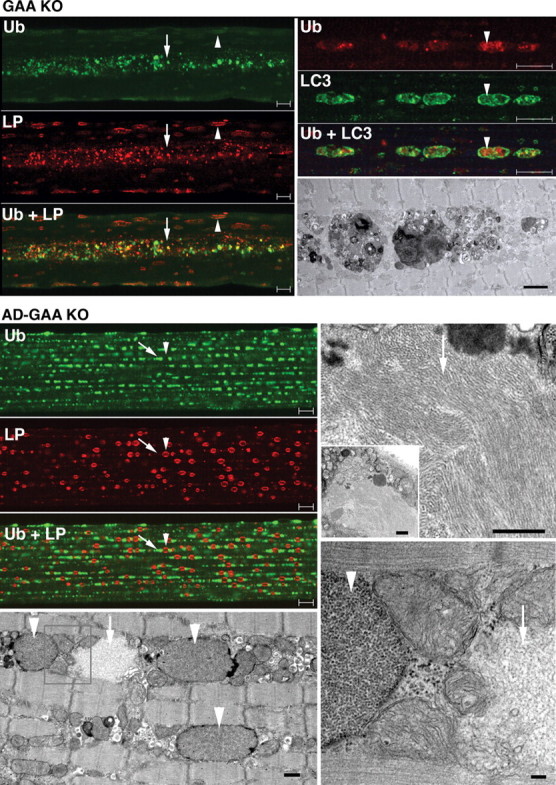
Distribution of Ub-proteins in fast (psoas) muscle of GAA KO and AD-GAA KO. Left top panel: GAA KO; single fiber stained for ubiquitin (Ub) and LAMP-1 (LP) showing the confinement of Ub-positive structures to the area of autophagic buildup (arrow); in contrast, LAMP-1 staining is present throughout the fiber—in the autophagic area and on the enlarged lysosomes (arrowhead). Bar: 10 µm. Right panel: single fiber stained for Ub and LC3 showing the presence of Ub-positive structures within LC3-positive autophagosomes. Bar: 10 µm. The EM panel shows autophagic buildup. Bar: 2 µm. Left panel: AD-GAA KO; single fiber stained for Ub and LAMP-1 showing Ub-positive structures (arrow) dispersed in a linear array throughout the fiber in a repeating pattern of inclusions flanking lysosomes (arrowhead). Bar: 10 µm. EM (left bottom panel) shows glycogen-filled lysosomes (arrowheads) and an electron-light, irregularly shaped structure (arrow), which likely represents a Ub-positive inclusion. Bar: 500 nm. At higher magnification (right EM panels) the filamentous inclusions (arrows) appear very different from the membrane-bound lysosomal glycogen (arrowhead). Bars: 500 nm (top); 100 nm (bottom).
Immunostaining of fast GAA KO fibers for P62/SQSTM1, an autophagic substrate that links Ub-proteins to autophagosomes (25–27), revealed accumulation of this protein only in the regions of the autophagic buildup in the core of the GAA KO fibers (Fig. 5). In contrast, P62-positive structures in atrophying AD-GAA KO fast fibers were dispersed throughout the fibers, and their distribution resembled that of Ub-proteins (Fig. 5). These data provide further evidence of the local functional deficiency of autophagy in the GAA KO.
Figure 5.

Distribution of P62/SQSTM1 protein in fast (psoas) muscle of GAA KO and AD-GAA KO. Single fibers stained for P62 and for LAMP-1 (LP). P62 protein accumulates in the area of autophagic buildup in muscle from GAA KO. In AD-GAA KO P62-positive structures are dispersed throughout the fiber [this pattern was observed in profoundly atrophic fibers (∼50% of the total); the remaining fibers showed no staining for P62]. The recording parameters were optimized for each genotype. The WT P62 image was recorded at a higher gain than the other two images to ensure the absence of cytoplasmic staining. Bar: 10 µm.
Both slow and fast GAA KO muscle exhibit an induction of autophagy, but in slow muscle fibers there is no functional deficiency of autophagy
In WT muscle, the abundance of both LC3 forms is higher in slow than in fast muscle fibers (Fig. 6A). In the GAA KO, LC3-II, the autophagosomal membrane-associated form, increases dramatically in both fiber types, an indication of an increase in the number of autophagosomes. In addition, there is more newly synthesized unprocessed LC3-I form in GAA KO muscle, an indication of an induction of autophagy (Fig. 6A).
Figure 6.
