

Abstract
Probabilistic models of sequence evolution are in widespread use in phylogenetics and molecular sequence evolution. These models have become increasingly sophisticated and combined with statistical model comparison techniques have helped to shed light on how genes and proteins evolve. Models of codon evolution have been particularly useful, because, in addition to providing a significant improvement in model realism for protein-coding sequences, codon models can also be designed to test hypotheses about the selective pressures that shape the evolution of the sequences. Such models typically assume a phylogeny and can be used to identify sites or lineages that have evolved adaptively. Recently some of the key assumptions that underlie phylogenetic tests of selection have been questioned, such as the assumption that the rate of synonymous changes is constant across sites or that a single phylogenetic tree can be assumed at all sites for recombining sequences. While some of these issues have been addressed through the development of novel methods, others remain as caveats that need to be considered on a case-by-case basis. Here, we outline the theory of codon models and their application to the detection of positive selection. We review some of the more recent developments that have improved their power and utility, laying a foundation for further advances in the modeling of coding sequence evolution.
Keywords: maximum likelihood, phylogenetics, evolutionary models, selection
INTRODUCTION
Models of molecular sequence evolution have diverse applications, from phylogenetics to genome annotation and comparative genomics. Although early models of sequence evolution achieved significant simplification by assuming that nucleotide sites evolve independently [1, 2], more realistic models have been proposed to take a range of different sources of context dependency into account [3, 4]. One of the most obvious sources of context dependency derives from the triplet nature of the genetic code. It is this dependency that is accounted for by codon models, which treat triplets of nucleotides as the states of the probabilistic process describing the evolution of a protein-coding sequence. Models that account for this have been shown to provide a substantially improved fit to protein-coding sequences [5, 6].
The degeneracy of the genetic code suggests a natural classification of codon substitutions, depending on whether they change or do not change the encoded amino acid (nonsynonymous and synonymous substitutions, respectively). The proposition that synonymous substitutions are generally neutral leads to the description of codon evolution as a combination of substitutions at the nucleotide level, and selective constraints operating at the protein level [7]. Codon models have distinct parameters describing the mutational and selective components of the substitution process, providing a means to assess the selective forces acting on a protein. Many of the applications of codon models relate directly to this capacity to quantify the strength of selection. Three types of selective pressures are frequently considered. When the rate at which nonsynonymous substitutions accumulate is lower than the rate of synonymous substitution, this is normally taken to imply that, on average, nonsynonymous substitutions have a negative effect on fitness. This is frequently referred to as purifying selection. A nonsynonymous substitution rate which is equal to the synonymous rate is consistent with neutral evolution, or the absence of selective constraints acting to preserve features of the encoded protein, when the rate of nonsynonymous substitution is greater than the rate of synonymous substitution this implies that, on average, nonsynonymous mutations confer a selective advantage and increase in frequency through positive selection. Thus, a comparison of the rate of nonsynonymous substitution to synonymous substitutions can provide evidence that a protein is evolving adaptively.
Codon models are very frequently used to identify protein-coding sequences evolving under purifying or positive selection pressure. Evidence of purifying selection pressure is useful for distinguishing coding from noncoding genomic regions, when orthologous sequences are available [8]. Any putative protein-coding sequence with a nonsynonymous substitution rate significantly lower than the synonymous rate can be inferred to be translated, and is likely to be functional. The ratio of nonsynonymous to synonymous substitutions also allows the strength of selection to be quantified and compared between different genes or gene copies. On the other hand, the identification of sequences or subsets of sites within a sequence evolving adaptively can provide information about key evolutionary processes and innovations. Because the effects of selection are very different at distinct sites in a sequence the question of how best to model site-to-site variation in parameters describing selective constraints receives particular attention. We review two trends in this regard, distinguished by whether sites are considered to belong to distinct site classes with fixed parameter values or whether the parameters themselves are described by random variables. These alternatives are referred to as fixed effects likelihood (FEL) and random effects likelihood (REL) models, respectively.
In this review, we outline the development of phylogenetic models of codon evolution with a focus on the use of these models to identify sites or lineages evolving adaptively. We consider recent advances leading to improved model realism, including the development of novel techniques to model sequence heterogeneity and a movement to integrate population genetics concepts into models of molecular evolution. Several caveats for the application of codon models are considered and we outline some of the solutions that have been proposed to address these concerns.
Probabilistic models of molecular sequence evolution
The modeling of molecular evolution has been facilitated largely by the development of an efficient means of calculating the likelihood [9] of an alignment, given a hypothesis of the phylogenetic tree relating the taxa in the alignment and an appropriate probabilistic model of the substitution process. These models generally take the form of a continuous time Markov process in which mutations are modeled along the branches of a phylogenetic tree, according to a rate matrix, Q, with elements (qij) describing the instantaneous rate of substitution from state i to state j, where the states may be nucleotides, amino acids or codons. Elements vary with model definition, yet typically make use of three parameter types: (i) frequency parameters, (ii) exchangeability parameters and (iii) rate heterogeneity parameters [7]. Both nucleotide and amino acid models of molecular evolution, and the methodological details of maximum likelihood phylogenetics, have been extensively reviewed elsewhere [1, 2, 10–13]. Codon models have previously been reviewed [1, 13], though not some of the more recent developments in the field.
Codon models have an alphabet of 64 states, typically reduced to 61 after removal of the three stop codons in the common genetic codes. The most general parameterization is [14]:
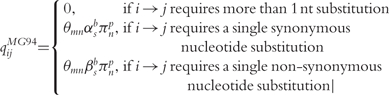 |
where following Kosakovsky Pond et al. [14], θmn describes the relative exchangeability of nucleotide m for n (θAG = 1). 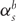 is a parameter proportional to the rate of synonymous substitution, indexed by site in the alignment, s, and branch of the phylogeny, b, in the formulation presented here. Similarly,
is a parameter proportional to the rate of synonymous substitution, indexed by site in the alignment, s, and branch of the phylogeny, b, in the formulation presented here. Similarly, 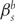 is a parameter proportional to the rate of nonsynonymous substitution, similarly indexed, and
is a parameter proportional to the rate of nonsynonymous substitution, similarly indexed, and 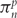 is the equilibrium frequency (usually estimated directly from the empirical data rather than optimized as a model parameter) of the target nucleotide n (i.e. the nucleotide that differs between codons i and j) at codon position p (p ranges from position 1 to position 3 of the triplet codon) [15]. The parameters θmn are typically chosen from the range of standard nucleotide models, with the HKY85 [16] and general time reversible (GTR or REV) models [17] being popular. It is worth noting that the appropriate model choice depends primarily on data set size rather than on biological considerations: for large data sets the more parameter-rich models (e.g. GTR) tend to be appropriate, while small data sets necessitate the use of less detailed models.
is the equilibrium frequency (usually estimated directly from the empirical data rather than optimized as a model parameter) of the target nucleotide n (i.e. the nucleotide that differs between codons i and j) at codon position p (p ranges from position 1 to position 3 of the triplet codon) [15]. The parameters θmn are typically chosen from the range of standard nucleotide models, with the HKY85 [16] and general time reversible (GTR or REV) models [17] being popular. It is worth noting that the appropriate model choice depends primarily on data set size rather than on biological considerations: for large data sets the more parameter-rich models (e.g. GTR) tend to be appropriate, while small data sets necessitate the use of less detailed models.
