

Abstract
To understand the role of the immune system in limiting HIV type 1 replication, it is critical to know to what extent the rapid turnover of productively infected cells is caused by viral cytopathicity or by immune-mediated lysis. We show that uncultured peripheral blood mononuclear cells of many patients contain cytotoxic T lymphocytes (CTL) that lyse target cells—at plausible peripheral blood mononuclear cell-to-target ratios—with half-lives of less than 1 day. In 23 patients with CD4 counts ranging from 10 to 900 per μl, the average rate of CTL-mediated lysis corresponds to a target cell half-life of 0.7 day. We develop mathematical models to calculate the turnover rate of infected cells subjected to immune-mediated lysis and viral cytopathicity and to estimate the fraction of cells that are killed by CTL as opposed to virus. The models provide new interpretations of drug treatment dynamics and explain why the observed rate of virus decline is roughly constant for different patients. We conclude that in HIV type 1 infection, CTL-mediated lysis can reduce virus load by limiting virus production, with small effects on the half-life of infected cells.
Keywords: virus dynamics, mathematical model, antiviral treatment, immune response
Recent studies of HIV type 1 (HIV-1) dynamics have shown that the initial rate of virus decline during drug treatment is approximately 30% per day, leading to the interpretation that productively infected cells have a half-life of about 2 days (1–6). There is little variation in this half-life, which ranges from about 1 to 4 days in patients with CD4 cell counts between 20 and 500 μl (Fig. 1). There is no correlation between the rate of viral turnover and CD4 cell count or virus load. The important question is whether the observed turnover rate of infected cells is caused by viral cytopathicity or immune-mediated clearance mechanisms. If we postulate that cytotoxic T lymphocyte (CTL)-mediated lysis determines the life span of a productively infected cell (7, 8), then it is surprising to find so little variation in the observed half-life of infected cells and no correlation with the disease stage (CD4 cell count) of a patient. On the other hand, if cell death is only due to viral cytopathicity, then immune-mediated lysis can have no effect on reducing virus production. We will analyze whether CTL-mediated lysis could be fast enough to account for the observed turnover rates, and we will develop a mathematical model to quantify the effect of CTL killing on infected cell half-life and virus production.
Figure 1.
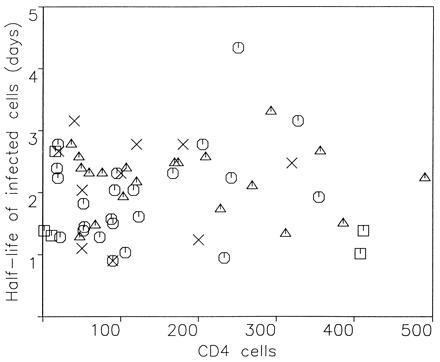
The turnover rate of productively infected cells from 59 HIV-1-infected patients treated with antiviral drugs. The average death rate of productively infected cells, calculated from the exponential decline of plasma virus load after treatment, is 0.37 ± 0.18 per day, corresponding to a half-life of 1.9 days. The minimum half-life is about 1 day. There is no correlation between infected cell half-life and CD4 cell count. Symbols represent patients from different studies: ▵, ref. 1; ○, ref. 2; ×, ref. 4; □, ref. 5.
In HIV-1-infected patients, antiviral CTL arise early in infection (9–11) and are present within uncultured circulating peripheral blood mononuclear cells (PBMC) as a subset of CD8+ cells (12–27). The rate of CTL-mediated killing should influence both the half-life of virus-infected cells and the amount of virus production. We determined the in vitro half-life of target cells [human leukocyte antigen (HLA) class I matched B cells which bear sufficient peptide for T-cell recognition] from assays of specific cytotoxicity by fresh PBMC. Fig. 2A shows time-resolved decay curves of target cells in the presence of PBMC from HIV-1-positive patient 84 (who has been infected for about 5 years and has currently a CD4 cell count of 540 per μl) at various PBMC-to-target ratios. From the slope of the decay curve, we can calculate the death rate of target cells. At a PBMC-to-target ratio of 64:1, the death rate of target cells corresponds to a half-life of 12 hr. Fig. 2B shows the half-lives of target cells for different PBMC-to-target ratios.
Figure 2.