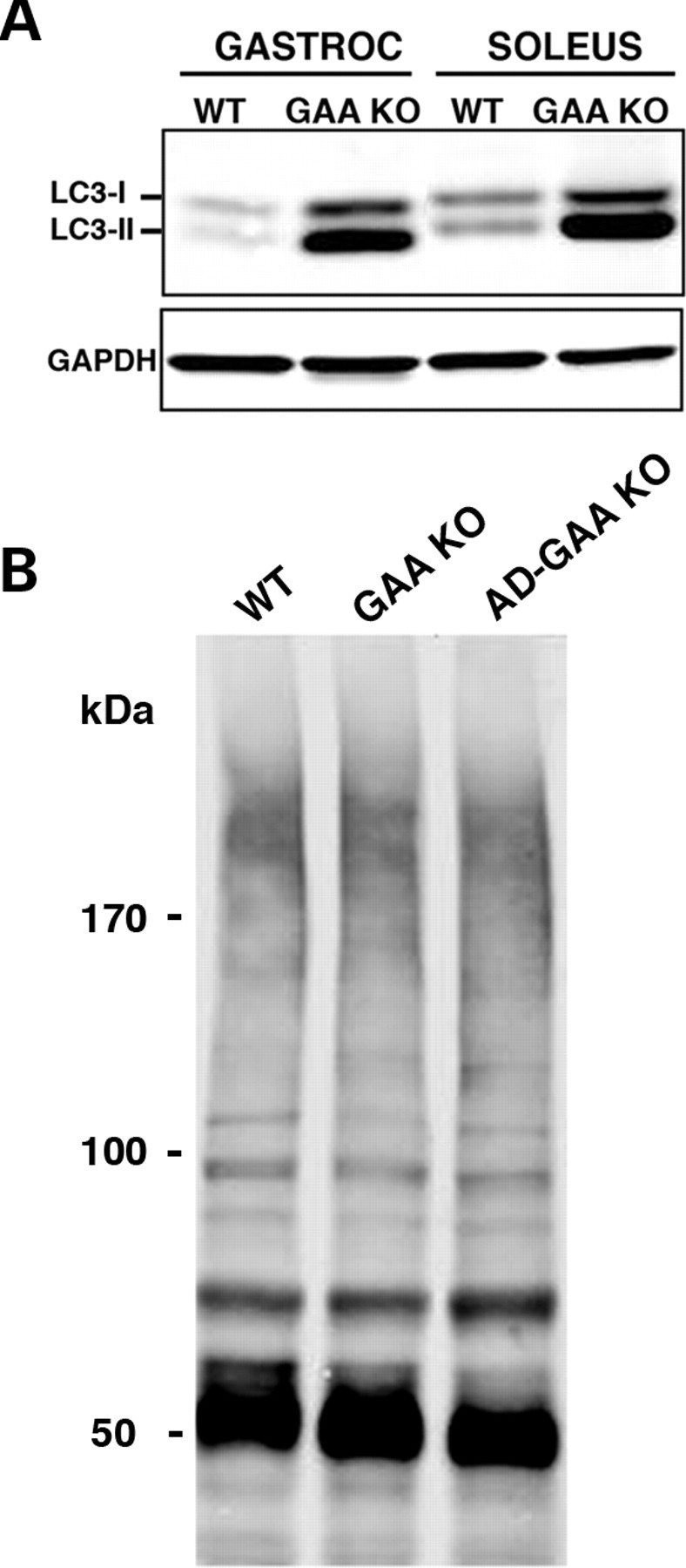
(A) Basal level of autophagy in gastrocnemius and soleus muscles from WT and GAA KO. Western blotting of protein lysates with LC3 antibody shows a dramatic increase in the levels of both LC3-I and LC3-II in GAA KO. At least five WT and five GAA KO animals were used for these experiments. (B) Western blotting of protein lysates from soleus (slow) muscle with FK2 antibody. No difference in the levels of Ub-proteins is observed among the three genotypes in 4-month-old mice. Data shown are representative of at least three independent experiments.
Unlike fast GAA KO myofibers, however, slow GAA KO myofibers, which do not exhibit autophagic buildup (10,12), showed no increase in Ub-proteins as detected by immunoblot (Fig. 6B) or by immunostaining of single fibers (data not shown) derived from mice of up to 6 months of age. Thus, the induction of autophagy, observed in both slow and fast GAA KO fibers, is accompanied by a functional deficiency of autophagy—massive autophagic buildup and the accumulation of Ub-proteins—only in fast muscle.
We observed no appreciable increase in the amount of Ub-proteins in slow fibers in either GAA KO or in AD-GAA KO up to 6 months of age (Fig. 6B), suggesting that autophagic flux is complete at this stage of the disease, but we cannot exclude the possibility that later in life, a functional deficiency could develop.
