

Abstract
A family of nanoscale-sized supramolecular cage compounds with a polyhedral framework is prepared by self-assembly from tritopic building blocks and rectangular corner units via noncovalent coordination interactions. These highly symmetrical cage compounds are described as face-directed, self-assembled truncated tetrahedra with Td symmetry.
The number of ways in which more than three identical multitopic subunits can be regularly arranged on the surface of a sphere is represented by the Platonic and Archimedean solids. Platonic solids embody a family of five convex uniform polyhedra that are made of the same regular polygons, whereas the family of Archimedean solids consists of 13 convex uniform polyhedra that are made of at least two different regular polygons (1). The three coordinate directions for the Platonic as well as for the Archimedean solids are equivalent, thus making them ideal models for the construction of spherical molecular hosts (2).
Covalently bonded cage compounds such as cyclophanes (3), cubanes (4, 5), adamantane (6–8), and dodecahedrane (9, 10) are difficult to synthesize and usually give low yields. Nature on the other hand demonstrates the ability of biological systems to form large supramolecular arrays from small building blocks, giving rise to a wide variety of structures and functions (11–13). Utilization of this biomimetic motive in the synthesis of larger synthetic entities provides an alternative to classical organic synthesis, offering the advantage of self-correction and a highly convergent synthetic protocol (14–16).
Trying to imitate and use this biological motive for potential applications in chemistry and materials science, chemists apply slightly different approaches. Besides the use of hydrogen-bonded systems, the coordination-driven self-assembly of supramolecular species has become an important method (17–24), because it allows for the incorporation of metal centers of various geometries and functionalities. Furthermore, the thermodynamic and kinetic stability of transition metal complexes can have a considerable advantage over pH- and temperature-sensitive biological systems.
Although this coordination-driven methodology has been widely used in the formation of self-assembled infinite networks, grids, and helical species (25), only a few discrete three-dimensional supramolecular entities have been reported to date. Most systems described in the literature are tetrahedral M4L6 (26–29) or cubic M8L8 ensembles (30, 31) that have the shape of Platonic solids. For the larger and more complex Archimedean solids only a few examples are known (32–37). Such discrete three-dimensional supramolecular species possess large void cavities and are therefore important in studies on molecular recognition, inclusion phenomena, and catalysis, because they provide an ideal environment for the encapsulation of smaller molecules or for stabilizing reactive species (38–40). Therefore, we now report the formation and characterization of diverse truncated tetrahedra, derived from the face-directed self-assembly of the tritopic tris(pyridylethynyl)benzene or tris(p-cyanophenylethynyl)benzene linkers with cis-platinum and cis-palladium bistriflate salts. In addition, we report the formation of the complementarily self-assembled system via reaction of cis-bis(p-pyridyl)porphyrin with a tritopic trisplatinum linker.
Experimental Section
General Methods.
IR spectra were recorded on a Mattson Polaris Fourier transform–IR spectrometer (Mattson Instruments, Madison, WI). UV-Vis spectra were obtained by using a Hewlett–Packard 8452A UV-Vis spectrophotometer. NMR spectra were recorded on a Varian XL-300 spectrometer. The 1H NMR spectra were recorded at 300 MHz (CDHCl2 δ 5.32 or CD2HNO2 δ 4.33 ppm). The 13C{1H} NMR spectra were recorded at 75 MHz (CD2Cl2 δ 54.0 or CD3NO2 δ 62.8 ppm). The 19F NMR spectra were recorded at 282 MHz (CFCl3 δ 0.0 ppm). The 31P{1H} NMR spectra were recorded at 121 MHz (external 85% aqueous H3PO4 δ 0.0 ppm). Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Mass spectra of 5, 7, 11, 12 and 14 were analyzed with a Micromass (Manchester, U.K.) Quatro II with ionization performed under electrospray conditions (flow rate: 7.7 μl/min; capillary voltage: 3.0 kV; cone: 50 V; extractor: 9 V). About 15 individual scans were averaged for the mass spectra. The calibration of the mass range of 500 to 4,000 atomic mass units was done with a 1:1 mixture of an isopropanol/H2O solution of NaI (2 μg/μl) and CsI (0.01 μg/μl). Samples were prepared as 20 pmol/μl solutions in acetone/CH2Cl2 1:1 just before the analysis.