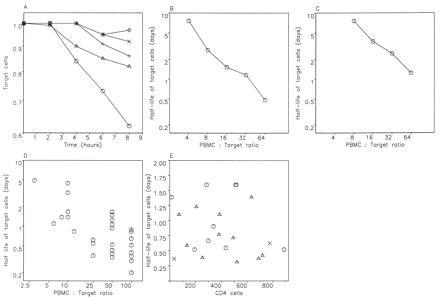
The average lifetime of target cells can be calculated from assays of specific cytotoxicity. (A) Time-resolved decay curve of peptide-sensitized target cells from HIV-1-infected, asymptomatic patient 84. The assays of fresh killing were performed using PBMC separated directly. Targets were HLA B8-matched B cells, untreated or prepulsed with 1 μM p17-3 (GGKKKYKL), an epitope in p17 to which this patient makes a predominant response. Targets were chromium-labeled, washed, and plated at 5000 cells per well in duplicate wells of a 96-well plate, and PBMC, medium, or Triton X (5%) was added to a total volume of 150 ml. Percentage-specific lysis was calculated as 100 × (experimental release − medium release)/(maximal release − medium release), and net specific lysis was determined by subtraction of background lysis against control targets. The decay rate of target cells can be directly obtained from the slope of the decay curves. There is an initial 2-hr time delay during which no killing can be observed. This may be a consequence of the effector cells needing time to attach to and kill the first targets. Between 2 and 8 hr, there is a roughly constant killing rate. PBMC-to-target ratios are 64:1 (○), 32:1 (▵), 16:1 (+), 8:1 (×), and 4:1 (◊). Different amounts of PBMC are added to the same number of targets. At a ratio of 64:1, target cell half-life is 11.6 hr (in the first 8 hr) and 8.7 hr (if calculated from the slope between 2 and 8 hr). (B) Half-life of target cells (calculated from 0 to 8 hr) versus PBMC-to-target ratio gives roughly a straight line in a double logarithmic plot with a slope of about −1. This suggests that the killing rate of target cells is simply proportional to the number of effector cells in the assay. (C) Half-life of HIV-infected target cells subjected to uncultured CTL-mediated lysis. Effector cells were freshly isolated PBMC from the B8-positive donor SC3. Targets were B8-matched C8166 cells used 48 hr after infection with 10 TCID50 of HIV IIIB. Targets were chromium-labeled, washed, and plated at 5000 cells per well as described in A. (D) Half-lives of target cells derived from published studies of lysis by fresh PBMC from HIV-1-infected patients (12–18). Points represent individual patients. (E) Half-life of target cells in assays of fresh cytotoxicity versus CD4 cell count of 23 patients. For each patient, we show the maximum response among the anti-Gag (○), anti-Pol (▵), or anti-Nef (×) CTL at a PBMC-to-target ratio of 50:1. (The anti-Tat response was never the maximum response in any of the patients.) The average rate of CTL-mediated lysis is 1.03 ± 0.57 per day, which corresponds to a half-life of 0.68 days. (At a PBMC-to-target ratio of 25:1, the average rate of CTL-mediated lysis is 0.62 ± 0.37 per day, which corresponds to a half-life of 1.1 day. Data not shown.) There is no correlation between CTL-mediated lysis and CD4 cell count. Effector cells were freshly isolated PBMC that were assessed for anti-HIV-1 CTL activity against 51Cr-labeled autologous B lymphoblastoid cell lines infected with recombinant vaccinia virus expressing HIV-1 (Lai) Gag, Pol, Env, Nef, or Tat at effector-to-target ratios of 50:1 and 25:1 (14, 15). Controls were target cells with medium alone for spontaneous release, and with detergent for maximal release of 51Cr. Spontaneous release averaged 17%. Split-sample validity testing showed <10% variation in CTL activity against the HIV-1 protein-expressing targets. Anti-HIV-1 CTL activity was not detected in PBMC from HIV-1 seronegative subjects.
Clearly, the above data are results from in vitro measurements, and it is uncertain how accurately they reflect the rate of CTL-mediated lysis in vivo. We argue, however, that using uncultured PBMC and adding a small amount of target cells may provide conditions that are as close as possible to the in vivo situation. The fraction of cells infected with HIV-1 and expressing HIV-1 RNA in lymph nodes can be as high as 3–6% in asymptomatic subjects (28, 29), although this may vary depending on the lymphoid microenvironment (30). This implies an overall ratio of lymphocytes to infected target lymphocytes of at least 15–30:1. In patients with lower viral burden, the ratio will be higher. Therefore, by using PBMC-to-target ratios of 25:1 or larger, we may simulate the in vivo situation.