Suppression of autophagy in WT muscle (AD-WT) results in low level accumulation of Ub-proteins and altered distribution of lysosomes in fast muscle fibers
As in the AD-GAA KO, autophagy was completely suppressed in skeletal muscle of the AD-WT (data not shown). Suppression of autophagy in normal healthy muscle did not result in any visible clinical abnormalities during the 7 months of observation. These mice performed no worse than the WT in the wire hang task and had no significant reduction in weight at 4–6 months of age; 31.6 ± 5.6 g for AD-WT (n = 10) and 33.0 ± 4.8 g for WT (n = 11).
There was, however, an atrophy of fast AD-WT myofibers compared with the WT, as shown by the reduction in the cross-sectional area: 343 ± 137 µm2 (n = 229) and 582 ± 227 µm2 for AD-WT and WT, respectively (P < 0.05). The atrophy in fast muscle fibers was associated with an increase in the amount of accumulated autophagic substrates, as demonstrated by immunostaining of isolated fibers for Ub and P62 (Fig. 7). These data are consistent with the results obtained in autophagy-deficient brain and liver (16,23,24). The cytoplasmic inclusions of Ub-proteins in the AD-WT were similarly distributed but much less numerous than in AD-GAA KO (compare Fig. 4 and Fig. 7A). In slow muscle fibers, however, we did not observe an increase in Ub-proteins in the AD-WT (data not shown), indicating that the consequences of loss of autophagy in normal muscle are dependent on fiber type.
Figure 7.

Accumulation of Ub-proteins and P62 in AD-WT gastrocnemius muscle. Single fibers stained for Ub and LAMP-1 (LP) show a typical Ub pattern (A) and a rare pattern of large deposits (C) of ubiquitin-positive material. The overall level of Ub-positive staining is much lower than in the AD-GAA KO shown in Figure 4. The WT Ub image (D) was recorded at a higher gain than the AD-WT images (A and C) to ensure the absence of cytoplasmic staining. (B) EM shows a filamentous inclusion, which resembles those shown in Figure 4. Bars: 500 nm; 100 nm (inset). (E) Single fiber stained for P62 and LAMP-1 showing the similarity between P62 and Ub staining patterns. Bars: 10 µm.
Unexpectedly, in fast muscle fibers of the AD-WT, the lysosomal size and density appear increased, and the distribution of the lysosomes and microtubules are strikingly altered compared with the WT: lysosomes and microtubules appear to be arranged in a more linear fashion than in the WT (Fig. 8). It has been shown that intact microtubules are essential for autophagosomal formation and movement (28–30). In an analogous fashion, we show here that intact autophagy is essential for the normal disposition of microtubules in skeletal muscle.
Figure 8.

Altered distribution of microtubules and lysosomes in AD-WT muscle. Single fibers from fast WT and AD-WT (gastrocnemius) muscle stained for alpha-tubulin (α tub) and LAMP-1 (LP) showing the realignment of microtubules and lysosomes in AD-WT. Arrowhead points to a string of microtubules and lysosomes. Bar: 10 µm. EM of fast (psoas) muscle from AD-WT shows vesicular structures in the intermyofibrillar space. The vesicles likely represent lysosomes. Bar: 1 µm.
DISCUSSION
The genetics, biochemistry, clinical presentations, and many other facets of Pompe disease are very well described, but important questions have remained unanswered: how is glycogen delivered to the lysosomes; and what is the mechanism of the profound skeletal muscle damage. The answers to both of these questions have implications for the proper design of therapy.
Glycogen has been thought to reach the lysosomes via the autophagic pathway. This assumption is based on data showing autophagic delivery of glycogen to the lysosomes in neonatal tissues (reviewed in 31,32). In this early postnatal period, when there is a high demand for glucose, ‘glycogen autophagy’ and lysosomal degradation of glycogen to glucose constitute a mechanism of supplying this sugar. It has been suggested that hepatic glycogen autophagy is a selective and highly regulated process in the newborn. Several pathways, including the cyclic AMP/cyclic AMP-dependent protein kinase and the phosphoinositides/TOR pathways are implicated in the control of this process (33–35). Earlier studies suggested that autophagic degradation and rapid mobilization of glycogen not only in liver, but also in skeletal muscle, is an important source of energy in the first critical period of extra-uterine life in newborn rats (36). However, little is known about the role of glycogen autophagy in adult animals. We have now demonstrated that the AD-GAA KO and GAA KO mice contain a comparable amount of glycogen in muscle, indicating that macroautophagy in muscle is not the major route of glycogen delivery to the lysosome in adult animals. This is true of both slow and fast muscles. Microautophagy, the direct transport of substrates to the lysosome by invagination of the lysosomal membrane, may be a plausible alternative. Thus, the findings indicate that suppression of autophagy is not an option for substrate deprivation therapy in Pompe disease.