Materials.
Commercial reagents were American Chemical Society reagent grade or higher and were used without further purification.
1,3,5-Tris(4′-pyridylethynyl)benzene, 1 (41); 1,3,5-tris (4′-cyanophenylethynyl)benzene, 2 (42); (dppf)Pd(H2O)2(OTf)2, 3 (43), (dppf)Pt(H2O)2(OTf)2, 4 (43); (Et3P)2Pd(H2O)2(OTf)2, 9 (44); (Et3P)2Pt(H2O)2(OTf)2, 10 (44); 1,3,5-tris[(trans-(PEt3)2Pt(OTf))ethynyl]benzene, 14 (45); and 5,10-di (4′-pyridyl)-15,20-diphenylporphyrin, 13 (46) were prepared according to published procedures.
General Procedure for the Formation of Truncated Tetrahedra 5–8, 11, and 12.
To a solution of the appropriate Pt or Pd acceptor unit 3, 4, 9 or 10 was added the appropriate planar donor unit 1 or 2 in a 3:2 ratio, and the product was crystallized as described in the experimental details that are published as supplementary material on the PNAS web site (www.pnas.org).
[(dppf)Pd(OTf)2]]6[1,3,5-Tris(4′-Pyridylethynyl)Benzene]4 (5).
Yield: 16.8 mg (88%). Mp = 172–180 (dec.). For spectral and physical data see the supplementary material.
[(dppf)Pd(OTf)2]6[1,3,5-Tris(4′-Cyanophenylethynyl)Benzene]4 (6).
Deep red solid. Yield: 11.9 mg (87%). Mp = 203–205 (dec.). For spectral and physical data see the supplementary material.
[(dppf)Pt(OTf)2]6[1,3,5-Tris(4′-Pyridylethynyl)Benzene]4 (7).
Yellow solid. Yield: 11.7 mg (82%). Mp = 187–190 (dec.). For spectral and physical data see the supplementary material.
[(dppf)Pt(OTf)2]6[1,3,5-Tris(4′-Cyanophenylethynyl)Benzene]4 (8).
Diamond-shaped orange crystals. Yield: 11.1 mg (75%). Mp = 224–227 (dec.). For spectral and physical data see the supplementary material.
[(Et3P)2Pd(OTf)2]6[1,3,5-Tris(4′-Pyridylethynyl)Benzene]4 (11).
Yield: 24.8 mg (89%). Mp = 168–170 (dec.). For spectral and physical data see the supplementary material.
[(Et3P)2Pt(OTf)2]6[1,3,5-Tris(4′-Pyridylethynyl)Benzene]4 (12).
White crystalline solid. Yield: 24.1 mg (87%). Mp = 254–259 (dec.). For spectral and physical data see the supplementary material.
[5,10-Di(4′-Pyridyl)-15,20-Diphenylporphyrin]6[1,3,5-Tris[(trans-(PEt3)2 Pt(OTf))Ethynyl]Benzene]4 (15).
To a solution of 9.8 mg (0.016 mmol) of 13 in 5 ml of CH2Cl2 was slowly added a solution of 20.0 mg (0.011 mmol) of 14 in a mixture of CHCl3 and CH3OH (3:1, 10 ml). The resulting mixture was stirred for 10 h at room temperature. Recrystallization gave a deep red solid. Yield: 26 mg (87%). Mp = 247–250 (dec.). For spectral and physical data see the supplementary material.