Although the above experiments were carried out with peptide pulsed B cell lines as targets, we have also measured the death rate of HIV-1-infected CD4 cells in the presence of uncultured PBMC from the HLA-B8 positive patient SC3 (an asymptomatic individual who has been infected for at least 3 years and has a CD4 cell count of 440 per μl). Effector cells were freshly isolated PBMC; targets were B8-matched C8166 cells used 48 hr after infection with 10 TCID50 of HIV IIIB. At a PBMC-to-target ratio of 64:1, we find 17% specific lysis after 8 hr, corresponding to a target cell half-life of 1.2 days. At a ratio of 32:1, we obtain a half-life of 2.4 days (Fig. 2C).
Results derived from published studies of fresh responses by many other workers (12–18) can also be used to calculate target cell half-lives (Fig. 2D). In some patients, the half-life of target cells is less than 12 hr at PBMC-to-target ratios of 50:1 or greater. On the other hand, many HIV-1-infected individuals do not show strong responses and target cell half-life by CTL killing would exceed 48 hr.
To explore a possible correlation between the rate of CTL-mediated lysis and disease stage of a patient, we calculated the half-life of target cells in the presence of fresh PBMC from 23 patients with CD4 cell counts ranging from 10 to 900 per μl. CTL activity was determined against targets expressing five different HIV proteins: Gag, Pol, Env, Nef, or Tat (25). Fig. 2E shows the calculated half-life of target cells with respect to the strongest response among the Gag-, Pol-, Nef-, or Tat-specific CTL in each patient. There is no correlation between the CD4 count of a patient and the half-life of target cells. At a PBMC-to-target ratio of 50:1, the average rate of CTL-mediated lysis is 1.0 ± 0.57 per day, which corresponds to a half-life of about 0.7 day. Note that the lack of correlation between viral turnover and CD4 count (Fig. 1) is paralleled by a lack of correlation between the fresh CTL response and CD4 count (Fig. 2E).
In contrast to the anti-Gag, -Pol, -Nef, or -Tat responses, which are mostly due to CTL activity, the anti-Env response is likely to contain a large proportion of antibody-dependent cell cytotoxicity (25–27). The calculated average rate of anti-Env-mediated lysis in vitro is 1.24 ± 0.66 per day, corresponding to a half-life of about 0.6 day. However, it is not clear how important antibody-dependent cell cytotoxicity is in vivo in the presence of a vast excess of serum Ig, which would compete with the specific antibodies for Fc receptors.
Given these findings, we will now explore whether CTL-mediated lysis can make a significant contribution to the turnover rate of HIV-infected cells in vivo. If HIV directly kills productively infected cells, does CTL-mediated destruction play a role in the overall decay rate of infected cells? We develop a mathematical model to relate the expected lifetime of an infected cell to the rate of CTL-mediated clearance mechanisms and virus cytopathicity and to calculate the amount of virus production inhibited by CTL. Consider a model with three compartments of infected cells representing different stages of the virus life cycle: y1 are newly infected cells, which are not yet producing free virus and are not targets for CTL killing; y2 are cells that are still not producing virus but can be killed by CTL, whereas y3 are cells that are producing free virus and can be killed by CTL.
There are different ways to specify the dynamics of the infected cell life cycle. In model 1, we assume that the transitions from y1 to y2 and from y2 to y3 occur at fixed times t1 and t2 in the life cycle of an infected cell. This means a cell gets invaded by virus at time t = 0. At time t1, enough new viral proteins have been produced such that the cell becomes a potential target for CTL-mediated killing. The death rate of a target cell due to CTL is given by α. At time t2, the cell starts to produce free virus particles. This increases the death rate by an amount c, which is due to virus cytopathicity. Thus we have a stepwise model for cellular decay. Between time 0 and t1, the death rate is 0; between times t1 and t2, the death rate is α; after time t2, the death rate is α + c. Another possibility (model 2) is to assume that the transitions occur at constant rates: y1 turns into y2 at rate a; y2 turns into y3 at rate b; and y3 cells die at rate c. As before, the CTL response eliminates y2 and y3 cells at rate α. Both models can be used to calculate the lifetime of infected cells, the average duration of virus production of a single cell, and the fraction of cells killed by CTL as opposed to virus (Fig. 3). Although it is likely that a cell becomes a target for CTL before onset of virus production (31), this assumption is not essential and can easily be reversed.
Figure 3.
