

Abstract
The cDNAs of two new human membrane-associated aspartic proteases, memapsin 1 and memapsin 2, have been cloned and sequenced. The deduced amino acid sequences show that each contains the typical pre, pro, and aspartic protease regions, but each also has a C-terminal extension of over 80 residues, which includes a single transmembrane domain and a C-terminal cytosolic domain. Memapsin 2 mRNA is abundant in human brain. The protease domain of memapsin 2 cDNA was expressed in Escherichia coli and was purified. Recombinant memapsin 2 specifically hydrolyzed peptides derived from the β-secretase site of both the wild-type and Swedish mutant β-amyloid precursor protein (APP) with over 60-fold increase of catalytic efficiency for the latter. Expression of APP and memapsin 2 in HeLa cells showed that memapsin 2 cleaved the β-secretase site of APP intracellularly. These and other results suggest that memapsin 2 fits all of the criteria of β-secretase, which catalyzes the rate-limiting step of the in vivo production of the β-amyloid (Aβ) peptide leading to the progression of Alzheimer's disease. Recombinant memapsin 2 also cleaved a peptide derived from the processing site of presenilin 1, albeit with poor kinetic efficiency. Alignment of cleavage site sequences of peptides indicates that the specificity of memapsin 2 resides mainly at the S1′ subsite, which prefers small side chains such as Ala, Ser, and Asp.
Keywords: membrane aspartic proteases, Alzheimer's disease
Many proteases are intimately involved in the regulation of cellular and physiological functions. There are five well studied human aspartic proteases. Pepsin and gastricsin participate in digestion in the stomach whereas cathepsins D and E function in intracellular protein degradation in lysosomes and endosomes. Human cathepsin D has been implicated in the metastasis of breast cancer and in Alzheimer's disease. Renin catalyzes the conversion of angiotensinogen to angiotensin 1, a clinically important step in hemostasis. The intimate involvement of human aspartic proteases in physiology and disease illustrates their central role in biology and medicine.
The emergence of human gene sequences in the expressed sequence tag (EST) database represents an important new resource for identifying novel human enzymes. We report here the cloning of two new human aspartic proteases, memapsin 1 (M1) and memapsin 2 (M2), initially identified from the human EST database. These proteases are unique among aspartic proteases in that they are membrane-anchored. The presence of M2 in the brain led us to test its capacity to hydrolyze the β-amyloid precursor protein (APP). Detailed enzymic and cellular studies suggest that M2 fits all of the criteria of β-secretase. Like M2, APP is a type I integral transmembrane protein and is known to be processed in vivo at three sites. The evidence suggests that cleavage at the α-secretase site by a membrane-associated metalloprotease is a physiological event. This site is located in APP 12 residues away from the luminal surface of the plasma membrane. Cleavage of the β-secretase site (28 residues from the plasma membrane's luminal surface) and the γ-secretase site (in the transmembrane region) results in a 40/42-residue β-amyloid peptide (Aβ), whose elevated production and accumulation in the brain are the central events in the pathogenesis of Alzheimer's disease (for review, see ref. 1). Presenilin 1, another membrane protein found in human brain, controls the hydrolysis at the APP γ-secretase site and has been postulated to be itself the responsible protease (2). Presenilin 1 is expressed as a single chain molecule, and its processing by a protease, presenilinase, is required to prevent it from rapid degradation (3, 4). The identity of presenilinase is unknown. The in vivo processing of the β-secretase site is thought to be the rate-limiting step in Aβ production (5) and, thus, is a strong therapeutic target. The production of recombinant M2 in Escherichia coli and the information on its specificity reported here should be useful for the design and testing of potential inhibitor drugs.
Materials and Methods
Cloning of Memapsin 2.
Three new EST sequences homologous to human aspartic proteases were found in EST database (www.ncbi.nlm.nih.gov) (EST AA136368, from human pregnant uterus; EST AA207232, from human neurepithelium; EST R55398, from human breast) using human aspartic protease motifs as a query. The corresponding bacterial strains 947471, 214526, and 392689 containing the EST sequences were obtained from the American Type Culture Collection. The completed sequences from these clones assembled into ≈80% of prepro-M2 cDNA. The 5′ region of M2 cDNA was obtained by using 5′ rapid amplification of cDNA ends PCR, which was carried out by using human pancreas Marathon-Ready cDNA (CLONTECH) and human pancreatic λ-gt10 and λ-gt11 libraries as templates and M2 specific primers. Prepro-M1 cDNA was cloned by using a similar procedure.
Tissue Distribution of Memapsin 1 and 2 mRNA.
Multiple human tissue cDNA panels (CLONTECH) were used as template for PCR amplification (30 cycles) of a 0.56-kb memapsin 1 cDNA fragment using primers M1F1 (5′-GACGTCACATGTAAG-TACACACAAGGAAGC) and M1R2 (5′-CAGAGATGCGCGGGCCACAGCTTCCACCAC), and of a 0.82-kb memapsin 2 cDNA fragment using primers M2F1 (5′-GGTAAGCATCCCCCATGGCCCCAACGTC-3′) and M2R2 (5′-CCAATTCGTTTTCGGGCCCGATCAAAGACAACG-3′). The amplified fragments were compared in agarose gel electrophoresis.