The need to expand our understanding of the disease pathogenesis and the mechanisms of muscle damage in Pompe disease has become particularly clear in light of the limited success of the recently developed ERT in reversing pathology in skeletal muscle (12,13,37). The long-held view of the pathogenesis is that expansion and rupture of lysosomes result in the release of glycogen and lytic enzymes into the cytoplasm, which causes a loss of myofibrils and contractile function (38,39). However, studies in a mouse model of the disease (40) have shown that the enlarged lysosomes in skeletal muscle cannot adequately account for the reduction in mechanical performance, and that the presence of large inclusions containing degraded myofibrils contributes to the impairment of muscle function (41,42). In another GAA KO mouse model (17), we showed the enormous extent of these inclusions in fast muscle fibers, their effect on muscle architecture, the problem they pose for ERT, and their considerable autophagic component, justifying the term ‘autophagic buildup’ (43,44).
It remained unclear, however, whether the increased number of autophagosomes in fast fibers is the result of an induction of autophagy, an impairment of autophagosomal–lysosomal fusion, or a combination of both. Distinguishing between these two opposite scenarios would greatly influence the development of therapeutic approaches. We did observe induction of autophagy in both fast and slow GAA KO fibers (although more robust in fast fibers) as evidenced by increased levels of LC3-I and LC3-II compared with WT levels.
The consequences of the induction of autophagy in the diseased muscle, however, are profoundly different depending on the fiber type. Counter-intuitively, the induction of autophagy in fast GAA KO muscle is associated with a functional deficiency of autophagy. In these fibers, there is also a problem with the other end of the autophagic process, namely impaired fusion with the lysosome. Failure to fuse with lysosomes, referred to as incomplete autophagic flux (45), results in accumulation of autophagic substrates, such as potentially toxic Ub-proteins. The accumulation of such Ub-proteins observed in GAA KO fast fibers is clear evidence of incomplete autophagic flux. This conclusion is strengthened by the finding that P62 also accumulates in these fibers. P62/SQTSM1, a scaffold protein that binds both LC3 and ubiquitinated substrates, is itself an autophagic substrate and thus accumulates only when autophagy is blocked (25,26,46). The autophagic dead-end in muscle is in effect a functional autophagic deficiency. The deficiency is, however, local and limited to the core of the fiber, and as such is less detrimental than the complete genetic deficiency and dispersed cytoplasmic accumulation of Ub-proteins in the AD-GAA KO fast fibers. Thus, the additional burden imposed by complete autophagy deficiency in the AD-GAA KO exaggerates the problem already present in the GAA KO. The elimination of toxic Ub-proteins was traditionally assigned exclusively to the proteasomal system, but is now recognized as a critical function of constitutive autophagy in addition to its major roles in the cellular responses to starvation or stress (reviewed in 1,47,48). This recognition was made possible by data showing accumulation of these proteins when autophagy was suppressed in liver or brain (16,23,24).
Malfunctioning of the endocytic pathway that terminates at the lysosome is also likely to contribute to the pool of Ub-proteins in the GAA KO. It has been shown that Ub-proteins destined for lysosomal degradation are delivered to this organelle not only by the autophagic, but also by the endocytic pathways. For example, multiple plasma membrane proteins and cell surface receptors are mono-ubiquitinated and sorted into the lumen of multivesicular bodies for delivery to lysosomes. Lysosomal contribution to the accumulation of Ub-proteins in GAA KO is supported by the fact that complete autophagic deficiency in healthy muscle (AD-WT) is much less harmful than the complete autophagic deficiency combined with abnormal lysosomes (AD-GAA KO). Lysosomal degradation of cell-surface receptors serves as a mechanism for transient downregulation of these polypeptides (49–51), and the failure to efficiently downregulate these molecules in GAA KO may result in unrestrained signaling with profound consequences. This possibility needs to be further investigated.
Muscle wasting in GAA KO mice occurs despite the absence of the transcriptional changes shown to be associated with muscle atrophy due to a variety of conditions, such as cancer cachexia, uremia, diabetes, etc. (22,52–56). Thus, the mechanism of muscle loss in Pompe disease and possibly other metabolic myopathies is quite distinct from the ‘common atrophy program’ (22).
This distinct mechanism may involve the accumulation of undegraded aggregate-prone Ub-proteins: in the GAA KO this pathology is observed before the first clinical symptoms, but expands with age, paralleling disease progression. It has been shown that cytoplasmic accumulation of large ubiquitin-containing aggregates and inclusion bodies in autophagy-deficient hepatocytes and neurons resulted in severe liver injury and neurodegeneration (16,23,24). Progressive neuronal loss in autophagy-deficient neurons was associated with increase in size and number of these inclusions, suggesting a strong correlation between the accumulation of the Ub-proteins and disease severity. In both autophagy-deficient brain and liver, these inclusions occurred despite the apparently normal proteasome function.
