

Abstract
The stimulation and maintenance of the pulmonary alveolar type II cell's capacity to biosynthesize, store, and secrete surfactant proteins (SPs) are modulated to a great extent by growth factors, extracellular matrix (ECM) components, and hormones. It is possible that differences in ECM composition, as exist between type I and II cells normally or as might occur with excessive cell surface shedding during inflammation or injury states, may specifically alter SP expression. Here, isolated type II cells were exposed to the model sulfated ECM heparin; desulfated heparin; and/or fibroblast growth factor (FGF)-1, -2, or -7 for 24 h to examine by quantitative real-time polymerase chain reaction their effects on SP gene expression. Aquaporin 5 (AQP-5) gene expression was also examined as a phenotypic marker for the type I cell. SP-B mRNA abundance was increased 4- to 8-fold by all three FGFs. Heparin at low concentrations (5 μg/ml) or desulfated heparin at high concentrations (500 μg/ml) enhanced the effects of FGF-2 and -7, while high heparin concentrations (500 μg/ml) were inhibitory. In contrast, SP-B mRNA abundance was increased by heparin in a dose- and sulfation-dependent manner when used in combination with FGF-1. SP-C and AQP-5 mRNA levels were increased by heparin alone in a dose- and sulfation-dependent manner, while all FGFs lacked effect on SP-C or AQP-5 mRNA levels. These data indicate that heparin can be stimulatory to SP gene expression depending on concentration, degree of sulfation, and surrounding FGF environment, and that heparin plays a significant role in modulating alveolar epithelial cell phenotype in vitro.
Keywords: extracellular matrix, fibroblast growth factor, keratinocyte growth factor, surfactant protein-B, surfactant protein-C
The pulmonary alveolar surface is lined primarily by two epithelial cell types, the alveolar type I cell and the alveolar type II cell. The squamous type I cell accounts for ∼ 40% of the alveolar epithelial cells by number and covers 95% of the alveolar surface (1). In contrast, the cuboidal type II cell accounts for ∼ 60% of alveolar epithelial cells by number, yet covers only 5% of the alveolar surface (1). Their striking morphologic differences reflect their specialized and intricately complementary functions within the deep lung. The squamous (0.2 μm) type I cell, with its large surface area, facilitates the efficient diffusion of respiratory gases across the lung epithelium. To maintain this surface area available for gas exchange, pulmonary surfactant, which consists of a mixture of phospholipids and associated proteins, is synthesized and secreted by the type II cell to lower surface tension in the alveoli and prevent their collapse (2). The type II cell, with its apical Na+ channels and basolateral Na+ pumps, also actively transports ions, thereby establishing and maintaining an osmotic gradient for fluid resorption from the airspaces (3). In concert, the type I cells express the membrane-bound water channel protein aquaporin 5, providing the pathway for water to follow the osmotic gradient out of the alveolus (4).
The type II cells are the progenitors of type I cells in vivo, replacing injured or damaged type I cells, and are capable of proliferating to maintain stable cell populations in the alveolus (5, 6). Similarly, isolated type II cells transdifferentiate into type I cells in vitro, acquiring a type I morphology and phenotype (7, 8). With time in culture, isolated type II cells cease to express type II–specific genes such as surfactant apoproteins, and begin to express type I–specific cell markers such as aquaporin 5 (7). The factors that modulate this phenotypic and functional transition are multiple and complex, and involve the capacity of these cells to respond to their extracellular matrix (ECM) environment and soluble growth factors, cytokines, and hormones (9–14).
The ECM composition of the basement membrane zones (BMZ) that lie beneath these cells has been shown to be distinctly and quantitatively different, with that of the type II being under-sulfated compared with that of the type I (15–17). This observation led to the proposal that such “under-sulfation” could influence, perhaps permitting or promoting, responsiveness to growth factors and thus account in part for their respective phenotypes and division of labor (16–19). In support of this notion, desulfated or otherwise low sulfate–containing ECMs have been shown to promote FGF-1– and FGF-2–stimulated DNA synthesis in isolated type II cells, while heavily sulfated components of ECMs (e.g., heparin) inhibit or reduce this response (18, 19). More recently, FGF-2 gene and protein expression and gene expression of FGF-1 and FGF-R2, but not FGF-R1, have been shown to be down-regulated by heparin (20). These collective observations demonstrate the important role that ECMs and their sulfated groups play in modulating alveolar epithelial cell function.