Results and Discussion
Although biological systems are formed by many weak hydrogen bonding and van der Waals interactions, metal-ligand bonding interactions are stronger and highly directional. Therefore, the structural and functional characteristics of metal containing self-assembled supramolecular entities depend highly on the symmetry and functionality of the individual building blocks. In addition, entropic factors favor the formation of polyhedra with a minimum number of subunits, because the entropy loss is significantly smaller than for larger entities or oligomers (47, 48). The modular self-assembly approach with defined rigid subunits allows for a rational design of discrete supramolecular entities (Fig. 1), while at the same time offering a wide variety of combinatorial assemblies based on a library of appropriate building blocks (17–24). Thus, for example, combination of four tritopic 60° building blocks with six linear linkers gives rise to the formation of a tetrahedron whereas combination of the same 60° building blocks with a 109° angular linker gives rise to a trigonal bipyramid.
Figure 1.

Molecular polyhedra by combination of tritopic and ditopic subunits.
The shape of a truncated tetrahedron, the smallest Archimedean solid, is formally derived from a tetrahedron by capping the four corners (Fig. 2a). Based on our design strategy, compounds resembling truncated tetrahedra can be made via face directed self-assembly of planar tritopic 120° subunits with angular ditopic 90° corner units (Fig. 2b). The ratio of faces to corners must be 2:3 with a total number of 10 subunits to obtain a closed structure.
Figure 2.

(a) Derivation of a truncated tetrahedron from a tetrahedron. (b) Self-assembly process of a tritopic ligand (red) with an angular metal bis(triflate) (blue) to form a truncated tetrahedron.
Addition of the tris(pyridyl) ligand 1 or the tris(cyano) ligand 2, dissolved in CH2Cl2, to a CH2Cl2 or nitromethane solution of the angular palladium or platinum bis(triflates) 3 or 4 in a 2:3 ratio, respectively, resulted in the quantitative formation of single products. The palladium compounds 5 and 6 gave pink microcrystalline solids, whereas the corresponding platinum complexes 7 and 8 gave stable yellow solids (Scheme S1). In a similar fashion, reaction of the nonchelated bis(triethylphosphino) substituted metal bis(triflates) 9 and 10 with 1,3,5-tris(4′-pyridylethynyl)benzene (1) yielded the analogous metal complexes 11 and 12 as white crystalline powders (Scheme S2). Although the ferrocenyl-substituted systems were remarkably soluble in organic solvents, such as CH2Cl2 or acetone, the bis(triethylphosphino)-substituted systems were soluble only in more polar solvents.
Scheme 1.

Scheme 2.

These polyhedral compounds have been fully characterized by analytical and spectroscopic means as described in the Experimental Section. In particular, all new complexes gave elemental analyses that are consistent with the proposed structures. Analysis of the NMR spectra of compounds 5-8 and 11-12 revealed the highly symmetrical nature of these complexes. The six compounds showed characteristic high field-shifted sharp singlets in the 31P-NMR spectra with appropriate 195Pt satellites for 7, 8, and 12. For the pyridyl-coordinated palladium systems 5 and 11 the observed high field shifts were quite large (Δδ = 17.1 ppm and Δδ = 19.2), whereas the shifts observed for the platinum compounds 7, 8, and 12 as well as for the cyano-coordinated system 6 (Δδ = 5.1, 0.9, 4.1, and 5.0 ppm) were much smaller. Also diagnostic for the end-on coordination of the organic linkers were the respective 1H and 13C{1H} NMR spectra. Besides the characteristic signals for the bis(diphenylphosphino)ferrocenyl or bis(triethylphosphino) ligands in the proton NMR, the two sets of aromatic resonances for the cyanobenzene units of 6 and 8 at δ = 7.58 and 7.59 ppm, respectively, were virtually identical and partially overlapping with other aromatic signals. For the pyridyl-coordinated complexes 5, 11, and 12 the protons in the α position to the coordinating nitrogen were significantly shifted downfield (0.1–0.9 ppm), whereas the corresponding α proton of 7 was shifted up field by about 0.2 ppm. In the case of the bis(triethylphosphino)-substituted complexes 11 and 12 a considerable downfield shift of 0.4 ppm was observed for the protons in the β position, whereas for the bis(diphenylphosphino)ferrocenyl substituted complexes 6 and 7 a high-field shift of 0.4 and 0.23 ppm, respectively, was observed as a result of shielding provided by the phenyl rings of the ferrocenyl ligands. The 19F NMR spectra for all six complexes displayed sharp singlets in the region of −79 to −77 ppm, which is characteristic for free triflate anions. No indication for the inclusion of triflate ions inside the tetrahedral cavity of these complexes was found on the 19F NMR time scale (Table 1).