An alternative parameterization of the codon model [18] is obtained by letting substitution rates be proportional to the frequency of the target codon rather than the position-specific frequency of the target nucleotide as in the Muse–Gaut (MG) [15] implementation. We use the terms Goldman–Yang (GY) and MG to denote these two alternative model assumptions. In the Goldman and Yang [18] parameterization, codon frequencies are estimated from the data either as the product of the frequencies of the 3 nt that make up the codon (referred to as F1 × 4), or as a product of position-specific nucleotide frequencies (F3 × 4). These estimates of equilibrium codon frequencies are more robust than an empirical count, since a very large amount of data would be required to provide a good estimate of the true equilibrium codon frequencies. However, actual counts can also be used, or codon frequencies can be treated as free parameters to be estimated. We evaluated the relative performance of the MG [15] versus GY [18] model assumptions, by fitting them to published datasets (Table 1) and comparing log likelihoods. Both implementations used the general time reversible model as the underlying nucleotide model, empirically estimated position-specific nucleotide frequencies (individually for MG and multiplied together as F3 × 4 estimates for GY) a discrete distribution for , corresponding to M3 in Ref. [19] and constant . In all cases the MG model assumption outperforms the GY assumption (Table 1). This is consistent with recent Bayesian model comparison results [20], in which the mechanistic assumptions of these models, and their respective performances, are discussed in detail.
Table 1:
Performance comparison of MG [15] and GY [18] codon model assumptions on previously published datasets
Detecting selection affecting specific sites
As formulated above, both the rate of synonymous substitution, , and the nonsynonymous substitution rate, , are allowed to vary across branches, b, and sites, s [19, 21–24]. In real protein-coding sequences, evolutionary rates vary substantially by position along the sequence and may vary by lineage; however, models allowing independent rates for all sites and all lineages would be severely over-parameterized. The choice of how to model heterogeneity in evolutionary rates provides an important distinction between alternative implementations of codon models. Earlier models treated evolutionary rates as random quantities described by parameterized distributions [19, 25], while some more recent models treat rates as a fixed effect but allow distinct classes of sites with independent parameters (including rate parameters). These two alternative approaches to modeling rate variation have been referred to as REL and FEL methods, respectively [26–28]. In both cases a nonsynonymous rate (dN) significantly greater than the synonymous rate (dS), or alternatively, ω = dN/dS significantly greater than one, points to positive Darwinian selection [29]. Sites at which this is the case are of particular interest and several implementations of codon models are extremely widely used to identify these sites (Box 1).
Box 1: Software implementations of codon models of evolution.
| Software | Description | URL | Platforms supported | |
| PAML | Phylogenetic statistical hypothesis testing | [97] | http://abacus.gene.ucl.ac.uk/software/paml.html | Linux Windows Mac OSX |
| HyPhy | Phylogenetic statistical hypothesis testing using existing and custom models | [98] | http://www.hyphy.org/ | Linux Windows Mac OSX |
| PyEvolve | Phylogenetic statistical hypothesis testing using existing and custom models | [99] | http://cbis.anu.edu.au/software.html | Linux Windows Mac OSX |
| Datamonkey | Free web server implementation of various models in the HyPhy package | http://www.datamonkey.org/ | Web server | |
| Selecton | Free web server for sitewise detection of selection | [100] | http://selecton.tau.ac.il/ | Web server |
| ADAPTSITE | Positive selection using counting methods | [101] | http://www.cib.nig.ac.jp/dda/yossuzuk/welcome.html | Linux Mac OSX |
| BEAST | Estimation of population parameters given uncertainty in the inference of phylogenies | [102] | http://beast.bio.ed.ac.uk/ | Linux Windows Mac OSX |
| MrBayes | Bayesian phylogenetics software with codon models for positive selection detection | [48] | http://mrbayes.csit.fsu.edu/ | Linux Windows Mac OSX |
The simplest random effects models use a constant rate of synonymous substitution and approximate the sitewise variation in nonsynonymous rates using a three category discrete distribution accounting for purifying selection (ω < 1), neutrality (ω = 1) and positive selection (ω > 1) [30]. A test of positive selection can be obtained by comparing the likelihood under a model where the positive selection component (ω > 1) of this distribution has zero weight to the likelihood without this constraint (corresponding to a comparison of Wong et al.apos;s [30] models M2a and M1a). Since these models are nested, the distribution of the likelihood ratio test (LRT) statistic (Box 2) approximates a χ2 distribution with degrees of freedom equal to the difference in the number of free parameters between the two models. For the above test, the difference in number of free parameters is two, one rate parameter (ω > 1), and a parameter describing the proportion of sites under positive selection and a conservative test can be obtained by comparing against a χ2 distribution with two degrees of freedom. In fact, in this particular case, the LRT statistic is asymptotically approximated by an equal mixture of a χ2 distribution with zero degrees of freedom and a χ2 distribution with one degree of freedom (see [31, 32] for a discussion of one-tailed LRTs in phylogenetics). In addition to this approximation of the heterogeneity of ω across sites, other distributions have been proposed [19, 33]. Wong et al.apos;s [30] M1a versus M2a, and Swanson et al.apos;s [33] M8a versus M8, which models ω as a mixture of a beta distribution and point mass at ω = 1 in the null, and with ω > 1 in the alternate [33], are recommended for tests of positive selection. The characteristics of these and other suitable REL models are presented elsewhere [19, 23, 30].