Not surprisingly, ECMs are also involved in the capacity of type II cells to synthesize and secrete surfactant proteins (SPs) (9, 10, 12, 14, 21–23). Type II cells tend to lose their cuboidal morphology and functional capabilities (i.e., surfactant production/secretion) with time in culture, which can be mitigated by their attachment to floating ECM gels (23), reconstituted basement membranes (12), or preconditioned/synthesized substrata, such as the Engelbreth-Holm-Swarm sarcoma ECM (22). These conditions help support and maintain type II cell morphology and function. These results, in conjunction with evidence that under-sulfated ECMs could further contribute to type II cell proliferation in vitro (18, 19), led us to hypothesize that the heavily sulfated BMZ associated with type I cells could promote the differentiation of type II cells into type I cells and, in the process, diminish or alter surfactant protein gene expression. Soluble high-molecular-weight heparin was used as a model for testing this basic hypothesis, appreciating at the same time that it is equally applicable for modeling the bioactivity of shed cell-surface sulfated proteoglycan ectodomains (24–26) which, in vivo, might also be expected to alter the responsiveness of type II cells in vivo to heparin-binding growth factors, such as FGFs. To address the hypothesis, FGF-1, FGF-2, and FGF-7 were chosen as relevant growth factors due to their known distribution in the lung (27, 28) and capacity to influence SP expression (29–31). The effects of high and low concentrations of heparin and desulfated heparin on type II cell differentiation and response to FGFs were then examined on isolated rat type II cells attached to collagen matrices for 24 h in the presence or absence of FGF-1, FGF-2, or FGF-7.
MATERIALS AND METHODS
Cell Culture and Treatment
Alveolar type II cells were isolated from pathogen-free 200- to 250-g Fischer F-344 CDF rats (Charles River Laboratory, Wilmington, MA) following the procedure of Dobbs (32) with minor modifications (19). All procedures were approved by the North Carolina State University Institutional Animal Care and Use Committee. Freshly isolated type II cells were suspended in a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 nutrient mixture (F12), supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml amphotericin B, and 50 μg/ml gentamicin, and seeded at a density of 1 × 105 cells/cm2 on tissue culture–treated 6-well plates (Costar, Corning, NY) that had been previously coated with 7.5 μg/cm2 type I collagen from rat tail (BD Biosciences, Bedford, MA). Cell cultures were incubated at 37°C in a humidified, 7.5% CO2 atmosphere and allowed to attach to the collagen-coated growth surfaces for a period of 24 h. After the 24-h attachment period in serum-supplemented media, attached cells were washed once with DMEM:F12 (1:1) and then treated in serum-free, hormonally defined medium (SFHDM) as described by Kawada and coworkers (22), but without epidermal growth factor. Briefly, the medium consisted of DMEM and F12 mixed 1:1 (vol/vol) and supplemented with 5 μg/ml insulin, 5 μg/ml transferrin, 1 μM hydrocortisone, 0.5 mM cAMP, 25 nM selenious acid, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml amphotericin B, and 10 μg/ml gentamicin.
Effects of Heparin and FGFs
To determine the effects of heparin and FGFs on surfactant protein and aquaporin 5 gene expression, cells were cultured in SFHDM and treated with FGF-1, FGF-2, or FGF-7 alone and in the presence of increasing concentrations of heparin. Cells were treated with or without the following factors: 5 μg/ml (“low dose”) or 500 μg/ml (“high dose”) of high-molecular-weight heparin (13,500–15,000 MW; Calbiochem Corp., La Jolla, CA), 100 ng/ml FGF-1 (bovine; R&D Systems, Inc., Minneapolis, MN), 100 ng/ml FGF-2 (bovine; R&D Systems, Inc.), or 20 ng/ml FGF-7 (recombinant human; R&D Systems, Inc.). These doses of FGF-1, -2, and -7 were determined in previous studies in our laboratory to be optimal for stimulating DNA synthesis in type II cells. After treatment with FGFs ± heparin in SFHDM for 24 h, cell cultures were terminated by removing the media and lysing the cells with RLT lysis buffer (QIAGEN, Valencia, CA). Cells treated with SFHDM without the addition of heparin or any growth factors served as the control for all relative gene expression determinations.
Effects of the Sulfation Level of Heparin
To determine the effects of the sulfation level of heparin on surfactant protein and aquaporin 5 gene expression, cells were cultured in SFHDM and treated with FGF-1, FGF-2, or FGF-7 alone and in the presence of sulfated or desulfated heparin. Desulfated heparin was obtained by subjecting high-molecular-weight heparin to solvolytic desulfation with dimethyl sulfoxide (33) resulting in > 85% desulfation as previously described (19). Cells were treated with or without the following factors: 500 μg/ml of high-molecular-weight heparin, 500 μg/ml of desulfated high-molecular-weight heparin, 100 ng/ml FGF-1, 100 ng/ml FGF-2, or 20 ng/ml FGF-7. After treatment with FGFs ± heparin/desulfated heparin in SFHDM for 24 h, cell cultures were terminated in RLT as previously described.