Table 1.
Chemical shifts of important signals in the NMR spectra of compounds 5–8, 11, 12, and 15
| Compound |
31P 1H NMR
|
1H NMR
|
19F NMR
|
||
|---|---|---|---|---|---|
| δ, ppm | Δδ, ppm | δ, ppm | Δδ, ppm | δ, ppm | |
| 5 | 34.1 | −17.0 | 8.70 (py-Hα) | 0.08 | −77.7 |
| 7.02 (py-Hβ) | −0.40 | ||||
| 6 | 46.1 | −5.0 | — | — | −77.7 |
| 7 | 6.0 | −5.1 | 8.42 (py-Hα) | −0.20 | −77.5 |
| 7.19 (py-Hβ) | −0.23 | ||||
| 8 | 10.2 | −0.9 | — | — | −77.7 |
| 11 | 30.9 | −25.1 | 8.96 (py-Hα) | 0.34 | −77.6 |
| 7.73 (py-Hβ) | 0.31 | ||||
| 12 | 5.3 | −4.1 | 9.32 (py-Hα) | 0.9 | −79.4 |
| 7.84 (py-Hβ) | 0.4 | ||||
| 15 | 19.8 | −1.8 | 9.20 (py-Hα) | 0.14 | −73.3 |
| 8.64 (py-Hβ) | 0.46 | ||||
Taking advantage of the modularity of our synthetic approach, formation of a complementary truncated tetrahedron was accomplished by an analogous face-directed assembly of cis-bis(p-pyridyl)porphyrin 13 with the tris(platinum)triflate 14. Unlike self-assemblies 5–12, the organic bis(pyridyl) compound acts as the corner unit and the metal triflate acts as the planar tritopic building block. Thus, reaction of four equivalents of 14 with six equivalents of 13 in a mixture of CH3Cl and MeOH gave the porphyrin-containing complex 15 in good yield, which was isolated as a deep red microcrystalline solid (Scheme S3). In analogy to complexes 5-12, the formation of a single highly symmetrical compound was observed by NMR. The 31P NMR exhibited a sharp singlet at δ = 19.8 ppm with corresponding 195Pt satellites. Coordination of the pyridyl ligand of 13 was indicated by a 5.8 ppm high-field shift of the phosphorus resonance in 15. A corresponding and quite characteristic strong deshielding effect also was observed in the proton NMR for both the α and β protons of the pyridyl ligand, giving rise to an AA′BB′-spin system at δ = 9.20 and 8.64 ppm, respectively. All other chemical shifts and the integrations of the proton signals were in accordance with the requirements for 15.
Scheme 3.

A noteworthy feature of complexes 5-8, 11, and 12 is the relatively high number of water molecules occluded in the solid state. Even upon heating at 60°C under vacuum for several days the water could not be removed. We therefore conclude that the water is likely to be interacting with the cationic platinum via the oxygen lone pairs and moreover it fills the void space inside the three-dimensional assembly to stabilize the polyhedral structure, very similar to the host-guest interaction observed by Fujita et al. (33, 34, 36).