Box 2: Statistical concepts and methods.
| Concept | Description |
| Likelihood | The likelihood of a model (usually expressed as a function of its set of parameters) is the probability of observing the data, given the model parameters. |
| Maximum likelihood estimates (MLEs) | MLEs are fixed parameter values that maximize the likelihood of observing the data. Maximum likelihood (ML)-based methods use these fixed estimates, rather than averaging over multiple parameter values, as in Bayesian methods. |
| LRT | A classical hypothesis test for comparing the ML of a simpler model, L0 (e.g. excluding positive selection) which is nested within a more general model, L1 (e.g. including positive selection). The LRT provides an indication of whether the simpler model is inadequate to explain the data. Under L0, the LRT test statistic (2*(lnL1 − lnL0)) is χ2 distributed with degrees of freedom equal to the number of extra free parameters in L1 (see text for discussion). |
| AIC | An alternative ML-based model comparison technique that allows for comparison of non-nested models and that penalizes models according to the number of parameters, k (AIC = 2*k − 2 ln(L)). |
| Bayesian inference | Bayesian inference methods account for uncertainty in parameter estimates by performing a weighted average over possible parameter values. Bayesian methods require prior distributions for all parameters, which are typically assumed to be uninformative. |
| NEB | A post hoc analysis that uses the MLEs from a ML-based method to infer posterior probability distributions for parameters of interest. NEB assumes the MLEs are correct. |
| BEB | Similar to NEB in that MLEs are used to infer posterior probability distributions, but takes uncertainty in the MLEs into account. |
| MCMC | A sampling approach that can be used to approximate a probability distribution that is mathematically intractable. MCMC is often used in a Bayesian setting to estimate posterior probability distributions of model parameters. |
The LRT provides a test of whether some sites in a sequence alignment are affected by diversifying selection, but does not identify specific sites at which ω is greater than one. In the case of REL models, empirical Bayesian methods (Box 2) have been used [25, 34]. Naïve empirical Bayesian (NEB) methods make use of maximum likelihood estimates of parameters describing the distribution of ω, inferred from the whole sequence, as a prior in order to estimate a posterior probability for each of the sites belonging to a site class in which ω > 1 [25]. The posterior probability that a site s, belongs to rate category k, is the product of the likelihood of the data at site s given the category k, L(Ds|ωk), and the prior (P(ωs = ωk), set to the inferred proportion of sites belonging to category k), normalized by the sum of site-specific likelihood for each category:
The posteriors, for each site and each rate category, are used to determine which sites are evolving under positive selection, given a posterior probability threshold. Since NEB uses the maximum likelihood parameter estimates of the distribution describing the sitewise variation in synonymous and nonsynonymous substitution rates as a prior, it is susceptible to inaccuracies in these estimates [35]. Potential solutions to this problem include a full Bayesian method for calculating posterior probabilities using Markov chain Monte Carlo (MCMC) [36, 37], and an approximate method, which accounts for some of the uncertainty in the maximum likelihood parameter estimates through averaging over a prior (Bayes empirical Bayes or BEB) [34, 38].
FEL methods provide an alternative to the REL models discussed above. These models treat sites as belonging to distinct classes, with independent parameters. In the extreme case, each site belongs to a separate class, requiring the estimation of a large number of free parameters, with the concomitant danger of over-parameterization [1]. When reasonable partitions of sites can be specified a priori, FEL models that allow separate parameters between these site classes may be useful [28]. For example, the a priori partitioning of MHC sites involved in the binding of foreign peptides [28] from other sites, allowed for the evaluation of positive selection independently for these classes. The degree to which parameters (including relative branch lengths, nonsynonymous substitution rate, synonymous substitution rate, transition–transversion rate ratio and codon frequencies) should be shared across these classes can be decided using model comparison techniques [26, 28]. Bao et al. [26] compared the use of the Akaike information criterion (AIC) [39], AICc [40] and a backward elimination procedure based on the LRT and found, from simulation, that the latter appeared to provide the best means of choosing the appropriate model. However, even when an a priori partitioning of sites into classes, based on protein folding, is available this is likely to model only a small proportion of the sitewise variation in the substitution process. Furthermore, power may be affected by sites which are conserved, yet occur within putatively positively selected protein domains [28].
An alternative to the use of a priori site classes, which may fail to model site-to-site variation adequately, and models that treat sites as completely independent, resulting in over-parameterization, can be obtained by the use of techniques that allow site classes to be inferred from the data. One approach is likelihood-based clustering (LiBaC) that allocates sites to classes by maximizing a mixture log likelihood, which takes account of uncertainty in site allocation by averaging over classes of sites. This model is implemented with an expectation–maximization (EM) algorithm that successively optimizes model parameters and adjusts a site allocation vector (a vector indicating site class membership for each site), given the proposed parameters. The LiBaC method showed significantly improved performance over REL models, especially when relative branch lengths differed between partitions [41]. Perhaps a limitation of the model is that the number of distinct site classes is chosen beforehand, by comparing the log likelihoods of REL models with different numbers of discrete components. An alternative to this is the use of Dirichlet process models, which have been applied in the contexts of modeling sitewise variation in the amino acid replacement process [42] or selection pressure using codon models [43]. Neither the site class membership nor the number of site classes is fixed, but is rather modeled as a mixture distribution using a Dirichlet process. This approach was adopted within a MCMC sampling framework to detect positive selection. The implementation accounted for uncertainty in the phylogeny and branch lengths resulting in a more conservative identification of positive selection than methods that first estimate these parameters and treat them as fixed in subsequent analysis [43].
Detecting selection affecting specific lineages
Codon models have been implemented that allow variable rates of evolution between lineages. Codon-based models that allow the rates of evolution to vary between lineages have been applied to assess evidence of adaptation affecting specific taxa or sets of taxa [44, 45]. In the earliest implementations, a separate value of ω was fitted along focal lineages which were specified a priori [44, 45], but without provision for site-to-site variation in selective pressure. Subsequent models that allowed site- and branch-specific selective pressure [23] were found to be subject to high false positive rates [46], although a modification of the original model showed improved levels of false positives [24]. Given a biologically plausible scenario, the prior specification of branches may be acceptable, but in many cases there may be no prior expectation regarding the selective processes that have affected different branches of the phylogeny. In such cases, each branch can be allowed an independent value (or distribution) of ω, but this approach requires corrections for multiple testing [21] with a corresponding loss of power. Given large phylogenies typically used in scans for lineage-specific positive selection, it is computationally impossible to test all possible alternatives of branch-specific selective regimes. Kosakovsky Pond et al. [22] provide a genetic algorithm alternative for model selection, in which AIC is used as a measure of fitness to select models with the most likely distribution of selective regimes among branches of the phylogeny [22]. The strength of this approach is that a larger population of potential models can be evaluated without the need for prior specification of lineages with alternate selective regimes. It has, however, been criticized for not providing a hypothesis test for the presence of positive selection [21]. The issue boils down to whether the traditional hypothesis testing framework, with its notion of statistical significance, is a more appropriate one in which to address questions such as these, than the alternative approach provided by criteria, such as the AIC. The latter attempts to quantify the strength of evidence for multiple models, none of which is assumed to be ‘correct’, rather than to measure the significance with which a specific (and possibly a priori unlikely) null hypothesis can be rejected. Both methodologies are statistically sound, and the debate about their relative merits has been conducted in a much broader context [47].