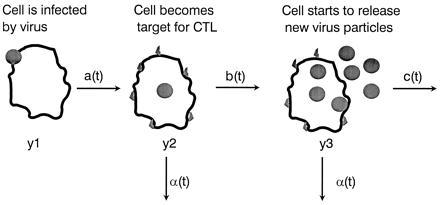
A mathematical model to explore the effect of CTL-mediated killing on the half-life of infected cells and virus production: y1 are newly infected cells that do not produce virus and are not killed by CTL, y2 are cells that can be killed by CTL, and y3 are cells that produce free virus and can be killed by CTL. The transition rates from y1 to y2 and from y2 to y3 are given by a(t) and b(t), respectively, where t is time since infection of the cell. The rate of CTL killing is given by α(t), and the rate of cell death due to virus is given by c(t). This is a general framework. We analyzed two specific cases. In model 1, we assumed that at time t1 after infection, a cell becomes a target for CTL killing, and at time t2, it starts to produce new virus particles. Death rate due to CTL killing is α, and death rate due to virus is c. These assumptions lead to an average lifetime of infected cells of T = t1 + (1/α) − c/[α(α + c)] e−α(t2−t1). In the absence of CTL-mediated lysis (α = 0), the average lifetime is T = t2 + (1/c). The of an infected cell is T1/2 = t1 + (1/α) log 2 if t2 > t1 + (1/α) log 2 or T1/2 = t2 + [1/(α + c)][log 2 − α(t2 − t1)] if t2 < t1 + (1/α) log 2. Note that the relation between half-life and average lifetime is not simply T1/2 = T log 2, because infected cell death is a combination of different exponential declines. The average duration of a cell producing new virus is Tv = [1/(α + c)] e−α(t2−t1). In the absence of CTL, this increases to T0 = 1/c. The fraction of cells that are killed by CTL before virus production sets in is f = 1 − e−α(t2−t1). The total fraction of cells killed by CTL as opposed to viral cytopathicity is F = 1 − [c/(α + c)] e−α(t2−t1). Assuming constant viral production after time t2, the fraction of virus production inhibited by CTL-mediated lysis can be defined as 1 − Tv/T0, which is equivalent to F. In model 2, we assumed that y1 turns into y2 at a constant rate, a; y2 turns into y3 at rate b; y3 cells are killed by virus at rate c, and y2 and y3 cells are killed by CTL at rate α. In this model, the average lifetime of a cell is T = (1/a) + [1/(α + b)] + [b/(α + b)][1/(α + c)] and the average duration of a cell producing virus is Tv = [b/(α + b)][1/(α + c)]. In the absence of a CTL response, α = 0, this duration increases to T0 = 1/c. The fraction of cells killed by CTL is F = 1 − [b/(α + b)][c/(α + c)], which is again equivalent to the total amount of virus production inhibited by CTL.
Between the two limiting cases, described by models 1 and 2, lies a spectrum of models where the transition from y1 to y2 (and from y2 to y3) is not given by a simple one-step process with an exponential distribution and also does not occur after a fixed length of time. We considered a Poisson process, where a number, n, of events have to accumulate (at certain rates) before the transition occurs. If n = 1, we obtain model 1, and if n is very large, we obtain model 2. For intermediate values of n, the resulting decay is not strictly exponential but for practical purposes may be indistinguishable from an exponential decay.
Table 1 gives numerical values for the effect of CTL killing. For a cytopathic virus, the half-life of infected cells does not vary much in the presence of weak or strong CTL responses, but the fraction of infected cells killed by CTL and the amount of virus production inhibited by CTL can be greatly affected by the rate of CTL-mediated lysis. For example, a response equivalent to 10% lysis in 4 hr can eliminate about 70% of infected cells; the remaining 30% is eliminated by virus cytopathic effects. For a noncytopathic virus, differences in CTL activity lead to large variation in infected cell half-life. Responses equivalent to 10% lysis in 4 hr eliminate 99% of infected cells.
Table 1.