The composition of these complexes was determined by electrospray MS, which allows for the identification of larger aggregates and assemblies up to 10,000 a.u. (49). As an example, we discuss the spectrum of complex 7 in a 1:1 mixture of acetone and CH2Cl2. The isotopically unresolved signals centered at m/z = 3,756, 2,454, 1,804, 1,413, and 1,153 correspond to the (M − 2 OTf)2+, (M − 3 OTf)3+, (M − 4 OTf)4+, (M − 5 OTf)5+, and (M − 6 OTf)6+ species, respectively, resulting from the loss of the appropriate number of triflate counterions. Fig. 3 shows an expansion plot of the 3+ charge-state of complex 7 along with the theoretical isotopic distribution of the predicted elemental composition of a 3+ molecular ion less three triflate counterions. These data provide an experimental molecular weight of 7,808.85, which agrees well with the theoretical molecular weight of 7,806.33, and therefore are in accord with the proposed composition of the truncated tetrahedral structure of complex 7. Fragmentation of 7 also was observed with the major fragments consisting of different combinations of the precursors 1 and 4. Although all complexes with a pyridyl framework gave consistent electrospray MS data, the signals for the palladium complexes were less intense compared with the platinum complexes. In contrast, no signals at all were observed for the cyano-coordinated complexes 6 and 8, probably because of the much weaker late transition metal-nitrile bond energies (44), which likely decreased the lifetime of the charged species in the mass spectrometer because of dissociation under the harsh conditions.
Figure 3.

Electrospray MS of the truncated tetrahedron 7, theoretical (Upper) and experimental (Lower) isotopic distribution of the (M-3OTf)3+ charge state.
An estimate of the size of the polyhedral complexes was derived from Extensible Systematic Force Field calculations (50) performed for the palladium complex 6. The energy minimized structure showed a molecule with overall Td symmetry and N-Pd-N bond angles for the square planar metal corners of 83.4° (Fig. 4). Taking the van der Waals radii into account, the interior diagonal Pd-Pd distances were calculated to be 21.7 Å and the Pd-Pd distances for the triangular facial windows as 14.2 Å. From a space-filling model an approximate diameter of 35 Å was derived for the overall size of this truncated tetrahedron.
Figure 4.

Space-filling model of the truncated tetrahedron 6 derived from Extensible Systematic Force Field simulations by using the insight ii 97.0 software package (50).
For the ferrocenyl-substituted polyhedra 5-8, the self-assembly reactions also were accompanied by a change in the absorbance spectra. When the donor ligands 1 or 2 replaced the weakly donating water ligands, a series of blue shifts in the UV/Vis spectra were observed (Fig. 5). Although for the less strongly binding unit 2 these effects were weak, the spectra of the 1,3,5-tris(4′-pyridylethynyl)benzene (1)-coordinated systems nevertheless showed significant changes. The insets in Fig. 5 show a superposition of the UV spectra of the bis(triflate) palladium complex 3 and the cyano-linked complex 6 (left) as well as the pyridyl-linked complex 5 (right). The coordination of the strongly donating pyridyl ligand resulted in a significant blueshift of the band around 550 nm, presumably a CT band, and in a complete quenching of absorption at 380 nm, presumably the palladium d-d transition (43). An analogous behavior also was observed for the platinum complexes 7 and 8, but was much less pronounced. For all four compounds, the absorption spectra are dominated by the absorption of the organic linker with an additive increase of the molar extinction coefficient, especially for the palladium compounds 5 and 6, compared with the free ligands 1 and 2.
Figure 5.
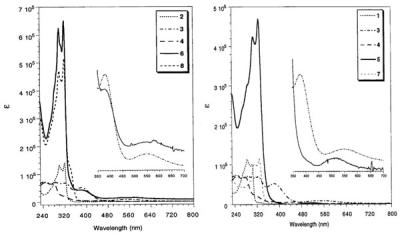
Absorption spectra of the cyano-linked complexes 6 and 8 (Left) and the pyridyl-linked complexes 5 and 7 (Right).
Conclusions
A variety of bis(diphenylphosphino)ferrocene- and bis(triethylphosphino)-substituted macrocyclic three-dimensional cage compounds were prepared via a rational coordination directed self-assembly paradigm. Despite extensive attempts we have not been successful to date in growing suitable x-ray quality crystals of these compounds. This may be related to the large cavity size of these molecules as well as the high solvent content of the crystals. However, the physical and spectral data, and in particular the mass spectral data, are consistent with the proposed truncated tetrahedral structures based on our design principles.