Caveats for application of codon models
Phylogenetic uncertainty
Thus far, we have assumed that the phylogeny on which we model molecular evolution is an accurate representation of the evolutionary history of the taxa. In general, even if we make the assumption that a true phylogenetic relationship between a set of sequences exists (i.e. the same relationship can be used to describe the entire length of the alignment, which is not true when recombination has occurred), then that relationship is unknown and must be inferred from the data. The usual approach is to infer a single tree and then base subsequent inference on that tree. Empirical results have indicated that inference of codon model parameters and/or positive selection is not very sensitive to tree topology, as long as ‘a reasonably good phylogeny’ (i.e. one that has been estimated from the data) is used [19].
Nevertheless, it is inevitable that using an incorrect tree would have some effect on inference. It is possible to account for uncertainty in tree topology using a Bayesian approach in which parameter estimates are averaged over the space of possible trees, weighted according to their individual posterior probabilities. A number of Bayesian phylogenetic software packages exist, of which MrBayes [48] is perhaps the most the commonly used one that supports codon models. Such methods relax the assumption that a specific tree is correct, but not the assumption that a correct, though unknown, tree exists. Perhaps a more important motivation for using the Bayesian approach is that, in addition to averaging over multiple plausible tree topologies, it also averages over multiple plausible parameter values, providing a natural way to take uncertainty in parameter estimates into account. The use of this approach for inferring positive selection was first demonstrated by Huelsenbeck and Dyer [36], and extended through the use of a Dirichlet process model [43] (as discussed above). Further evaluation of Bayesian models [37], demonstrated that some form of Bayesian approach (either a full Bayesian approach or the BEB method described above) is important for medium-sized data sets (e.g. 30 taxa, 100 codons, with a tree length of up to two substitutions per codon). However, the extra computational cost of the fully Bayesian approach as compared to BEB is unlikely to be justified except when analyzing data sets with very low divergence and hence low information content.
Recombination
When the sequences under consideration have recombined, it is no longer the case that there is a single phylogeny describing their evolution. Instead, different phylogenies are required to describe the evolutionary relationships of the segments of the alignment defined by recombination breakpoints. The standard practice of assuming that a single topology describes the entire alignment can lead to arbitrarily high false positive rates when inferring positive selection from recombining sequences [49, 50].
We illustrate this in Figure 1, which shows how the false positive rate increases as a function of recombination rate. These results were obtained using Codonrecsim [49] to simulate 100 neutrally evolving data sets (ω = 1 at all sites) with codon frequency parameters and transition–transversion rate ratio matched to the Hepatitis D virus antigen (HDV) data set used in one of the previous studies [49]. For each replicate data set, positive selection was inferred if there was a significant improvement in the likelihood of the selection model (M2a) over the neutral model (M1a). As the recombination rate (ρ) is increased, the false positive rate rapidly increases from below the acceptable level of 5% towards 100% (Figure 1). For a data set with parameters as described above, the significance level (α = 5%) is a reasonable estimate of the false detection rate only for ρ < 0.004. For ρ > 0.024, positive selection is more likely to be inferred than not, even if all sites evolve neutrally.
Figure 1:

The relationship between the recombination rate and the rate of false inference of positive selection in an example data set consisting of 10 taxa (using a population-scaled substitution rate of μ = 3.6, and a population-scaled recombination rate varying from ρ = 0 to 0.1 with increments of 0.002). Recombination rates are measured as ρ = 2Nr, where N is the effective population size and r is the number of recombination events per inter-codon link per lineage per generation. Hence, a recombination rate of ρ = 0.004 means that a given inter-codon link at a given lineage experiences, on average, one recombination event every 500N generations.
To illustrate the extent of the problem in real sequences, we constructed 10-taxon intra-subtype HIV-1 data sets of the env and gag genes and applied this simulation methodology. At the recombination rates estimated by the program LAMARC [51], we obtained false inference of positive selection in, respectively, 100 and 92 out of 100 replicates. This means that the apparent signature of positive selection in these sequences may have arisen as a result of recombination in the absence of positive selection, and that the recombination problem is therefore of practical rather than just theoretical importance.
One solution to this problem is a population genetics approximation to the coalescent which co-estimates recombination rate and selective pressure [52]. An alternative solution involves identification of recombination breakpoints and estimation of a separate phylogeny for each recombinant partition. The parameters of the codon models are then estimated in the usual way, except that phylogenies and branch lengths are partition-specific, while the remaining parameters are shared across all segments [53]. It also proved important to incorporate synonymous rate variation (see below) in this case, because recombination can cause an apparent variation in the synonymous rate [53]. Fortunately, the recombination breakpoints detected by off-the-shelf recombination detection tools appear to be sufficient to address the false positive problem, and may even give better results than when the true recombination breakpoints are known due to the fact that the recombination breakpoints that are difficult to detect typically have little effect on inference and can be ignored in order to estimate phylogenies on longer, more informative segments [53]. A similar approach [54] incorporates the breakpoints estimated, using a genetic algorithm for recombination detection (GARD), into a FEL selection analysis, suitable for the detection of individual adaptively evolving sites in recombined sequences.
Synonymous rate variation
In the application of codon models to detect selection, the synonymous substitution rate is often assumed to be constant across all sites in the alignment. Synonymous substitution rates may vary either because of site-to-site variation in mutation rates or because of selection acting to preserve functional motifs at the nucleotide sequence level. The latter, in particular, is now recognized as pervasive in protein-coding sequences [55]. Synonymous changes in protein sequences can cause, among many other effects, changes in the stability of mRNA secondary structure and formation or disruption of motifs that affect splicing (e.g. exonic splice enhancers) [56]. Codon models to detect synonymous rate variation applied to various published datasets revealed significant evidence of synonymous rate variation in 9 out of 10 cases tested and highlighted the likelihood of misleading inference of selection when this is ignored [57]. Indeed, a recent evaluation of a likelihood-based clustering method [41] demonstrated the false inference of positive selection with typical positive selection models (M2, M3, M8) when nonselective components (synonymous rate, transition/transversion rate ratio, relative branch lengths) of the substitution process are constant across sites. Selection on synonymous changes has been modeled explicitly when such selection is constant across all sites [58], (e.g. site-independent codon usage bias), but remains an issue for selection acting on only a subset of sites.