E. coli Expression of the Protease Domain of pro-Memapsin 2.
A fragment of M2 cDNA encoding the estimated pro and protease domain (without the C-terminal extension) were PCR amplified and inserted into the BamHI site of vector pET11a. The resulting vector, pET11-PM2pd, expresses a protein, pro-M2pd, whose sequence starts from Ala−8P to Ala326 [pepsin residue numbering (Fig. 1)]. The expression and purification of M2pd was carried out in E. coli strain BL21(DE3) as previously described (6) with minor modifications as follows: inclusion bodies dissolved in 8 M urea were rapid-diluted into 20 mM Tris base, and the pH was adjusted to 8.0.
Figure 1.

Predicted amino acid sequence of human prepro-memapsin 1 (GenBank accession no. AF200192) and prepro-memapsin 2 (GenBank accession no. AF200193) aligned against the sequence of human pre-pepsinogen. Amino acid codes are according to standard International Union of Pure and Applied Chemistry nomenclature. The beginning of pro and mature protease regions of pepsinogen are marked by residue numbers 1P and 1, respectively. Two active-site aspartic acids in D(T/S)G motifs are marked by ■ and conserved Tyr75 by ♦. Residues in the predicted transmembrane domains are in boldface.
Activation of pro-Memapsin 2 and Enzyme Kinetics.
Incubation in 0.1 M sodium acetate (pH 4.0) for 16 h at 22°C autocatalytically converted pro-M2pd to M2pd. For initial hydrolysis tests, two synthetic peptides were separately incubated with pro-M2pd in 0.1 M Na acetate (pH 4.0) for different periods ranging from 2 to 18 h. The incubated samples were subjected to HPLC/mass spectroscopy for the identification of the hydrolytic products (average error in mass determination was 0.02%). For kinetic studies, the identified HPLC (Beckman System Gold) product peaks were integrated for quantitation. The Km and kcat values for presenilin 1 and Swedish-APP peptides (Table 1) were measured by steady-state kinetics. The individual Km and kcat values for APP peptide could not be measured accurately by standard methods, so its kcat/Km value was measured by competitive hydrolysis of mixed substrates against presenilin 1 peptide (7).
Table 1.
Alignment on amino acids appearing in eight subsites of memapsin substrates
| Peptide | Subsites
|
|||||||
|---|---|---|---|---|---|---|---|---|
| P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | |
| APP β | E | V | K | M | D | A | E | F |
| Swedish APP β | E | V | N | L | D | A | E | F |
| PS-1 | L | V | N | M | A | E | G | D |
| M2-Pro | R | G | S | M | A | G | V | L |
| G | T | Q | H | G | I | R | L | |
| M2 | S | S | N | F | A | V | G | A |
| G | L | A | Y | A | E | I | A | |
| Insulin B-chain | H | L | C* | G | S | H | L | V |
| C* | G | E | R | G | F | F | Y | |
| E | A | L | Y | L | V | C* | G | |
| Nch† | G | V | L | L | S | R | K | |
| V | G | S | G | V | L | L | ||
| V | G | S | G | V | ||||
C is cysteic acid.
† Nch sequence VGSGVLLSRK.
Mammalian Cell Transfections.
Pro-M2 cDNA was cloned into the EcoRV site of vector pSecTag A (Invitrogen). Human APP cDNA was PCR amplified from human placenta λ-gt11 library (CLONTECH) and was cloned into the NheI and XbaI sites of pSecTag A. The procedure for transfection into HeLa cells and vaccinia virus infection for T7-based expression are essentially the same as described (8).
Pulse–Chase and Immunodetection.
Transfected cells were metabolically labeled with 200 μCi of 35S methionine and cysteine (TransLabel; ICN) in 0.5 ml of serum-free/methionine-free media for 30 min, were rinsed with 1 ml of media, and were replaced with 2 ml of DMEM/10% FCS. To block vesicle acidification, bafilomycin A1 was included in the media (9). At different time points (chase), media was removed and the cells were harvested and lysed in 50 mM Tris, 0.3 M NaCl, 5 mM EDTA, and 1% Triton X-100 (pH 7.4) containing 10 mM iodoacetamide, 10 μM TPCK, 10 μM TLCK, and 2 μg/ml leupeptin. The supernatant (14,000 × g) of cell lysates and media were immunoadsorbed onto antibody bound to protein G Sepharose (Sigma). Anti-APP N-terminal domain antibody (Chemicon) was used to recover the Nβ-fragment of APP, and anti-Aβ1–17 antibody (Chemicon, recognizing the N-terminal 17 residues of Aβ) was used to recover the 12 kDa βC-fragment. The former antibody recognized only denatured protein, so media was first incubated in 2 mM dithiothrietol and 0.1% SDS at 55°C for 30 min before immunoadsorption. Samples were cooled and diluted with an equal volume of cell lysis buffer before addition of anti-APP N-terminal domain (Chemicon). Beads were washed, were eluted with loading buffer, were subjected to SDS/PAGE (NOVEX, San Diego), and were visualized by autoradiogram or PhosphorImaging (Molecular Dynamics) on gels enhanced with Amplify (Amersham). Immunodetection of the Nα fragment was accomplished by transblotting onto a PVDF membrane and detecting with anti-Aβ1–17 and chemiluminescent substrate (Amersham).