The association of skeletal muscle weakness with the accumulation of high molecular weight proteins and Ub-containing protein inclusions has been shown for sporadic inclusion-body myositis and hereditary inclusion body myopathies (IBM) (57). A similar association has been recently shown for IBM caused by mutations in valosin-containing protein p97/VCP (58). In these cases, the accumulation of the Ub-proteins appears to be a consequence of abnormal proteasome function.
Finally, accumulation of Ub-proteins due to an autophagic block is associated with neuronal cell death in a deficiency of another lysosomal enzyme, multiple sulfatase deficiency (5), suggesting that abnormal autophagy and toxicity from ubiquitinated material may also be a general mechanism of cellular damage not only for metabolic myopathies, but also for other lysosomal storage disorders.
MATERIALS AND METHODS
The following primary antibodies were used for immunostaining of fixed fibers: rabbit antiserum to bovine CI-MPR (a gift from Dr Stuart Kornfeld, Washington University School of Medicine, St Louis, MO); rabbit anti-LC3B (microtubule-associated protein 1 light chain 3) (Sigma, St Louis, MO); rat anti-mouse LAMP-1 (Lysosomal-Associated Membrane Protein 1) (BD Pharmingen, San Diego, CA); mouse anti-mono- and poly-ubiquitinated conjugates (clone FK2) and mouse anti-poly-ubiquitinated conjugates (clone FK1; IgM immunoglobulin subclass) (both from BIOMOL International, L.P., Philadelphia, PA); mouse monoclonal anti-alpha tubulin (Sigma); rabbit anti-phospho-FKHRL1/FOXO3A (Upstate, Lake Placid, NY); mouse monoclonal anti-P62/SQSTM1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); mouse monoclonal anti-GAPDH antibody (Abcam, Cambridge, MA) served as a loading control. Alexa Fluor-conjugated antibodies (Molecular Probes, Eugene, OR) were used as secondary antibodies.
Generation of mice
Atg5 conditional knockout mice (24), crossed with transgenic mice expressing Cre recombinase under the control of human skeletal actin promoter (HSA) (59) were intercrossed to produce mice deficient for Atg5 specifically in skeletal muscle (HSA-Cre:Atg5flox/flox). These muscle-specific autophagy-deficient mice were then crossed with GAA KO mice to produce (after several rounds of crossing) GAA KO mice deficient for Atg5 in skeletal muscle (HSA-Cre:Atg5flox/flox: GAA−/−; AD-GAA KO). The AD-GAA KO colony contained 140 mice. The clinical signs of muscle disease were monitored in approximately 100 mice. Genomic DNA was isolated from tail clips using iPrep™ ChargeSwitch® gDNA tissue Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The presence of Cre recombinase is indicated by a 400 bp PCR product made with a primer pair: Cre sense/antisense: 5′ccggtgaacgtgcaaaacagcctcta 3′/5′ cttccagggcgcgagtggatagc 3′. The Atg5 wild-type, Atg5flox/+ and Atg5flox/flox alleles were detected as described (24). The GAA wild-type, GAA +/− and GAA−/− alleles were detected as described (17).
Isolation of fixed single muscle fibers and immunofluorescence microscopy
White gastrocnemius, soleus and psoas muscles were removed immediately after sacrifice and pinned to Sylgaard-coated dishes for fixation with 2% paraformaldehyde in 0.1 M phosphate buffer for 1 h, followed by fixation in methanol (−20° C) for 6 min. Single fibers were obtained by manual teasing. Fibers were placed in a 24-well plate in Blocking Reagent (Vector Laboratories, Burlingame, CA) for 1 h. The fibers were then permeabilized, incubated with primary antibody overnight at 4°C, washed, incubated with secondary antibody for 2 h, washed again and mounted in Vectashield (Vector Laboratories, Burlingame, CA) on a glass slide. The fibers were analyzed using a Zeiss 510 META confocal microscope. The white part of gastrocnemius and psoas muscles in mice are a good source of glycolytic fast-twitch type II fibers (referred to as fast fibers), whereas soleus muscle is a good source of oxidative slow-twitch type I fibers (referred to as slow fibers) (60,61). At least three animals from each genotype were used to obtain single muscle fibers for immunostaining. For each immunostaining and for confocal analysis, at least 20 fibers were isolated from each of the three muscle groups.