Recent modifications and extensions of codon models
Independence of sites
Traditionally, models of sequence evolution have included the assumption that distinct sites in a sequence alignment evolve independently. This significantly reduces model complexity and allows the likelihood to be calculated as the product over likelihoods of individual sites. However, site independence is an assumption that in many cases is grossly at odds with biological expectations. In real data, the rate of evolution is often autocorrelated along the sequence, with regions of conservation and rapid evolution. The combination of hidden Markov models to model rate variation along the sequence and phylogenetic models describing sequence evolution across taxa was introduced to model autocorrelation of evolutionary rates [4, 59] and subsequently adapted for use with codon models [60]. Interestingly, in an analysis of HIV-1 genes, dependency between adjacent sites was greater for synonymous than for nonsynonymous substitutions [60], potentially the result of tertiary structure, where nonsynonymous mutations co-vary at sites that are adjacent in the tertiary structure of the protein [12] and not necessarily adjacent in the primary sequence.
Models that allow autocorrelated rates are useful for accounting for local features of a sequence, allowing distinct functional classes and rate classes that are modeled as a Markov chain of hidden states. Functional interactions between sites can also be nonlocal, so that the rate and nature of sequence change at one site may depend on the state present at another site, which is not necessarily adjacent or close to the site under consideration. Examples of the latter include models of Watson–Crick pairs in RNA stem regions [61–63] or of interacting amino acids that maintain the stability of proteins [64–66]. The most general models of sequence evolution that set aside the assumption of independence of sites model whole sequences as the states in the continuous-time Markov process, resulting in rate matrices of extremely high dimension [64] (4N × 4N, where N is the sequence length). Robinson et al. [64] proposed a method to estimate such a model using a MCMC method to sample model parameters and to sample from possible sequence histories, proposed under a simpler evolutionary model (see [67] for an efficient method to sample from evolutionary histories). In a related development, Poon et al. [68] set out to identify interacting sites by reconstructing ancestral states under a codon model that assumed site independence and then estimated a Bayesian network from an array of branch-specific synonymous and nonsynonymous substitutions [68]. Interacting sites within the network are identified as those at which substitutions repeatedly co-occur along the same branch. This method has facilitated the identification of clusters of spatially separated codons that show dependent or compensatory mutations in HIV-1 [68].
Models with context dependency have also been used to account for elevated rates of mutation from cytosine to thymine at CpG dinucleotides [3, 69, 70], the result of spontaneous deamination of methylated cytosine [71]. These models, which account for CpG-context dependence both within codons, and across codon boundaries, have demonstrated the significance of CpG hypermutation within protein-coding genes [3, 70]. In the context of viral evolution CpG gain and loss is modeled [72] since unmethylated CpG dinucleotides may trigger a host immune response [73], or increased methylation at CpG sites may negatively affect viral gene expression [74]. Thus these models need to consider selective constraints operating at the amino acid or codon (codon usage bias) levels as well as the mutational effects and potentially selective effects of CpG dinucleotides at the nucleotide level.
Linking models of sequence evolution to population genetics
The models of sequence evolution presented above are typically applied to alignments of coding sequences from multiple species and encompassing evolution over long timescales. These models are concerned with the rates at which specific substitutions occur and, in turn, these rates are influenced by the rate of mutation and by selection. Recently there has been an upsurge of interest in relating parameters of sequence evolution models to the quantities that are of interest in population genetics, such as fitness and associated fixation probability. This work builds on a model of codon evolution that included separate terms for the probability of a mutation and the probability of fixation of a newly arisen mutant allele [75]. In the original application, this approach was used to model position-specific frequencies of amino acids in order to estimate evolutionary distances more accurately. More recently, a conceptually similar approach has been applied to estimate the distributions of selection coefficients associated with mutations in 5S ribosomal RNA [76, 77] and of substitutions between synonymous codons [58]. These studies not only bridge the gap between sequence models and population genetics, but also include phenotype quantitatively in the sequence model, adding considerably to the scope and potential applications of sequence models.
Selection on codon usage can result from differences in translation speed and accuracy between synonymous codons (see [78] for a review). Standard codon models, which include parameters describing codon equilibrium frequencies, can be used to infer positive selection even with strong codon usage bias, regardless of whether the bias is mutational or due to selection [79]. The novel codon model, FMutSel, proposed by Yang and Nielsen [58], separates out the selective and mutational components of codon usage bias. By comparing the fit of this model to a restricted form that models only the amino acid frequencies, the authors estimate the strength of selection acting on codon usage in large sets of mammalian genes. FMutSel shows a significantly improved fit to real data over standard codon models, but is parameter rich, requiring separate parameters to be estimated for every codon in its full form; however, use of empirical rather than estimated parameters appears to have only a marginal effect on model likelihoods and parameter estimates [58].
Integrating empirical and mechanistic models of codon evolution
Although codon models generally fit protein-coding sequence data far better than nucleotide models, at the basic level they take no account of the nature of the encoded amino acids and instead treat all nonsynonymous substitutions as selectively equivalent. Given the differences in the physical and chemical properties of amino acids, this omission leaves a substantial proportion of the evolutionary process unmodeled. In functionally conserved regions of an amino acid sequence, for example, replacement of one amino acid with another with similar properties may have a much smaller negative effect on fitness than replacement with a radically different amino acid. Models of amino acid sequences based on observed replacements in closely related and reliably aligned sequences have been in widespread use since the since the 1970s [80]. Because they are based on real data these empirical models naturally account for aspects of the amino acid properties that affect the likelihood of amino acid replacement. The empirical models also reflect mutational differences between pairs of amino acids implied by the genetic code; however, in this framework it is not possible to separate out the mutational and selective forces.