Effect of CTL-mediated lysis on the half-life of infected cells, T1/2, and the fraction, F, of cells killed by CTL according to model 1
| Rate of CTL killing, per day | Cytopathic virus
early target formation
|
Cytopathic virus late target
formation
|
Noncytopathic virus
|
|||
|---|---|---|---|---|---|---|
| T1/2, days | F | T1/2, days | F | T1/2, days | F | |
| 0 (0%) | 2.7 | 0 | 2.7 | 0 | 70 | 0 |
| 0.12 (2%) | 2.2 | 0.29 | 2.3 | 0.24 | 6.2 | 0.92 |
| 0.31 (5%) | 1.7 | 0.54 | 1.9 | 0.45 | 3.1 | 0.97 |
| 0.63 (10%) | 1.2 | 0.75 | 1.6 | 0.64 | 2 | 0.99 |
| 1.34 (20%) | 0.8 | 0.91 | 1.3 | 0.8 | 1.4 | 0.99 |
| 3.06 (40%) | 0.5 | 0.99 | 1.1 | 0.92 | 1.1 | 0.998 |
We assume that virus production starts t2 = 1 day after infection of a cell. For the cytopathic virus, the death rate of virus producing cells is c = 0.4 per day; for the noncytopathic virus, this death rate is c = 0.01 per day. For the cytopathic virus, we explore early target formation (t1 = 0.3 day) and late target formation (t1 = 0.9 day). For the noncytopathic virus, the value of t1 makes almost no difference, and we choose t1 = 0.9. The rate of CTL killing, α, is given per day (in brackets the equivalent percentage specific lysis in a 4-hr assay). The results show that for a cytopathic virus, differences in the rate of CTL-mediated lysis have only small effects on the half-life of infected cells (ranging from 0.5 to 2.7 days), whereas for the noncytopathic virus, there is large variation in infected cell half-life (ranging from 1.1 to 70 days). In both cases, CTL-mediated lysis can kill a large fraction, F, of infected cells and, therefore, significantly reduce virus production.
The above models also provide new insights into the dynamics of virus decline after drug treatment. Before drug treatment, the virus population is at steady state and infected cells, y1, are produced at a constant rate, β (which depends on the abundance of infectable cells, virus load, cytokine levels, etc.). The effect of drug treatment is to reduce β to zero. Reverse transcriptase inhibitors prevent the infection of new cells, whereas protease inhibitors render virus particles, which are being produced from already infected cells, noninfectious. Therefore reverse transcriptase inhibitors bring β to (almost) 0 as soon as they achieve a high enough concentration within the patient, whereas protease inhibitors allow for a short time infection of new cells by virus particles that have been produced before the drug was given (5, 6). If the free virus half-life is short, then the difference is negligible. In both models 1 and 2, this leads to an (approximately) exponential decline in plasma virus, v, after an initial shoulder. In model 1, the slope of the exponential decay is given by α + c (Fig. 4). Thus, the rate of CTL-mediated killing should influence the rate of free virus decline. In model 2, however, the slope of the exponential decline is given by the smallest value among a, α + b, and α + c, which means that the slowest step in the life cycle of an infected cell (including the effect of α) determines the rate of virus decline during drug treatment. If the time between infection of the cell and expressing enough viral protein to become a target for CTL is rate limiting, then the observed exponential slope of virus decline is given by a and does not depend on the rate of CTL-mediated killing. Even if a is larger than b or c, it is still possible that in most patients a is smaller than b + α and c + α, and therefore again the observed rate of virus decline reflects the initial phase of the viral life cycle and is unaffected by CTL-mediated lysis.
Figure 4.
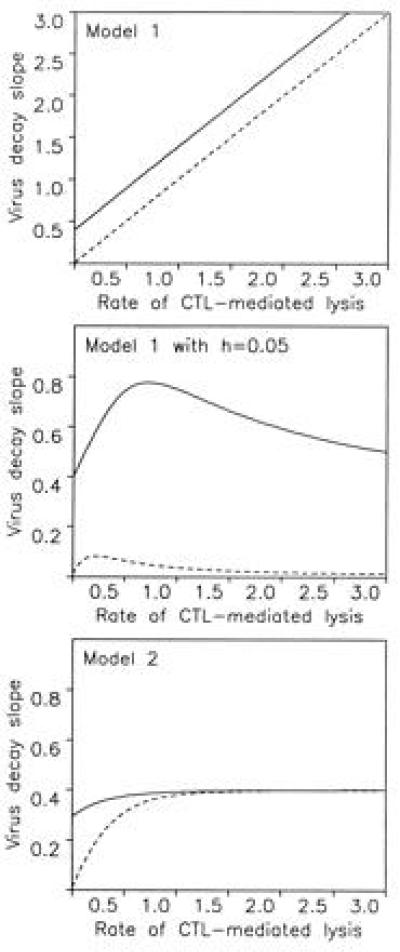
The models provide a new interpretation of the slope of virus decay in drug treatment studies (1–6). We assume that before therapy, new infections occur at a constant rate β. This leads to an equilibrium distribution of y1, y2, and y3 cells and free virus v. Drug treatment reduces β to zero, which leads to a decay of free virus and infected cells. (A) In model 1, the equilibrium distribution of infected cells is y1(t) = 1 for t < t1, y2(t) = exp[−α(t − t1)] for t1 < t < t2, and y3(t) = exp[−α(t2 − t1) − (α + c)(t − t2)] for t2 < t, where t is the time since infection of the cell. Before drug treatment, the total amount of virus producing cells is Y3(0) = ∫t2∞ y3(t)dt = [1/(α + c)]exp[−a(t2 − t1)]. During drug treatment, this cell population declines as Y3(T) = Y3(0) for