Western blot of muscle tissues
Whole muscle tissues were homogenized in RIPA buffer (PBS containing 1% NP40, 0.5% Sodium deoxycholate, 0.1% SDS and the following protease inhibitors: 4 µmol Pefabloc, 10 µg/mL Leupeptin, 10 µg/mL Aprotinin (Roche Diagnostics, Indianapolis, IN) and 5 µg/mL E64 (Calbiochem, San Diego, CA). Samples were centrifuged for 30 min at 13 000 rpm at 4°C. Alternatively, detergent-soluble and -insoluble fractions from muscle tissues were obtained as previously described (24). Protein concentrations of the supernatants of the total lysates or soluble fractions were measured using Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA). Equal amounts of protein were run on SDS–PAGE gels (Invitrogen) followed by electro-transfer onto nitrocellulose membranes (Invitrogen). Membranes were blocked in 1:1 PBS and Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE), incubated with primary antibodies overnight at 4°C, washed, incubated with the secondary antibodies, and washed again. Blots were scanned on an infrared imager (LI-COR Biosciences).
Glycogen measurement and light microscopy
Glycogen concentration in skeletal muscle was evaluated by measuring the amount of glucose released after treatment of tissue extracts with Aspergillus niger amyloglucosidase as described (13). Tissues were fixed in 3% glutaraldehyde (EM grade, Electron Microscopy Sciences, Hatfield, PA) in 0.2 M Sodium Cacodylate buffer for 4 h at 4°C, washed in 0.1 M Sodium Cacodylate buffer and stored at 4°C in the same buffer. Samples were then imbedded in paraffin, sectioned and stained with periodic acid-Schiff (PAS) by standard methods.
Measurement of the cross-sectional area of the fibers
PAS-stained cross-sections from the psoas muscle were used for the measurements. Bright field images of cross-sections were obtained on a Leica DMR microscope (objective = 16X/0.5 NA) with a QImaging Qcolor3 digital camera. Two representative frames per slide were analyzed for fiber diameter. Images were first opened in Adobe Photoshop CS2, and a brush tool (matched to the background color of the bright field images) was used to draw lines between adjacent fibers. These images were then quantitated with ImageJ (written at NIH by Wayne Rasband and freely available at http://rsbweb.nih.gov/ij/) and then converted to binary form to measure the cross-sectional area of the fibers. Student’s t-tests were used to compare fiber diameters between genotypes.
Electron microscopy
Immediately after dissection, the clamped muscles were injected with ice-cold 5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) buffered with 0.1 M sodium phosphate, using a 27-gauge hypodermic needle. The muscles were then immersed and kept in the fixative for 2 h. They were subsequently rinsed and kept in ice-cold buffer overnight. Small blocks of muscle were then cut, osmicated and dehydrated before Epon embedding. The blocks were sectioned and observed in a Jeol 1200 equipped with an XR-100 CCD camera (Advanced Microscopy Techniques Corporation, Danvers, MA).
Quantitative real-time PCR
Total RNA was extracted from gastrocnemius muscle using TRIzol reagent (Gibco-BRL, Grand Island, NY) according to a standard procedure. The RNA cleanup protocol from the Qiagen RNeasy Mini Kit (Valencia, CA) was used to eliminate short transcripts. Five micrograms of total RNA was used for cDNA synthesis using the High Capacity cDNA Archive Kit from Applied Biosystems (Foster City, CA). The cDNA was then diluted 1:5 and 1 µL of the diluted cDNA was used to perform real-time PCR in 20 µL reactions in 96-well optical plates according to the manufacturer’s instructions. The following FAM-MGB-labeled TaqMan Gene Expression Assays were used: atrogin-1/Fbxo32 (Mm01207875_m1); cathepsin L/Ctsl (Mm00515597_m1); FoxO1 (Mm00490672_m1); MuRF-3/Trim54 (Mm01220477_g1). Mouse GAPDH was used as an endogenous control. Fold changes from expression levels in WT muscle samples were calculated using the formula 2−ΔΔCt.
Muscle strength measurement
Muscle strength was measured by the wire hang test (also known as inverted screen test). The animals were placed on lattice covers which were held horizontally. The lattice cover was then turned upside down, and the time at which the animals lost grip was recorded. The animals were individually tested three to four times every day of testing and allowed to rest between trials. All tested animals were females aged 4.5–5.5 months. Four AD GAA KO mice (n = 74 tests); three GAA KO mice (n = 57 tests); five WT mice (n = 28 tests); and four AD WT mice (n = 20 tests) were tested.
Animals used for weight comparison were 4–5-month-old males and 5–7-month-old females. Animals were starved overnight before sacrifice. Animal care and experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Data in text and figures are given as mean ± SE. The Student’s t-test was used for comparisons between the groups. Differences were considered significant at P < 0.05.