The combination of ferrocene ligands with late transition metal complexes provides an interesting link between the chemistry of ferrocenes and supramolecular self-assembly, thus opening this class of compounds to possible redox chemistry. Furthermore, modeling data show that these polyhedral complexes have large cavities capable of hosting larger guest molecules that are the subject of ongoing investigations.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-9818472) and the National Institutes of Health (5R01GM57052) for support (to P.J.S.) and the Alexander von Humboldt Foundation for a Feodor Lynen Fellowship (to M.S. and S.L.).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030264697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030264697
References
- 1.Holden A. Shapes, Space, and Symmetry. New York: Columbia Univ. Press; 1971. [Google Scholar]
- 2.MacGillivray L R, Atwood J L. Angew Chem Int Ed. 1999;38:1018–1033. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1018::AID-ANIE1018>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Diederich F N. Cyclophanes. Cambridge: Royal Society of Chemistry; 1991. [Google Scholar]
- 4.Eaton P E, Cole F W. J Am Chem Soc. 1964;86:3157–3160. [Google Scholar]
- 5.Fleischer E B. J Am Chem Soc. 1964;86:3889–3890. [Google Scholar]
- 6.Prelog V, Seiwerth R. Chem Ber. 1941;74:1769–1772. [Google Scholar]
- 7.Stetter H, Bänder O-E, Neumann W. Chem Ber. 1956;89:1922–1926. [Google Scholar]
- 8.Fort R, Schleyer P v R. Chem Rev. 1964;64:277–300. [Google Scholar]
- 9.Paquette L A, Ternansky R J, Balogh D W. J Am Chem Soc. 1982;104:4502–4503. [Google Scholar]
- 10.Ternansky R J, Balogh D W, Paquette L A. J Am Chem Soc. 1982;104:4503–4504. [Google Scholar]
- 11.Klug A. Angew Chem Int Ed Engl. 1983;22:565–582. [Google Scholar]
- 12.Horne R W. Virus Structure. New York: Academic; 1974. [Google Scholar]
- 13.Varner J E, editor. Self-Assembling Architecture. New York: Liss; 1988. [Google Scholar]
- 14.Terfort A, Bowden N, Whitesides G M. Nature (London) 1997;386:162–164. [Google Scholar]
- 15.Russell V A, Evans C C, Li W, Ward M D. Science. 1997;276:575–579. doi: 10.1126/science.276.5312.575. [DOI] [PubMed] [Google Scholar]
- 16.Conn M M, Rebek J., Jr Chem Rev. 1997;97:1647–1668. doi: 10.1021/cr9603800. [DOI] [PubMed] [Google Scholar]
- 17.Fujita M, Ogura K. Coord Chem Rev. 1996;148:249–264. [Google Scholar]
- 18.Fujita M. Chem Soc Rev. 1998;6:417–425. [Google Scholar]
- 19.Stang P J, Olenyuk B. Acc Chem Res. 1997;30:502–518. [Google Scholar]
- 20.Albrecht M. Chem Soc Rev. 1998;4:281–287. [Google Scholar]
- 21.Linton B, Hamilton A D. Chem Rev. 1997;97:1669–1680. doi: 10.1021/cr960375w. [DOI] [PubMed] [Google Scholar]
- 22.Jones C. Chem Soc Rev. 1998;4:289–299. [Google Scholar]
- 23.Olenyuk B, Fechtenkötter A, Stang P J. J. Chem. Soc. Dalton Trans. 1998. 1707–1728. [Google Scholar]
- 24.Caulder D L, Raymond K N. J. Chem. Soc. Dalton Trans. 1999. , 1185–1200. [Google Scholar]
- 25.Constable E C. Comprehensive Supramolecular Chemistry. 1996. chair ed. Lehn, J.-M., exec. eds. Atwood, J. L., Davis, J. E. D., MacNicol, D. D. & Vögtle, F. (Pergamon, Oxford), Vol. 9, Ch. 6, p. 213. [Google Scholar]