T < t2, and Y3(T) = Y3(0)exp[−(α + c)(T − t2)] for T > t2. Here T denotes time after start of drug treatment. Thus, virus decline occurs with a shoulder of length t2 followed by an exponential decline with slope α + c. (B) If we include a small fraction, h, of cells that are not exposed to CTL-mediated killing, the virus-producing cell population declines as Y3(T) = [(1 − h)/(α + c)]exp[−α(t2 − t1) − (α + c)(T − t2)] + (h/c)exp[−c(T − t2)]. In a patient with a weak CTL response (α ≈ 0), the exponential decline is c, and in a patient with a strong CTL response (α ≫ c), the decline is again roughly c. Thus, the rate of virus decline does not reflect the rate of CTL-mediated killing, α. (C) In model 2, virus-producing cells, Y3(T), decline as [(b − c)/a]e−at + [(α − a + c)/(α + b)]e−(α+b)t − [(α − a + b)/(α + c)]e−(α+c)t. This expression again describes an initial shoulder followed by an exponential decay. The slope of the exponential decay is determined by the smallest value among a, b + α, or c + α. If the rate, a, at which infected cells proceed to become targets for CTL killing is slow, then the exponential decay in treatment studies may simply reflect this process and not depend on the rate of CTL-mediated killing, α. In all models, free virus is produced from infected cells according to v̇ = kY3 − uv. If free virus turnover is fast, then v(T) is proportional to Y3(T) and the decline of Y3(T) can directly be interpreted as free virus decline; if not, then one more integration is necessary, but the conclusions are unaffected as long as u is not the slowest rate constant, which is very unlikely. For model 1, we chose c = 0.4, t1 = 0.5, t2 = 1, and h = 0.05. For model 2 we chose a = 0.4, b = 2, and c = 0.5 (continuous lines). Broken lines indicate noncytopathic virus with c = 0.01.
There is one additional explanation for why CTL-mediated killing may not affect the rate of virus decline during drug therapy. It is natural to assume that in a given patient infected cells are exposed to different rates of CTL-mediated lysis. This can be a consequence of spatial heterogeneity (31), cell tropism, or antigenic variation. Therefore, rather than assuming a fixed average rate, α, of CTL-mediated lysis for all cells, it may be better to consider a distribution of different α values. In the simplest model, we assume that in each patient, there is a small and variable fraction h of cells not exposed to CTL killing. Using the framework of model 1 and assuming that a fraction, h, of cells has death rate c and the remaining 1 − h cells have death rate c + α, we find that virus decay in treatment studies is given by [(1 − h)/(α + c)]exp[−α(t2 − t1) − (α + c)(T − t2)] + (h/c)exp[−c(T − t2)]. For a patient with a weak CTL response (α ≈ 0), the slope is c; for a patient with a strong CTL response (α ≫ c), the slope is again dominated by c. Therefore, if a small fraction of infected cells (less than 5–10%) escape from CTL surveillance, the resulting virus decay slopes are independent of the average rate of CTL-mediated killing in this patient. The intuitive reason behind this explanation is that virus dynamics experiments measure the turnover rate of cells that produce most of the plasma virus. But cells that are killed by CTL produce less plasma virus than cells that are not killed by CTL. Therefore, the virus decay slope will be biased toward the decay rate of cells that have escaped from CTL-mediated lysis. Clearly, the same argument applies to model 2.
Fig. 4 shows the slope of virus decline during drug therapy as a function of the rate of CTL-mediated lysis. For both model 1 with heterogeneity and model 2, we find that the virus decay slope is roughly constant provided that HIV-1 is cytopathic and would also kill cells in the absence of a CTL response. If HIV-1 were noncytopathic, there should be much smaller decay rates in patients with weak or absent CTL responses. The lack of extensive variation in virus decay slopes following drug therapy (Fig. 1) suggests that HIV-1 is cytopathic. Otherwise, one would have to argue that all individuals measured so far have a minimum CTL-mediated (or immune-mediated) clearance rate of infected cells that corresponds to a target cell lysis of 5–10% in 4 hr, which seems unlikely.
For comparison, in infections with the hepatitis B virus, which is considered to be noncytopathic, the half-life of productively infected cells varies from 10 to 100 days in different individuals (32).