Gene Expression Analysis
After lysis with RLT buffer, RNA was isolated using RNeasy Mini-Kits (QIAGEN) with the optional on-column DNase digestion, performed to minimize any genomic DNA carry-over, according to the instructions of the manufacturer. The RNA was eluted in 30 μl RNase-free diH2O and quantified by absorption at 260 nm. Two micrograms of total RNA from each sample was reverse transcribed using cloned AMV reverse transcriptase (Invitrogen, Carlsbad, CA) with oligo dT20 primers according to the instructions of the manufacturer. cDNA samples were diluted 1:5 with RNase-free diH2O and stored at −80°C until analyzed by qRT- PCR. Ten microliters of each diluted cDNA sample was PCR-amplified by TaqMan gene expression primer-probe assays for rat SP-A, SP-B, SP-C, SP-D, AQP-5, and β-actin (Applied Biosystems, Foster City, CA) using the TaqMan Universal PCR Master Mix (Applied Biosystems) and PCR reaction conditions according to the instructions of the manufacturer. Relative gene expression analysis was conducted using the 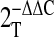 method (34) using β-actin as the reference gene.
method (34) using β-actin as the reference gene.
Statistics
Both experiments were repeated three times. All data were subjected to ANOVA using the general linear models (GLM) procedure in SAS System software (SAS Institute, Inc., Cary, NC). When significance differences among the means were detected in the data set by ANOVA, individual means were compared with Student-Newman-Keuls test using the GLM procedure in SAS software. Differences were considered significant at P < 0.05.
RESULTS
Effects of Heparin and FGFs
SP-B was the most dynamically regulated surfactant protein gene, with mRNA levels increased up to 10-fold compared with control in response to FGF-1/high heparin (Figure 1). There was a clear dose-dependent increase in SP-B mRNA levels with increasing concentration of heparin in FGF-1–supplemented media, while heparin in the absence of any growth factors was without effect on SP-B mRNA levels. The effect of heparin in the presence of FGF-2 or FGF-7 was less clear, but SP-B mRNA levels appeared to exhibit a biphasic response to heparin concentration. SP-B mRNA levels were increased 4-fold by FGF-2/low heparin and FGF-7/low heparin, which were significantly different from controls. In contrast, the high dose of heparin appeared to inhibit the actions of FGF-2 and FGF-7, as these combinations did not result in increased SP-B mRNA levels that were statistically different from controls (Figure 1). Thus, for all three FGFs, the low dose of heparin appeared to potentiate the stimulation of SP-B expression, while the high dose of heparin inhibited the actions of FGF-2 and -7, but further enhanced the action of FGF-1. SP-C mRNA levels were increased up to 2-fold by heparin in a clear dose-dependent manner in all media, while all three FGFs in the absence of heparin were without any specific effect on SP-C mRNA levels (Figure 2). Neither growth factor nor heparin treatment had significant effects on SP-A or SP-D (data not shown). Interestingly, AQP-5 mRNA levels were increased by heparin in a dose-dependent manner in all media, while all three FGFs were without effect on AQP-5 mRNA levels (Figure 3).
Figure 1.

SP-B mRNA abundance in type II cells cultured in serum-free, hormonally-defined media (SFHDM) and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin low concentration (5 μg/ml), or high concentration (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
Figure 2.

SP-C mRNA abundance in type II cells cultured in SFHDM and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin low concentration (5 μg/ml), or high concentration (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
Figure 3.

AQP-5 mRNA abundance in type II cells cultured in SFHDM and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin low concentration (5 μg/ml), or high concentration (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
Effects of the Sulfation Level of Heparin
There was a clear dose-dependent increase in SP-B mRNA levels, up to 7-fold, with increasing sulfation level of heparin in FGF-1–supplemented media, while heparin or desulfated heparin in the absence of any growth factors was without effect on SP-B mRNA levels (Figure 4). SP-B mRNA levels exhibited a biphasic response to the sulfation level of heparin in FGF-2– and FGF-7–supplemented media, as SP-B mRNA levels were increased between 3- and 4-fold by desulfated heparin, but only 2-fold by heparin. Treatment with desulfated heparin resulted in a greater elevation of SP-B mRNA levels in concert with FGF-1, -2, or -7 than resulted from FGF-1, -2, or -7 alone; thus, for all three FGFs, desulfated heparin appeared to potentiate the stimulation of SP-B expression, while sulfated heparin inhibited the actions of FGF-2 and -7, but further enhanced the action of FGF-1. SP-C mRNA levels were increased up to 2-fold by heparin in a sulfation-dependent manner in all media, while none of the FGFs in the absence of heparin or desulfated heparin were had a specific effect on SP-C mRNA levels (Figure 5). Again, SP-A and SP-D mRNA levels exhibited no significant differences from control in response to any of the treatments (data not shown). There was a clear dose-dependent increase in AQP-5 mRNA levels in response to the sulfation level of heparin (Figure 6). Desulfated heparin treatment resulted in an ∼ 2-fold increase in AQP-5 mRNA levels in all media, while fully sulfated heparin increased AQP-5 mRNA levels 3- to 4-fold in all media.