Considering the advantages of the mechanistic approach taken by codon models and the value of the empirical information contained in amino acid models, it is natural to investigate methods for combining these approaches. Kosiol et al. [81] proposed an empirical model for codons, and estimated nearly 2000 free parameters describing the empirical exchangeabilities of pairs of codons, assuming reversibility, from a large database of aligned-coding sequences. They incorporate these estimated codon exchangeabilities into the framework of standard mechanistic models of codon evolution and find that they generally provide a significantly improved fit to real data. In an alternative approach Doron-Faigenboim et al. [82], developed a method to incorporate a given empirical amino acid replacement matrix into the mechanistic framework. Again the combined empirical and mechanistic model provides a greatly improved fit to real data. In contrast to standard mechanistic codon models, both models are implemented such that they allow nonzero instantaneous rates of substitution between pairs of codons differing at more than 1 nt position. Although interpretation of the ω parameters is more difficult in the case of both of these models, it is possible to use the merged empirical and mechanistic models to infer positive Darwinian selection, though whether they have a power advantage over standard methods in this application is unclear. Another potential application of these models is to phylogenetic inference, where the improved model realism may offer a substantial advantage [81, 82]. In general, the use of codon models in phylogenetics incurs a significant computational cost due to the number of free parameters to be estimated and due to the size of the instantaneous rate matrix. Despite these costs, codon models show improved model fit over both standard nucleotide models, and nucleotide models which have codon position-specific nucleotide frequencies [6], suggesting that codon models are highly appropriate for phylogeny estimation.
CONCLUSIONS
The trend towards increasingly biologically realistic models and associated increase in the numbers of free parameters to estimate carries a risk of over-parameterization [1]. Perhaps a better alternative to maximum likelihood methods in this context is provided by Bayesian methods, which are generally better suited to large parameterizations [83]. The Dirichlet process [42, 43] that allows the data to influence the complexity of the model (in this case the number of distinct site classes) is useful in this regard. In addition to variable evolutionary rate parameters, models are being proposed that relax the assumption that all sites share a set of character state equilibrium frequencies. Relaxing this assumption can be useful, for example, to model distinct amino acid profiles [42], or fitness landscapes across sites [84]. Directional selection models, which have been applied in the context of the evolution of drug resistance [85], or antigenic drift associated with host-immune pressure [84, 86] also go beyond modeling only evolutionary rates and instead consider fitness effects associated with mutations involving specific amino acids. Nonetheless, these models generally still assume a fitness landscape that is constant in time, despite varying across sites. Stationarity is the assumption that the Markov process, along with its nucleotide, codon or amino acid frequencies, does not change over time. This implies that the process is at equilibrium, so that the observed frequencies of the states of the Markov process are also the equilibrium frequencies. Similarly, rates of substitution are assumed to be time homogenous. Nonstationary models have been applied in the nucleotide and amino acid contexts, specifically for the estimation of phylogenies when base compositions vary between closely related taxa [87–92]. Furthermore, selective constraints are likely to change over time [93–95], such that codon models incorporating variable selective pressure offer improved model fit [96]. Given that adaptive evolution, almost by definition, implies a process which is both directional and time heterogeneous, realistic descriptions of time-variable directional selective pressures will be of particular interest in models that seek to understand how adaptation shapes the evolution of molecular sequences.
Key Points.
Phylogenetic models of sequence evolution typically make use of the formalism of the continuous-time Markov process and are in widespread use in molecular evolution.
Models treating the 61 sense codons as the states of the process (codon models) have the advantage of capturing both the mutational process operating at the nucleotide level and selective processes operating at the protein level.
These models are used to detect selection within genes, at sites within genes, along lineages and at sites along lineages.
Caveats concerning the use of codon models to estimate evolutionary selective pressures acting on protein-coding sequences have recently been highlighted. Some of these, including uncertainty in phylogeny and parameter estimates, and sequence recombination have been addressed through the development of new methods.
Further developments have improved the biological realism of codon models, including integration of codon models with empirical amino acid models that help to account for amino acid properties.
More recently, models have been developed that allow interactions between sites and include parameters describing fitness. The latter go some of the way towards bridging the gap between models of sequence evolution and population genetics theory.
Future developments in codon models may include further relaxing assumptions such that temporal heterogeneity in both mutational and selective processes can be modeled.
FUNDING
South African National Bioinformatics Network.
Acknowledgements
We are grateful to anonymous reviewers for helpful comments on the article.
Biographies
Wayne Delport is a postdoctoral associate with an interest in modeling evolution, particularly with applications in human health and pathogen evolution.
Konrad Scheffler is a lecturer in Computer Science at the University of Stellenbosch, and focuses on probabilistic models of evolution.
Cathal Seoighe has been at the University of Cape Town since 2004 and was previously at the University of the Western Cape. He has research interests in molecular evolution, HIV-1 and mRNA splicing.
References
- Felsenstein J. Inferring Phylogenenies. Sunderland, MA: Sinauer Associates, Inc.; 2004. [Google Scholar]
- Huelsenbeck JP, Crandall KA. Phylogeny estimation and hypothesis testing using maximum likelihood. Ann Rev Ecol Syst. 1997;28:437–66. [Google Scholar]
- Siepel A, Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–88. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Combining phylogenetic and hidden Markov models in biosequence analysis. J Comput Biol. 2004;11:413–28. doi: 10.1089/1066527041410472. [DOI] [PubMed] [Google Scholar]
- Seo T-K, Kishino H. Synonymous substitutions substantially improve evolutionary inference from highly diverged proteins. Syst Biol. 2008;57:367–77. doi: 10.1080/10635150802158670. [DOI] [PubMed] [Google Scholar]
- Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- Whelan S, Lio P, Goldman N. Molecular phylogenetics: state-of-the-art methods for looking into the past. Trends Genet. 2001;17:262–72. doi: 10.1016/s0168-9525(01)02272-7. [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Makova KD, Li WH. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome Res. 2002;12:198–202. doi: 10.1101/gr.200901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–76. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Bollback JP. Application of the likelihood function in phylogenetic analysis. In: Balding DJ, Bishop M, Cannings C, editors. Handbook of Statistical Genetics. West Sussex, England: John Wiley & Sons, Inc; 2007. pp. 460–88. [Google Scholar]
- Lio P, Goldman N. Models of molecular evolution and phylogeny. Genome Res. 1998;8:1233–44. doi: 10.1101/gr.8.12.1233. [DOI] [PubMed] [Google Scholar]
- Thorne JL, Goldman N. Probabilistic models for the study of protein evolution. In: Balding DJ, Bishop M, Cannings C, editors. Handbook of Statistical Genetics. West Sussex, England: John Wiley & Sons; 2007. pp. 439–59. [Google Scholar]
- Yang Z. Computational Molecular Evolution. London: Oxford University Press; 2006. [Google Scholar]
- Kosakovsky Pond SL, Poon AFY, Frost SDW. Lemey P, Salemi M, Vandamme A-M. The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing. 2nd edn. Cambridge University Press; Estimating selection pressures on alignments of coding sequences. [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–24. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci. 1986;17:57–86. [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–36. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, et al. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue N, Lartillot N, Philippe H. Bayesian comparisons of codon substitution models. Genetics. 2008 doi: 10.1534/genetics.108.092254. doi:10.1534/genetics.108.092254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–28. doi: 10.1093/molbev/msm042. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 2005;22:478–85. doi: 10.1093/molbev/msi031. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–17. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–9. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–36. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Gu H, Dunn KA, et al. Methods for selecting fixed-effect models for heterogeneous codon evolution, with comments on their application to gene and genome data. BMC Evol Biol. 2007;7(Suppl. 1):S5. doi: 10.1186/1471-2148-7-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–22. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Yang Z, Swanson WJ. Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol. 2002;19:49–57. doi: 10.1093/oxfordjournals.molbev.a003981. [DOI] [PubMed] [Google Scholar]
- Muse SV. Estimating synonymous and nonsynonymous substitution rates. Mol Biol Evol. 1996;13:105–14. doi: 10.1093/oxfordjournals.molbev.a025549. [DOI] [PubMed] [Google Scholar]
- Wong WSW, Yang Z, Goldman N, et al. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–51. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota R, Waddell PJ, Hasegawa M, et al. Appropriate likelihood ratio tests and marginal distributions for evolutionary tree models with constraints on parameters. Mol Biol Evol. 2000;17:798–803. doi: 10.1093/oxfordjournals.molbev.a026358. [DOI] [PubMed] [Google Scholar]
- Whelan S, Goldman N. Distributions of statistics used for the comparison of models of sequence evolution in phylogenetics. Mol Biol Evol. 1999;16:1292–9. [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19:950–8. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Dyer KA. Bayesian estimation of positively selected sites. J Mol Evol. 2004;58:661–72. doi: 10.1007/s00239-004-2588-9. [DOI] [PubMed] [Google Scholar]
- Scheffler K, Seoighe C. A Bayesian model comparison approach to inferring positive selection. Mol Biol Evol. 2005;22:2531–40. doi: 10.1093/molbev/msi250. [DOI] [PubMed] [Google Scholar]
- Deely JJ, Lindley DV. Bayes empirical Bayes. J Am Stat Assoc. 1981;76:833–41. [Google Scholar]
- Akaike H. Information theory and an extension of the maximum likelihood principle. In: Petran BN, Csáki F, editors. International Symposium on Information Theory. 2nd edn. Akadémiai Kiadi, Budapest, Hungary: 1973. [Google Scholar]
- Hurvich CM, Tsai C-L. Regression and time series model selection in small samples. Biometrika. 1989;76:297–307. [Google Scholar]
- Bao L, Gu H, Dunn KA, et al. Likelihood-based clustering (LiBaC) for codon models, a method for grouping sites according to similarities in the underlying process of evolution. Mol Biol Evol. 2008;25:1995–2007. doi: 10.1093/molbev/msn145. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Jain S, Frost SW, et al. A Dirichlet process model for detecting positive selection in protein-coding DNA sequences. Proc Natl Acad Sci USA. 2006;103:6263–8. doi: 10.1073/pnas.0508279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol Biol Evol. 1998;15:1600–11. doi: 10.1093/oxfordjournals.molbev.a025888. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–18. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- Zhang J. Frequent false detection of positive selection by the likelihood method with branch-site models. Mol Biol Evol. 2004;21:1332–9. doi: 10.1093/molbev/msh117. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach. New York: Springer; 2002. [Google Scholar]
- Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164:1229–36. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner D, Nickle DC, Jensen MA, et al. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet Res. 2003;81:115–21. doi: 10.1017/s0016672303006128. [DOI] [PubMed] [Google Scholar]
- Kuhner MK. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22:768–70. doi: 10.1093/bioinformatics/btk051. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, McVean G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics. 2006;172:1411–25. doi: 10.1534/genetics.105.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Martin DP, Seoighe C. Robust inference of positive selection from recombining coding sequences. Bioinformatics. 2006;22:2493–99. doi: 10.1093/bioinformatics/btl427. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, et al. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23:1891–901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Parmley JL, Chamary JV, Hurst LD. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol. 2006;23:301–9. doi: 10.1093/molbev/msj035. [DOI] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Muse SV. Site-to-Site variation of synonymous substitution rates. Mol Biol Evol. 2005;22:2375–85. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol Biol Evol. 2008;25:568–79. doi: 10.1093/molbev/msm284. [DOI] [PubMed] [Google Scholar]
- Stern A, Pupko T. An evolutionary space-time model with varying among-site dependencies. Mol Biol Evol. 2006;23:392–400. doi: 10.1093/molbev/msj044. [DOI] [PubMed] [Google Scholar]
- Mayrose I, Doron-Faigenboim A, Bacharach E, et al. Towards realistic codon models: among site variability and dependency of synonymous and non-synonymous rates. Bioinformatics. 2007;23:i319–27. doi: 10.1093/bioinformatics/btm176. [DOI] [PubMed] [Google Scholar]
- Hudelot C, Gowri-Shankar V, Jow H, et al. RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol Phylogenet Evol. 2003;28:241–52. doi: 10.1016/s1055-7903(03)00061-7. [DOI] [PubMed] [Google Scholar]