FUNDING
This research was supported by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health. S.T. was supported in part by a CRADA between the NIH and Genzyme Corporation (Framingham, MA).
ACKNOWLEDGEMENTS
We wish to thank Dr Judith Melki (Molecular Neurogenetics Laboratory, INSERM, U798, Evry, France) and Dr Steven Burden (New York University Medical School, NY) for the HSA-Cre: Atg5flox/+ mice. We are grateful to Dr Andrew Engel (Mayo Clinic, Rochester, MN) for help with the EM protocol, which was designed to preserve lysosomal glycogen. We thank Dr J.H. Tao-Cheng (NINDS, NIH) for help with the EM and Dr Kristien Zaal (NIAMS) for help with fiber measurement.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Levine B., Klionsky D.J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klionsky D.J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 4.Kiselyov K., Jennigs J.J., Jr, Rbaibi Y., Chu C.T. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–262. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 6.Koike M., Shibata M., Waguri S., Yoshimura K., Tanida I., Kominami E., Gotow T., Peters C., von Figura K., Mizushima N., et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am. J. Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugie K., Noguchi S., Kozuka Y., Arikawa-Hirasawa E., Tanaka M., Yan C., Saftig P., von Figura K., Hirano M., Ueno S., et al. Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J. Neuropathol. Exp. Neurol. 2005;64:513–522. doi: 10.1093/jnen/64.6.513. [DOI] [PubMed] [Google Scholar]
- 8.Engel A.G., Hirschhorn R., Huie M.L. Acid maltase deficiency. In: Engel A.G., Franzini-Armstrong C., editors. Myology. New York: McGraw-Hill; 2004. pp. 1559–1586. [Google Scholar]
- 9.Nishino I. Autophagic vacuolar myopathies. Curr. Neurol. Neurosci. Rep. 2003;3:64–69. doi: 10.1007/s11910-003-0040-y. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda T., Roberts A., Ahearn M., Zaal K., Ralston E., Plotz P.H., Raben N. Autophagy and lysosomes in Pompe disease. Autophagy. 2006;2:318–320. doi: 10.4161/auto.2984. [DOI] [PubMed] [Google Scholar]
- 11.Raben N., Takikita S., Pittis M.G., Bembi B., Marie S.K.N., Roberts A., Page L., Kishnani P.S., Schoser B.G.H., Chien Y.H., et al. Deconstructing Pompe disease by analyzing single muscle fibers. Autophagy. 2007;3:546–552. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda T., Ahearn M., Roberts A., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H., Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raben N., Fukuda T., Gilbert A.L., de Jong D., Thurberg B.L., Mattaliano R.J., Meikle P., Hopwood J.J., Nagashima K., Nagaraju K., Plotz P.H. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N., Ohsumi Y., Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 15.Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raben N., Nagaraju K., Lee E., Kessler P., Byrne B., Lee L., LaMarca M., King C., Ward J., Sauer B., et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 18.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanida I., Ueno T., Kominami E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes M.D., Lecker S.H., Jagoe R.T., Navon A., Goldberg A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl Acad. Sci. USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lecker S.H., Jagoe R.T., Gilbert A., Gomes M., Baracos V., Bailey J., Price S.R., Mitch W.E., Goldberg A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 23.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 24.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 25.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Overvatn A., Bjorkoy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 26.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichimura Y., Kumanomidou T., Sou Y.S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 28.Fass E., Shvets E., Degani I., Hirschberg K., Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 29.Kochl R., Hu X.W., Chan E.Y., Tooze S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 30.Jahreiss L., Menzies F.M., Rubinsztein D.C. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotoulas O.B., Kalamidas S.A., Kondomerkos D.J. Glycogen autophagy in glucose homeostasis. Pathol. Res. Pract. 2006;202:631–638. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Schiaffino S., Mammucari C., Sandri M. The role of autophagy in neonatal tissues: just a response to amino acid starvation? Autophagy. 2008;4:727–730. doi: 10.4161/auto.6143. [DOI] [PubMed] [Google Scholar]
- 33.Kotoulas O.B., Kalamidas S.A., Kondomerkos D.J. Glycogen autophagy. Microsc. Res. Tech. 2004;64:10–20. doi: 10.1002/jemt.20046. [DOI] [PubMed] [Google Scholar]
- 34.Kondomerkos D.J., Kalamidas S.A., Kotoulas O.B. An electron microscopic and biochemical study of the effects of glucagon on glycogen autophagy in the liver and heart of newborn rats. Microsc. Res. Tech. 2004;63:87–93. doi: 10.1002/jemt.20000. [DOI] [PubMed] [Google Scholar]