- 26.Mann S, Huttner G, Zsolnai L, Heinze K. Angew Chem, Int Ed Engl. 1996;35:2808–2809. [Google Scholar]
- 27.Saalfrank R W, Burak R, Reihs S, Low N, Hampel F, Stachel H D, Lentmaier J, Peters K, Peters E M, von Schnering H G. Angew Chem Int Ed Engl. 1995;34:993–995. [Google Scholar]
- 28.Caulder D L, Powers R E, Parac T N, Raymond K N. Angew Chem Int Ed Engl. 1998;37:1840–1843. [Google Scholar]
- 29.Enemark E J, Stack T D P. Angew Chem Int Ed Engl. 1998;37:932–935. doi: 10.1002/(SICI)1521-3773(19980420)37:7<932::AID-ANIE932>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 30.Klausmeyer K K, Rauchfuss T B, Wilson S R. Angew Chem Int Ed Engl. 1998;37:1694–1696. doi: 10.1002/(SICI)1521-3773(19980703)37:12<1694::AID-ANIE1694>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 31.Heinrich J L, Berseth P A, Long J R. J. Chem. Soc. Chem. Commun. 1998. 1231–1232. [Google Scholar]
- 32.Hartshorn C M, Steel P J. J. Chem. Soc. Chem. Commun. 1997. 541–542. [Google Scholar]
- 33.Fujita M, Oguro D, Miyazawa M, Oka H, Yamaguchi K, Ogura K. Nature (London) 1995;378:469–471. [Google Scholar]
- 34.Takeda N, Umemoto K, Yamaguchi K, Fujita M. Nature (London) 1999;398:794–796. [Google Scholar]
- 35.Olenyuk B, Whiteford J A, Fechtenkötter A, Stang P J. Nature (London) 1999;398:796–799. doi: 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- 36.Kusukawa T, Fujita M. Angew Chem Int Ed Engl. 1998;37:3142–3144. doi: 10.1002/(SICI)1521-3773(19981204)37:22<3142::AID-ANIE3142>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Olenyuk B, Levin M D, Whiteford J A, Shield J E, Stang P J. J Am Chem Soc. 1999;121:10434–10435. [Google Scholar]
- 38.Kang J, Rebek J., Jr Nature (London) 1997;385:50–52. doi: 10.1038/385050a0. [DOI] [PubMed] [Google Scholar]
- 39.Meissner R S, Mendoza J D, Rebek J., Jr Science. 1995;270:1485–1488. doi: 10.1126/science.270.5241.1485. [DOI] [PubMed] [Google Scholar]
- 40.Atwood J L, editor. Inclusion Phenomena and Molecular Recognition. New York: Plenum; 1990. [Google Scholar]
- 41.Stang P J, Olenyuk B, Muddiman D C, Smith R D. Organometallics. 1997;16:3094–3096. [Google Scholar]
- 42.Gardener G B, Venkataraman D, Moore J S, Lee S. Nature (London) 1995;374:792–795. [Google Scholar]
- 43.Stang P J, Olenyuk B, Fan J, Arif A M. Organometallics. 1996;15:904–908. [Google Scholar]
- 44.Stang P J, Cao D H, Saito S, Arif A M. J Am Chem Soc. 1995;117:6273–6283. [Google Scholar]
- 45.Leininger S, Stang P J, Huang S. Organometallics. 1998;17:3981–3987. [Google Scholar]
- 46.Fleischer E B, Schachter A M. Inorg Chem. 1991;30:3763–3769. [Google Scholar]
- 47.Chi X, Guerin A J, Hunter C A, Sarson L D. J. Chem. Soc. Chem. Commun. 1995. 2563–2565. [Google Scholar]
- 48.Mammen M, Shakhnovich E I, Deutch J, M, Whitesides G M. J Org Chem. 1998;63:3821–3830. [Google Scholar]
- 49.Manna J, Kuehl C J, Whiteford J A, Stang P J, Muddiman D C, Hofstadler S A, Smith R D. J Am Chem Soc. 1997;119:11611–11619. [Google Scholar]
- 50.Molecular Simulations. insight ii 97.0. San Diego, CA: Molecular Simulations; 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.