- Jow H, Hudelot C, Rattray M, et al. Bayesian phylogenetics using an RNA substitution model applied to early mammalian evolution. Mol Biol Evol. 2002;19:1591–601. doi: 10.1093/oxfordjournals.molbev.a004221. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Mannino FV, Gravenor MB, et al. Evolutionary model selection with a genetic algorithm: a case study using stem RNA. Mol Biol Evol. 2007;24:159–70. doi: 10.1093/molbev/msl144. [DOI] [PubMed] [Google Scholar]
- Robinson DM, Jones DT, Kishino H, et al. Protein evolution with dependence among codons due to tertiary structure. Mol Biol Evol. 2003;20:1692–704. doi: 10.1093/molbev/msg184. [DOI] [PubMed] [Google Scholar]
- Rodrigue N, Lartillot N, Bryant D, et al. Site interdependence attributed to tertiary structure in amino acid sequence evolution. Gene. 2005;347:207–17. doi: 10.1016/j.gene.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Rodrigue N, Philippe H, Lartillot N. Assessing site-interdependent phylogenetic models of sequence evolution. Mol Biol Evol. 2006;23:1762–75. doi: 10.1093/molbev/msl041. [DOI] [PubMed] [Google Scholar]
- Rodrigue N, Philippe H, Lartillot N. Uniformization for sampling realizations of Markov processes: applications to Bayesian implementations of codon substitution models. Bioinformatics. 2008;24:56–62. doi: 10.1093/bioinformatics/btm532. [DOI] [PubMed] [Google Scholar]
- Poon AF, Lewis FI, Pond SL, et al. An evolutionary-network model reveals stratified interactions in the V3 loop of the HIV-1 envelope. PLoS Comput Biol. 2007;3:e231. doi: 10.1371/journal.pcbi.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttley GA. Modeling the impact of DNA methylation on the evolution of BRCA1 in mammals. Mol Biol Evol. 2004;21:1760–8. doi: 10.1093/molbev/msh187. [DOI] [PubMed] [Google Scholar]
- Hobolth A, Nielsen R, Wang Y, et al. CpG + CpNpG analysis of protein-coding sequences from tomato. Mol Biol Evol. 2006;23:1318–23. doi: 10.1093/molbev/msk017. [DOI] [PubMed] [Google Scholar]
- Sved J, Bird A. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc Natl Acad Sci USA. 1990;87:4692–6. doi: 10.1073/pnas.87.12.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen AK, Wiuf C, Christiansen FB. A codon-based model designed to describe lentiviral evolution. Mol Biol Evol. 1998;15:1069–81. doi: 10.1093/oxfordjournals.molbev.a026006. [DOI] [PubMed] [Google Scholar]
- Hoelzer K, Shackelton LA, Parrish CR. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008;36:2825–37. doi: 10.1093/nar/gkn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpaer EG, Mullins JI. Selection against CpG dinucleotides in lentiviral genes: a possible role of methylation in regulation of viral expression. Nucleic Acids Res. 1990;18:5793–7. doi: 10.1093/nar/18.19.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern AL, Bruno WJ. Evolutionary distances for protein-coding sequences: modeling site-specific residue frequencies. Mol Biol Evol. 1998;15:910–17. doi: 10.1093/oxfordjournals.molbev.a025995. [DOI] [PubMed] [Google Scholar]
- Thorne JL, Choi SC, Yu J, et al. Population genetics without intraspecific data. Mol Biol Evol. 2007;24:1667–77. doi: 10.1093/molbev/msm085. [DOI] [PubMed] [Google Scholar]
- Yu J, Thorne JL. Dependence among sites in RNA evolution. Mol Biol Evol. 2006;23:1525–37. doi: 10.1093/molbev/msl015. [DOI] [PubMed] [Google Scholar]
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–9. doi: 10.1016/s0959-437x(02)00353-2. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff MO, Schwartz RM, Orcutt BC. A model of evolutionary change in proteins. In: Dayhoff MO, editor. Atlas of Protein Sequence and Structure. Silver Spring, Maryland: National Biomedical Research Foundation; 1979. pp. 345–52. [Google Scholar]
- Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24:1464–79. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- Doron-Faigenboim A, Pupko T. A combined empirical and mechanistic codon model. Mol Biol Evol. 2007;24:388–97. doi: 10.1093/molbev/msl175. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Larget B, Miller RE, et al. Potential applications and pitfalls of Bayesian inference of phylogeny. Syst Biol. 2002;51:673–88. doi: 10.1080/10635150290102366. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Bazykin GA, Plotkin J, et al. Directionality in the evolution of influenza A haemagglutinin. Proc Biol Sci. 2008;275:2455–64. doi: 10.1098/rspb.2008.0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C, Ketwaroo F, Pillay V, et al. A model of directional selection applied to the evolution of drug resistance in HIV-1. Mol Biol Evol. 2007;24:1025–31. doi: 10.1093/molbev/msm021. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Poon AF, et al. A maximum likelihood method for detecting directional evolution in protein sequences and its application to Influenza A virus. Mol Biol Evol. 2008;25:1809–24. doi: 10.1093/molbev/msn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquart S, Lartillot N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol Biol Evol. 2006;23:2058–71. doi: 10.1093/molbev/msl091. [DOI] [PubMed] [Google Scholar]
- Blanquart S, Lartillot N. A site- and time-heterogeneous model of amino acid replacement. Mol Biol Evol. 2008;25:842–58. doi: 10.1093/molbev/msn018. [DOI] [PubMed] [Google Scholar]
- Foster PG. Modeling compositional heterogeneity. Syst Biol. 2004;53:485–95. doi: 10.1080/10635150490445779. [DOI] [PubMed] [Google Scholar]
- Galtier N, Gouy M. Inferring phylogenies from DNA sequences of unequal base compositions. Proc Natl Acad Sci USA. 1995;92:11317–21. doi: 10.1073/pnas.92.24.11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowri-Shankar V, Rattray M. On the correlation between composition and site-specific evolutionary rate: implications for phylogenetic inference. Mol Biol Evol. 2006;23:352–64. doi: 10.1093/molbev/msj040. [DOI] [PubMed] [Google Scholar]
- Mooers AO, Holmes EC. The evolution of base composition and phylogenetic inference. Trends Ecol Evol. 2000;15:365–9. doi: 10.1016/s0169-5347(00)01934-0. [DOI] [PubMed] [Google Scholar]
- Bazykin GA, Dushoff J, Levin SA, et al. Bursts of nonsynonymous substitutions in HIV-1 evolution reveal instances of positive selection at conservative protein sites. Proc Natl Acad Sci USA. 2006;103:19396–401. doi: 10.1073/pnas.0609484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazykin GA, Kondrashov FA, Ogurtsov AY, et al. Positive selection at sites of multiple amino acid replacements since rat-mouse divergence. Nature. 2004;429:558–62. doi: 10.1038/nature02601. [DOI] [PubMed] [Google Scholar]
- Messier W, Stewart CB. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–4. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Guindon S, Rodrigo AG, Dyer KA, et al. Modeling the site-specific variation of selection patterns along lineages. Proc Natl Acad Sci USA. 2004;101:12957–62. doi: 10.1073/pnas.0402177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Butterfield A, Vedagiri V, Lang E, et al. PyEvolve: a toolkit for statistical modelling of molecular evolution. BMC Bioinformatics. 2004;5:1. doi: 10.1186/1471-2105-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Doron-Faigenboim A, Erez E, et al. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007;35:W506–11. doi: 10.1093/nar/gkm382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Gojobori T, Nei M. ADAPTSITE: detecting natural selection at single amino acid sites. Bioinformatics. 2001;17:660–1. doi: 10.1093/bioinformatics/17.7.660. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]