Our analysis reveals that also low levels of CTL-mediated lysis can account for elimination of large fractions of infected cells. Therefore, it will be important to improve the sensitivity of ex vivo CTL measurements, either by longer CTL assays (8–24 hr) or direct staining and quantitation of antigen-specific CTL in PBMC samples (33). The more conventional assays involving culture, and sometimes cloning, of CTL probably measure a different parameter made up of the frequency of memory CTL and the effectiveness of T-cell help for CTL development. These memory cells are the source of the effector CTL measured in fresh PBMC, but the relationship between the two is complex.
With respect to HIV-1 disease progression, the crucial question is to what extent immune responses reduce virus load in infected patients (34). Our results suggest that CTL-mediated lysis is sufficiently fast to eliminate a large fraction of productively infected cells and thereby greatly reduce virus production. In addition, CD8+ T cells also release cytokines (35–37) that can block virus entry into cells (38–43). Both mechanisms are likely to reduce virus load and therefore slow down the rate of disease progression (44).
Acknowledgments
We thank Rolf Zinkernagel, Xiao-Li Huang, Zheng Fan, Philip Goulder, Matt Collin, Ann Edwards, Paul Giangrande, Sebastian Bonhoeffer, Barbara Bittner, the staff of the Department of Genito-Urinary Medicine at the Radcliffe Infirmary, and Haemophilia Centre, Churchill Hospital, Oxford, and the Pittsburgh, PA portion of the Multicenter AIDS Cohort Study. This work was supported by the Wellcome Trust, the Medical Research Council (U.K.) and the National Institutes of Health.
Footnotes
Abbreviations: CTL, cytotoxic T lymphocyte(s); PMBC, peripheral blood mononuclear cell(s).
References
- 1.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 2.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 3.Coffin J M. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 4.Nowak M A. Nature (London) 1995;375:193. doi: 10.1038/375193a0. [DOI] [PubMed] [Google Scholar]
- 5.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 6.Herz A V M, Bonhoeffer S, Anderson R M, May R M, Nowak M A. Proc Natl Acad Sci USA. 1996;93:7247–7251. doi: 10.1073/pnas.93.14.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wain-Hobson S. Nature (London) 1995;373:102. doi: 10.1038/373102a0. [DOI] [PubMed] [Google Scholar]
- 8.Zinkernagel R M. Science. 1996;271:173–179. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]
- 9.Borrow P, Lewicki H, Hahn B H, Shaw G M, Oldstone M B. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pantaleo G, Demarest J F, Soudeyns H, Graziosi C, Denis F, Adelsberger J W, Borrow P, Saag M S, Shaw G M, Sekaly R P, Fauci A S. Nature (London) 1994;370:463–467. doi: 10.1038/370463a0. [DOI] [PubMed] [Google Scholar]
- 11.Safrit J T, Andrews C A, Zhu T, Ho D D, Koup R A. J Exp Med. 1994;179:463–472. doi: 10.1084/jem.179.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker B D, Chakrabarti S, Moss B, Paradis T J, Flynn T, Durno A G, Blumberg R S, Kaplan J C, Hirsch M S, Schooley R T. Nature (London) 1987;328:345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- 13.Walker B D, Flexner C, Paradis T J, Fuller T C, Hirsch M S, Schooley R T, Moss B. Science. 1988;240:64–66. doi: 10.1126/science.2451288. [DOI] [PubMed] [Google Scholar]
- 14.Plata F, Autran B, Martins L P, Wain-Hobson S, Raphael M, Mayaud C, Denis M, Guillon J M, Debre P. Nature (London) 1987;328:348–351. doi: 10.1038/328348a0. [DOI] [PubMed] [Google Scholar]
- 15.Koenig S, Earl P, Powell D, Pantaleo G, Merli S, Moss B, Fauci A S. Proc Natl Acad Sci USA. 1988;85:8638–8642. doi: 10.1073/pnas.85.22.8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gotch F M, Nixon D F, Alp N, McMichael A J, Borysiewicz L K. Int Immunol. 1990;2:707. doi: 10.1093/intimm/2.8.707. [DOI] [PubMed] [Google Scholar]