Figure 4.

SP-B mRNA abundance in type II cells cultured in SFHDM and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin (500 μg/ml), or desulfated high-molecular-weight heparin (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
Figure 5.

SP-C mRNA abundance in type II cells cultured in SFHDM and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin (500 μg/ml), or desulfated high-molecular-weight heparin (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
Figure 6.

AQP-5 mRNA abundance in type II cells cultured in SFHDM and treated for 24 h ± FGF-1 (100 ng/ml), FGF-2 (100 ng/ml), FGF-7 (20 ng/ml), high-molecular-weight heparin (500 μg/ml), or desulfated high-molecular-weight heparin (500 μg/ml). Data plotted are means ± SEM, n = 3. Means sharing a letter are not significantly different (P > 0.05).
DISCUSSION
The observation that the regions or microdomains of the BM of the alveolus are quantitatively less sulfated beneath type II compared with type I cells (15–17) led to the proposal that sulfation of ECMs, in their soluble (shed) and insoluble (matrix-bound) forms, are key factors in controlling the responsiveness of alveolar epithelial cells to growth factors (16, 18–20). Evidence in support of this contention has been obtained with in vitro studies, where under-sulfated or desulfated ECMs promote DNA synthesis (18) while fully sulfated ECMs inhibit DNA synthesis (19, 20) as well as specific gene and protein expression (19) in cultured type II cells. It is now reported that the model sulfated ECM component heparin can alter the responsiveness of type II cells to FGF-1, FGF-2, or FGF-7 with differing effects on SP gene expression, and that heparin itself can exert effects, independent of FGFs, on SP-C mRNA levels. In this study, heparin was used as an additive in excess of existing heparan sulfate proteoglycans (HSPGs) in an attempt to model both the heavily sulfated BM of the pulmonary alveolus (6, 16, 17) and shed HSPG ectodomains (24–26). Heparin-like molecules act as low-affinity co-receptors that are required for the binding of FGF-2 to its high-affinity receptor (35, 36). Studies in other cell types have shown that heparin can either facilitate or inhibit responses to growth factors, such as the FGF family of growth factors, that use HSPGs as low-affinity co-receptors for binding to the plasma membrane (37, 38). We therefore predicted that heparin would influence the responsiveness of type II cells to FGFs and alter their expression of SP mRNAs in a manner dependent upon its concentration and sulfation level.
The only significant changes in SP mRNA abundance in the present study were detected for SP-B and SP-C. SP-B and SP-C are large, hydrophobic proteins that facilitate the formation of a phospholipid film at the air–liquid interface and are secreted by regulated exocytosis of lamellar bodies (26); however, mRNA levels of SP-B and SP-C were found to be regulated quite differently in the present study. SP-B mRNA levels were increased by all three FGFs studied, increasing up to 10-fold over control, which is in accord with results from other studies with FGF-1 (7, 39) and KGF/FGF-7 (7, 31, 39) in cultured type II cells. Data from fetal rat type II cells indicates that KGF may enhance SP-B mRNA stability, as the disappearance of SP-B mRNA in the presence of actinomycin D was prevented by KGF (40). When adult rat type II cells were treated with FGFs and heparin for 48 h, SP-B mRNA levels were increased up to 48-fold, likely due to greater accumulation over the longer treatment duration (K. A. Leiner, unpublished results). In contrast, in the present study, levels of SP-C mRNA were not altered by treatment with FGF-1, -2, or -7, but were found to be upregulated by heparin in the present study. Studies by other investigators on the regulation of SP-C mRNA levels by FGF-1 or FGF-7 have yielded conflicting reports. In some studies, FGF-7 was found to increase SP-C mRNA levels (7, 40, 41), while in others FGF-1 and FGF-7 were found to decrease or have no effect on SP-C mRNA levels (14, 39, 41). It seems likely that the varied experimental designs and treatment durations among those studies account for some of the observed variability in the response of SP-C mRNA levels to KGF. For example, when type II cells were co-cultured with lung fibroblasts for 72 h, or when type II cells alone were treated with KGF for 72 h, SP-C mRNA levels were significantly elevated; however, when type II cells were co-cultured with lung fibroblasts for only 24 h, there was no effect on SP-C mRNA levels (41). In the present study, which also examined SP-C mRNA levels at 24 h, FGF-7 had no effect on SP-C mRNA, which may be similar to the results of Shannon and colleagues (41), as fibroblasts express FGF-7 (42).