- 35.Kondomerkos D.J., Kalamidas S.A., Kotoulas O.B., Hann A.C. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol. Histopathol. 2005;20:689–696. doi: 10.14670/HH-20.689. [DOI] [PubMed] [Google Scholar]
- 36.Schiaffino S., Hanzlikova V. Autophagic degradation of glycogen in skeletal muscles of the newborn rat. J. Cell Biol. 1972;52:41–51. doi: 10.1083/jcb.52.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kishnani P.S., Nicolino M., Voit T., Rogers R.C., Tsai A.C., Waterson J., Herman G.E., Amalfitano A., Thurberg B.L., Richards S., et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffin J.L. Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Arch. Cell Pathol. Incl. Mol. Pathol. 1984;45:23–36. doi: 10.1007/BF02889849. [DOI] [PubMed] [Google Scholar]
- 39.Thurberg B.L., Lynch M.C., Vaccaro C., Afonso K., Tsai A.C., Bossen E., Kishnani P.S., O’Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for pompe disease. Lab. Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- 40.Bijvoet A.G., Van Hirtum H., Vermey M., Van Leenen D., Van der Ploeg A.T., Mooi W.J., Reuser A.J. Pathological features of glycogen storage disease type II highlighted in the knockout mouse model. J. Pathol. 1999;189:416–424. doi: 10.1002/(SICI)1096-9896(199911)189:3<416::AID-PATH445>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 41.Hesselink R.P., Wagenmakers A.J., Drost M.R., van der Vusse G.J. Lysosomal dysfunction in muscle with special reference to glycogen storage disease type II. Biochim. Biophys. Acta. 2003;1637:164–170. doi: 10.1016/s0925-4439(02)00229-6. [DOI] [PubMed] [Google Scholar]
- 42.Drost M.R., Hesselink R.P., Oomens C.W., van der Vusse G.J. Effects of non-contractile inclusions on mechanical performance of skeletal muscle. J. Biomech. 2005;38:1035–1043. doi: 10.1016/j.jbiomech.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 43.Fukuda T., Ewan L., Bauer M., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H., Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann. Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 44.Fukuda T., Roberts A., Plotz P.H., Raben N. Acid alpha-glucosidase deficiency (Pompe disease) Curr. Neurol. Neurosci. Rep. 2007;7:71–77. doi: 10.1007/s11910-007-0024-4. [DOI] [PubMed] [Google Scholar]
- 45.Klionsky D.J., Abeliovich H., Agostinis P., Agrawal D.K., Aliev G., Askew D.S., Baba M., Baehrecke E.H., Bahr B.A., Ballabio A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komatsu M., Waguri S., Koike M., Sou Y.S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 47.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 48.Klionsky D.J. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marmor M.D., Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–2070. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- 50.Dupre S., Urban-Grimal D., Haguenauer-Tsapis R. Ubiquitin and endocytic internalization in yeast and animal cells. Biochim. Biophys. Acta. 2004;1695:89–111. doi: 10.1016/j.bbamcr.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 51.Barriere H., Nemes C., Du K., Lukacs G.L. Plasticity of polyubiquitin recognition as lysosomal targeting signals by the endosomal sorting machinery. Mol. Biol. Cell. 2007;18:3952–3965. doi: 10.1091/mbc.E07-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K., et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 53.Jagoe R.T., Lecker S.H., Gomes M., Goldberg A.L. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J. 2002;16:1697–1712. doi: 10.1096/fj.02-0312com. [DOI] [PubMed] [Google Scholar]
- 54.Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S.H., Goldberg A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gomes M.D., Lecker S.H., Jagoe R.T., Navon A., Goldberg A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl Acad. Sci. USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mammucari C., Schiaffino S., Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–526. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 57.Askanas V., Engel W.K. Inclusion-body myositis and myopathies: different etiologies, possibly similar pathogenic mechanisms. Curr. Opin. Neurol. 2002;15:525–531. doi: 10.1097/00019052-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Weihl C.C., Miller S.E., Hanson P.I., Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet. 2007;16:919–928. doi: 10.1093/hmg/ddm037. [DOI] [PubMed] [Google Scholar]
- 59.Miniou P., Tiziano D., Frugier T., Roblot N., Le Meur M., Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27:e27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burkholder T.J., Fingado B., Baron S., Lieber R.L. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol. 1994;221:177–190. doi: 10.1002/jmor.1052210207. [DOI] [PubMed] [Google Scholar]
- 61.Hawes M.L., Kennedy W., O’Callaghan M.W., Thurberg B.L. Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol. Genet. Metab. 2007;91:343–351. doi: 10.1016/j.ymgme.2007.04.018. [DOI] [PubMed] [Google Scholar]