- 17.McElrath M J, Rabin M, Hoffman M, Klucking S, Garcia J V, Greenberg P D. J Virol. 1994;68:5074–5083. doi: 10.1128/jvi.68.8.5074-5083.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buseyne F, Blanche S, Schmitt D, Griscelli C, Riviere Y. J Immunol. 1993;141:3569–3581. [PubMed] [Google Scholar]
- 19.Nixon D F, Townsend A R M, Elvin J G, Rizza C R, Gallwey J, McMichael A J. Nature (London) 1988;336:484–487. doi: 10.1038/336484a0. [DOI] [PubMed] [Google Scholar]
- 20.Langlade-Demoyen P, Michel F, Hoffenbach A, Vilmer E, Dadaglio G, Garcia-Pons F, Mayaud C, Autran B, Wain-Hobson S, Plata F. J Immunol. 1988;141:1949–1957. [PubMed] [Google Scholar]
- 21.Koup R A, Sullivan J L, Levine P H, Brettler D, Mahr A, Mazzara G, McKenzie S, Panicali D. Blood. 1989;73:1909–1914. [PubMed] [Google Scholar]
- 22.Riviere Y, Tanneau-Salvadori F, Regnault A, Lopez O, Sansonetti P, Guy B, Kieny M-P, Fournel J-J, Montagnier L. J Virol. 1989;63:2270–2277. doi: 10.1128/jvi.63.5.2270-2277.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clerici M, Lucey D R, Zajac R A, Boswell R N, Gebel H M, Takahashi H, Berzofsky J A, Shearer G M. J Immunol. 1991;146:2214–2219. [PubMed] [Google Scholar]
- 24.Carmichael A, Jin X, Sissons P, Borysiewicz L. J Exp Med. 1993;177:249–256. doi: 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinaldo C, Huang X-L, Fan Z, Ding M, Beltz L, Logar A, Panicali D, Mazzara G, Liebmann J, Cottrill M, Gupta P. J Virol. 1995;69:5838–5842. doi: 10.1128/jvi.69.9.5838-5842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torpey D, Huang X-L, Armstrong J, Ho M, Whiteside T, McMahon D, Pazin G, Heberman R, Gupta P, Tripoli C. Clin Immunol Immunopathol. 1993;68:263–272. doi: 10.1006/clin.1993.1127. [DOI] [PubMed] [Google Scholar]
- 27.Rinaldo C, Beltz L A, Huang X L, Gupta P, Fan Z, Torpey D J. AIDS Res Hum Retroviruses. 1995;11:481–489. doi: 10.1089/aid.1995.11.481. [DOI] [PubMed] [Google Scholar]
- 28.Embretson J, Zupanic M, Ribas J L, Burke A, Racz P, Tenner-Racz K, Haase A T. Nature (London) 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 29.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. Nature (London) 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 30.Cheynier R, Henrichwark S, Hadida F, Pelletier E, Oksenhendler E, Autran B, Wain Hobson S. Cell. 1994;78:373–387. doi: 10.1016/0092-8674(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 31.Yang O, Kalams S, Rosenzweig M, Trocha A, Jones N, Koziel M, Walker B, Johnson R. J Virol. 1996;70:5799–5806. doi: 10.1128/jvi.70.9.5799-5806.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nowak M A, Bonhoeffer S, Hill A, Boehme R, Thomas H, McDowd H. Proc Natl Acad Sci USA. 1996;93:4398–4402. doi: 10.1073/pnas.93.9.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altman J, Moss P A H, Goulder P, Barouch D, McHyzer-Williams M, Bell J I, McMichael A J, Davis M M. Science. 1996;274:94–99. [PubMed] [Google Scholar]
- 34.Nowak M A, Bangham C R M. Science. 1996;272:74–79. doi: 10.1126/science.272.5258.74. [DOI] [PubMed] [Google Scholar]
- 35.Walker C M, Moody D J, Stites D P, Levy J A. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 36.Baier M, Werner A, Bannert N, Metzner K, Kurth R. Nature (London) 1995;378:563. doi: 10.1038/378563a0. [DOI] [PubMed] [Google Scholar]
- 37.Cocchi F, DeVico A L, Garzino D A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 38.Feng Y, Broder C C, Kennedy P E, Berger E A. Science. 1996;272:872–877. [Google Scholar]
- 39.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S C, Schall T J, Littman D R, Landau N R. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 40.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 41.Weiss R A, Clapham P R. Nature (London) 1996;381:647–648. doi: 10.1038/381647a0. [DOI] [PubMed] [Google Scholar]
- 42.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 43.Weiss R A. Science. 1996;272:1885–1886. doi: 10.1126/science.272.5270.1885. [DOI] [PubMed] [Google Scholar]
- 44.Mellors J W, Rinaldo C R, Jr, Gupta P, White J T, Kingsley L. Science. 1996;272:1167–1169. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]