Heparin was found to influence significantly the actions of FGF-1, FGF-2, and FGF-7 on SP-B mRNA abundance. Heparin potentiated the actions of FGF-2 and FGF-7 at the low concentration, but inhibited the actions of FGF-2 and FGF-7 at the high concentration. The highest levels of SP-B mRNA resulting from FGF-2 or FGF-7 treatment occurred in the presence of either the low dose of heparin or the high dose of desulfated heparin. This indicates that the sulfation level of heparin is critical to its interaction with FGF-2 or FGF-7, and since the desulfation of heparin is not complete (> 85% desulfation), the high dose of desulfated heparin may have approximated the sulfation level of the low dose of heparin. The high concentration of fully-sulfated heparin had no such stimulatory effect on SP-B mRNA in combination with FGFs 2 and 7, and in fact caused some inhibition of these growth factors' stimulation of SP-B. Low concentrations of heparin have been reported to enhance the binding of FGF-2 (37) and FGF-7 (38) to their receptors, while high concentrations of heparin were reported to be inhibitory (37, 38). Interestingly, heparin was found to potentiate the stimulatory actions of FGF-1 on SP-B mRNA levels in a dose-dependent manner at both concentrations of heparin tested, with the highest levels of SP-B mRNA resulting from FGF-1 in the presence of the high heparin concentration. Again, this effect was related to the sulfation level of heparin, as the high dose of desulfated heparin was very similar to the low dose of heparin in its ability to potentiate the actions of FGF-1 on SP-B mRNA levels, which were intermediate between the FGF-1 response in the absence of heparin and that with high heparin. It is possible that this differential modulation of the actions of FGF-1, FGF-2, or FGF-7 by heparin may be due to the differences in the receptors that bind each ligand, as FGF-1 and FGF-2 can exert their actions through multiple FGF receptor (FGFR) types, while FGF-7 binds only to a single form of the receptor that is uniquely expressed in epithelial cells, FGFR-2, IIIb (KGFR) (43). However, LaRochelle and coworkers (38) demonstrated that high heparin concentrations inhibited the binding and signaling of FGF-7 at the KGFR, but did not inhibit the binding and signaling of FGF-1 (which also binds to KGFR) in Chinese hamster ovary cells that were transfected with a KGFR-encoding cDNA. This is in accord with results presented here with the type II cell, indicating a dose-dependent increase in SP-B mRNA with increasing heparin concentration/sulfation level and FGF-1, and a biphasic response with increasing heparin concentration/sulfation level and FGF-7.
Heparin was also found to exert effects on SP-C gene expression independent of FGFs. The highest levels of SP-C mRNA resulted from the high concentration of heparin, and were not influenced by the presence or absence of FGFs in the media. As discussed above, this effect was dependent on the presence of sulfate groups on heparin. The high concentration of desulfated heparin yielded a very similar response to the low concentration of fully sulfated heparin, which was intermediate between no heparin and the high concentration of heparin treatments. Perhaps relatedly, the culture of Embryonic Day 13 lung explants in the presence of sodium chlorate, an inhibitor of proteoglycan (PG) sulfation, resulted in decreased expression of SP-C mRNA (44). SP-C expression was reduced by 80% in intact lung tips, and by 95% or more in lung mesenchyme/lung epithelium recombinant cultures (44). These data underscore the importance of sulfated PGs not only to normal lung development, but also to the induction of SP-C. But if SP-C expression, a marker for type II cell phenotype, is increased by exposure to heparin alone as in the current study, how does heparin, as a model for sulfated ECMs, also induce the predicted shift in phenotype toward the type I cell? One possibility is that the model itself is flawed by the likelihood that soluble heparin may interact with cell surfaces differently than insoluble (immobilized) forms. Previous evidence, however, suggested that adsorbed (immobilized) forms of heparin, heparan sulfate, and dermatan sulfate behaved identically to the soluble forms in DNA synthesis assays on isolated type II cells (18–20). These studies were done in the presence of various growth factors, whose signaling pathways are typically sensitive to heparin, and heparin in the absence of growth factor resulted in no visible changes in the signaling pathways examined (45). In the absence of growth factors it is possible that heparin signals via Toll-like receptors (TLR2 and/or 4), which have been shown to be activated by molecules related to heparin—hyaluronan (46), biglycan (47), or heparan sulfate (48)—which, in turn, activate the proinflammatory transcription factor NF-κB in macrophages and dendritic cells of the lung. This intriguing notion, however, should be tempered by the fact that these biologic responses may differ according to the degree and nature of the sulfation as well as the molecule itself (i.e., dermatan sulfate versus chondroitin sulfate versus heparin) (48), and only SP-D is increased in inflammation while SP-A, SP-B, and SP-C are decreased (49). Alternatively, heparin is known to gain access to the cytoplasm by high-affinity binding sites and endocytosis (50) or specific receptor (51), wherein it could modulate multiple signaling pathways. In spite of the lack of clarity on this specific issue, these collective observations serve to emphasize the need for more attention to the important role that ECMs in their various forms (sulfated/unsulfated, soluble/insoluble) play, directly or as co-factors, in modifying specific gene expression in the lung during development, homeostatic maintenance, and in response to injury or disease.
Aquaporin-5 mRNA expression was also examined to track the phenotypic changes in cultured alveolar epithelial cells in response to treatment with heparin and FGFs, as they transdifferentiate from type II cells into type I cells (4, 7). Borok and coworkers (7) demonstrated that AQP-5 mRNA expression increased with time in culture, while mRNAs for SP-B and SP-C decreased, as type II cells transitioned to a type I phenotype. These changes could be prevented and partially reversed by KGF, indicating that KGF plays a major role in modulating the alveolar epithelial cell phenotype (7). The data presented here indicate that sulfated components of the ECM, as modeled by heparin, also play an important role in modulating alveolar epithelial cell phenotype. Heparin was found to significantly upregulate AQP-5 expression, as much as 3-fold over control, in a dose-dependent and sulfate-dependent manner. We therefore conclude that sulfated heparin promoted the differentiation of type II cells into type I cells, as might be expected based on the in situ characteristics of the BMZ beneath type I and type II cells in the alveolus (15–17). This also makes sense in light of previous results indicating that heparin inhibits DNA synthesis and cell proliferation in cultured type II cells (19, 20), which may be losing their proliferative capacity as they acquire a type I phenotype. During the short culture duration employed in this study, alveolar epithelial cells retained the ability to respond to FGFs and heparin with increased SP mRNA abundance, and it is particularly interesting that heparin upregulated both AQP-5 and SP-C mRNAs, although the reason for this is uncertain but may reflect an intermediate stage of differentiation. Thus, sulfated ECMs, as modeled by heparin, play a major role in modulating alveolar epithelial cell phenotype as well as the actions of FGFs on the expression of SP-B and SP-C.
In conclusion, SP-B and SP-C mRNA levels are differentially regulated by heparin and FGF-1, FGF-2, and FGF-7. Low concentrations of heparin enhanced FGF-stimulated expression SP-B mRNA, while high concentrations further potentiated the actions of FGF-1 on SP-B, but inhibited the actions of FGF-2 and FGF-7. Desulfated heparin also enhanced FGF-stimulated expression of SP-B mRNA, similar to the effect observed with the low concentration of fully sulfated heparin. While all three FGFs lacked any effect on SP-C mRNA levels, heparin itself upregulated SP-C mRNA and AQP-5 mRNA levels in a dose-dependent and sulfation-dependent manner. These data indicate that heparin-like molecules can be either stimulatory or inhibitory of SP-B gene expression depending on concentration, degree of sulfation, and surrounding FGF-containing microenvironment, and that heparin-like molecules play a significant role in modulating alveolar epithelial cell phenotype, as may be relevant in the context of sulfation levels of BMZ microdomains or the shedding of syndecan-1 and syndecan-4 ectodomains into the extracellular fluid.
This study was supported by Public Health Service Grant HL-44497 and a grant from the State of North Carolina.
Originally Published in Press as DOI: 10.1165/rcmb.2006-0159OC on June 22, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respir Dis 1983;128:S42–S46. [DOI] [PubMed] [Google Scholar]
- 2.Wright JR, Dobbs LG. Regulation of pulmonary surfactant secretion and clearance. Annu Rev Physiol 1991;53:395–414. [DOI] [PubMed] [Google Scholar]
- 3.Mason RJ, Williams MC, Widdicombe JH, Sanders MJ, Misfeldt DS, Berry LC Jr. Transepithelial transport by pulmonary alveolar type II cells in primary culture. Proc Natl Acad Sci USA 1982;79:6033–6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobbs LG, Gonzalez R, Matthay MA, Carter EP, Allen L, Verkman AS. Highly water-permeable type I alveolar epithelial cells confer high water permeability between the airspace and vasculature in rat lung. Proc Natl Acad Sci USA 1998;95:2991–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adamson IY, Bowden DH. The type 2 cell as progenitor of alveolar epithelial regeneration: a cytodynamic study in mice after exposure to oxygen. Lab Invest 1974;30:35–42. [PubMed] [Google Scholar]
- 6.Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp Mol Pathol 1975;22:142–150. [DOI] [PubMed] [Google Scholar]
- 7.Borok Z, Lubman RL, Danto SI, Zhang XL, Zabski SM, King LS, Lee DM, Agre P, Crandall ED. Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin-5. Am J Respir Cell Mol Biol 1998;18:554–561. [DOI] [PubMed] [Google Scholar]
- 8.Danto SI, Shannon JM, Borok Z, Zabski SM, Crandall ED. Reversible transdifferentiation of alveolar epithelial cells. Am J Respir Cell Mol Biol 1995;12:497–502. [DOI] [PubMed] [Google Scholar]
- 9.Adamson IY, King GM, Young L. Influence of extracellular matrix and collagen components on alveolar type 2 cell morphology and function. In Vitro Cell Dev Biol 1989;25:494–502. [DOI] [PubMed] [Google Scholar]
- 10.Cott GR, Walker SR, Mason RJ. The effect of substratum and serum on the lipid synthesis and morphology of alveolar type II cells in vitro. Exp Lung Res 1987;13:427–447. [DOI] [PubMed] [Google Scholar]
- 11.McMillan EM, King GM, Adamson IY. Sex hormones influence growth and surfactant production in fetal lung explants. Exp Lung Res 1989;15:167–179. [DOI] [PubMed] [Google Scholar]
- 12.Shannon JM, Emrie PA, Fisher JH, Kuroki Y, Jennings SD, Mason RJ. Effect of a reconstituted basement membrane on expression of surfactant apoproteins in cultured adult rat alveolar type II cells. Am J Respir Cell Mol Biol 1990;2:183–192. [DOI] [PubMed] [Google Scholar]
- 13.Shannon JM, Mason RJ, Jennings SD. Functional differentiation of alveolar type II epithelial cells in vitro: effects of cell shape, cell-matrix interactions and cell-cell interactions. Biochim Biophys Acta 1987;931:143–156. [DOI] [PubMed] [Google Scholar]
- 14.Sugahara K, Mason RJ, Shannon JM. Effects of soluble factors and extracellular matrix on DNA synthesis and surfactant gene expression in primary cultures of rat alveolar type II cells. Cell Tissue Res 1998;291:295–303. [DOI] [PubMed] [Google Scholar]
- 15.Grant MM, Cutts NR, Brody JS. Alterations in lung basement membrane during fetal growth and type 2 cell development. Dev Biol 1983;97:173–183. [DOI] [PubMed] [Google Scholar]
- 16.Sannes PL. Differences in basement membrane-associated microdomains of type I and type II pneumocytes in the rat and rabbit lung. J Histochem Cytochem 1984;32:827–833. [DOI] [PubMed] [Google Scholar]
- 17.Van Kuppevelt TH, Domen JG, Cremers FP, Kuyper CM. Staining of proteoglycans in mouse lung alveoli. I. Ultrastructural localization of anionic sites. Histochem J 1984;16:657–669. [DOI] [PubMed] [Google Scholar]
- 18.Sannes PL, Khosla J, Cheng PW. Sulfation of extracellular matrices modifies responses of alveolar type II cells to fibroblast growth factors. Am J Physiol 1996;271:L688–L697. [DOI] [PubMed] [Google Scholar]
- 19.Sannes PL, Khosla J, Li CM, Pagan I. Sulfation of extracellular matrices modifies growth factor effects on type II cells on laminin substrata. Am J Physiol 1998;275:L701–L708. [DOI] [PubMed] [Google Scholar]
- 20.Li CM, Newman D, Khosla J, Sannes PL. Heparin inhibits DNA synthesis and gene expression in alveolar type II cells. Am J Respir Cell Mol Biol 2002;27:345–352. [DOI] [PubMed] [Google Scholar]
- 21.Edelson JD, Shannon JM, Mason RJ. Effects of two extracellular matrices on morphologic and biochemical properties of human type II cells in vitro. Am Rev Respir Dis 1989;140:1398–1404. [DOI] [PubMed] [Google Scholar]
- 22.Kawada H, Shannon JM, Mason RJ. Improved maintenance of adult rat alveolar type II cell differentiation in vitro: effect of serum-free, hormonally defined medium and a reconstituted basement membrane. Am J Respir Cell Mol Biol 1990;3:33–43. [DOI] [PubMed] [Google Scholar]
- 23.Shannon JM, Jennings SD, Nielsen LD. Modulation of alveolar type II cell differentiated function in vitro. Am J Physiol 1992;262:L427–L436. [DOI] [PubMed] [Google Scholar]
- 24.Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Biol 2000;148:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med 1998;4:691–697. [DOI] [PubMed] [Google Scholar]
- 26.Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J Biol Chem 1997;272:14713–14720. [DOI] [PubMed] [Google Scholar]
- 27.Panos RJ, Rubin JS, Csaky KG, Aaronson SA, Mason RJ. Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J Clin Invest 1993;92:969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sannes PL, Burch KK, Khosla J. The immunohistochemical localization of epidermal growth factor and acidic and basic fibroblast growth factors in post-natal developing and adult rat lungs. Am J Respir Cell Mol Biol 1992;7:230–237. [DOI] [PubMed] [Google Scholar]
- 29.Cardoso WV, Itoh A, Nogawa H, Mason I, Brody JS. FGF-1 and FGF-7 induce distinct patterns of growth and differentiation in embryonic lung epithelium. Dev Dyn 1997;208:398–405. [DOI] [PubMed] [Google Scholar]
- 30.Matsui R, Brody JS, Yu Q. FGF-2 induces surfactant protein gene expression in foetal rat lung epithelial cells through a MAPK-independent pathway. Cell Signal 1999;11:221–228. [DOI] [PubMed] [Google Scholar]
- 31.Xu X, McCormick-Shannon K, Voelker DR, Mason RJ. KGF increases SP-A and SP-D mRNA levels and secretion in cultured rat alveolar type II cells. Am J Respir Cell Mol Biol 1998;18:168–178. [DOI] [PubMed] [Google Scholar]
- 32.Dobbs LG. Isolation and culture of alveolar type II cells. Am J Physiol 1990;258:L134–L147. [DOI] [PubMed] [Google Scholar]
- 33.Nagasawa K, Inoue Y, Kamata T. Solvolytic desulfation of glycosaminoglycuronan sulfates with dimethyl sulfoxide containing water or methanol. Carbohydr Res 1977;58:47–53. [DOI] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 35.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 1991;252:1705–1708. [DOI] [PubMed] [Google Scholar]
- 36.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991;64:841–848. [DOI] [PubMed] [Google Scholar]
- 37.Fannon M, Forsten KE, Nugent MA. Potentiation and inhibition of bFGF binding by heparin: a model for regulation of cellular response. Biochemistry 2000;39:1434–1445. [DOI] [PubMed] [Google Scholar]
- 38.LaRochelle WJ, Sakaguchi K, Atabey N, Cheon HG, Takagi Y, Kinaia T, Day RM, Miki T, Burgess WH, Bottaro DP. Heparan sulfate proteoglycan modulates keratinocyte growth factor signaling through interaction with both ligand and receptor. Biochemistry 1999;38:1765–1771. [DOI] [PubMed] [Google Scholar]
- 39.Sugahara K, Rubin JS, Mason RJ, Aronsen EL, Shannon JM. Keratinocyte growth factor increases mRNAs for SP-A and SP-B in adult rat alveolar epithelial cells in culture. Am J Physiol 1995;269:L344–L350. [DOI] [PubMed] [Google Scholar]
- 40.Chelly N, Mouhieddine-Gueddiche OB, Barlier-Mur AM, Chailley-Heu B, Bourbon JR. Keratinocyte growth factor enhances maturation of fetal rat lung type II cells. Am J Respir Cell Mol Biol 1999;20:423–432. [DOI] [PubMed] [Google Scholar]
- 41.Shannon JM, Pan T, Nielsen LD, Edeen KE, Mason RJ. Lung fibroblasts improve differentiation of rat type II cells in primary culture. Am J Respir Cell Mol Biol 2001;24:235–244. [DOI] [PubMed] [Google Scholar]
- 42.Adamson IY, Young L. Alveolar type II cell growth on a pulmonary endothelial extracellular matrix. Am J Physiol 1996;270:L1017–L1022. [DOI] [PubMed] [Google Scholar]
- 43.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000;7:165–197. [DOI] [PubMed] [Google Scholar]
- 44.Shannon JM, McCormick-Shannon K, Burhans MS, Shangguan X, Srivastava K, Hyatt BA. Chondroitin sulfate proteoglycans are required for lung growth and morphogenesis in vitro. Am J Physiol 2003;285:L1323–L1336. [DOI] [PubMed] [Google Scholar]
- 45.Newman DR, Li C-M, Simmons R, Khosla J, Sannes PL. Heparin affects signaling pathways stimulated by FGF-1 and FGF-2 in type II cells. Am J Physiol Lung Cell Mol Physiol 2004;287:L191–L200. [DOI] [PubMed] [Google Scholar]
- 46.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005;11:1173–1179. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 2005;115:2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol 2002;168:5233–5239. [DOI] [PubMed] [Google Scholar]
- 49.Fujita M, Shannon JM, Ouchi H, Voelker DR, Nakanishi Y, Mason RJ. Serum surfactant protein D is increased in acute and chronic inflammation in mice. Cytokine 2005;31:25–33. [DOI] [PubMed] [Google Scholar]
- 50.Castellot JJ Jr, Wong K, Herman B, Hoover RL, Albertini DF, Wright TC, Caleb BL, Karnovsky MJ. Binding and internalization of heparin by vascular smooth muscle cells. J Cell Physiol 1985;124:13–20. [DOI] [PubMed] [Google Scholar]
- 51.Savage JM, Gilotti AC, Granzow CA, Molina F, Lowe-Krentz LJ. Antibodies against a putative heparin receptor slow cell proliferation and decrease MAPK activation in vascular smooth muscle cells. J Cell Physiol 2001;187:283–293. [DOI] [PubMed] [Google Scholar]