Cellular Localization of M2 and APP.
HeLa cells transfected with APP or M2 (see above) in four-well chamber slides were fixed with acetone for 10 min and were permeabilized in 0.2% Triton X-100 in PBS for 6 min. For localizing M2, polyclonal goat anti-pro-M2pd antibodies were purified on DEAE-Sepharose 6B and were affinity-purified against recombinant pro-M2pd immobilized on Affigel (Bio-Rad). Purified anti-pro-M2pd antibodies were conjugated to Alexa568 (Molecular Probes) according to the manufacturer's protocol. Fixed cells were incubated overnight with a 1:100 dilution of antibody in PBS containing 0.1% BSA and were washed four times with PBS. For APP, two antibodies were used: antibody Aβ1–17 (described above) and antibody Aβ17–42, which recognizes the first 26 residues after the α-secretase cleavage site (Chemicon). After four PBS washes, the cells were incubated overnight with an anti-mouse FITC conjugate at a dilution of 1:200. Cells were mounted in Prolong anti-fade reagent (Molecular Probes) and were visualized on a Leica (Deerfield, IL) TCS confocal laser scanning microscope.
Results
EST clones from three human cDNAs were identified from homology search based on aspartic protease motifs and were sequenced after 5′ rapid amplification of cDNA ends. The deduced protein sequences (Fig. 1) are characteristic of mammalian aspartic protease zymogens. Uniquely, each enzyme also has a C-terminal extension of over 80 residues (see Fig. 1 alignment with pepsin), which includes a putative transmembrane region. The enzymes are named memapsin 1 and memapsin 2 (for membrane-anchored aspartic protease of the pepsin family; GenBank accession nos. AF200192 and AF200193, respectively). In human tissues, spleen and prostate have the highest levels of M1 mRNA level [but is absent in the brain (Fig. 2A)] whereas pancreas and brain have the highest amount of M2 (Fig. 2B). To search for M2 substrates among membrane proteins of the brain, the estimated pro-M2 protease domain (pro-M2pd, without the C-terminal extension) was expressed in E. coli as inclusion bodies, refolded and purified. In acidic solutions, pro-M2pd spontaneously generated activity (see below) with the concomitant appearance of smaller fragments (Fig. 1, Fig. 3A, and Table 1), a common property for aspartic protease zymogens. Whether this activity is produced by the classical intramolecular activation of aspartic protease zymogens (10, 11) is presently unknown. For the current discussion, we will refer to this activity as that of M2 (M2pd).
Figure 2.

Tissue distribution of M1 (A) and M2 (B) mRNA determined by reverse transcription–PCR visualized in agarose gel electrophoresis. Control experiment contained no template DNA. Amplification of glyceraldehyde-3-phosphate dehydrogenase mRNA produced uniform intensity (data not shown).
Figure 3.

(A) Conversion of pro-M2pd (lane 1) at pH 4.0 to smaller fragments (lane 3) as shown by SDS/polyacrylamide electrophoretic pattern. Difference in migration between pro-M2pd and converted enzyme is evident in a mixture of the two (lane 2). Squares mark band positions. (B) Initial rate of hydrolysis of synthetic peptide swAPP (see Table 1) by M2pd at different pH. (C) Relative kcat/Km values for steady-state kinetic of hydrolysis of peptide substrates by M2pd (see text for peptides and values).
Modeling of M2 three-dimensional structure as a type I integral membrane protein suggested that its globular protease unit can hydrolyze a membrane-anchored polypeptide at a distance range of 20–30 residues from the membrane surface (results not shown). As a transmembrane protein of the brain, APP is a potential substrate. Its β-secretase site, located about 28 residues from the plasma membrane surface, is within in the range for M2 proteolysis. A synthetic peptide derived from this site (SEVKM/DAEFR) was hydrolyzed by M2pd at the β-secretase site (marked by a slash). A second peptide (SEVNL/DAEFR) derived from APP β-secretase site with the “Swedish mutation” (12), known to elevate the level of Aβ production in cells (13), was hydrolyzed by M2pd with much higher catalytic efficiency (Fig. 3C). Both substrates were optimally cleaved at pH 4.0 (Fig. 3B). A peptide derived from the processing site of presenilin 1 (SVNM/AEGD) was also cleaved by M2pd with poor kinetic parameters (Fig. 3C). A peptide derived from the APP γ-secretase site (KGGVVIATVIVK) was not cleaved by M2pd. Interestingly, pepstatin A inhibited M2pd poorly (IC50 ≈ 0.3 mM). The kinetic parameters indicate that both presenilin 1 (kcat, 0.67 s−1; Km, 15.2 mM; kcat/Km, 43.8 s−1⋅M−1) and native APP peptides (kcat/Km, 39.9 s−1⋅M−1) are very poor substrates whereas Swedish APP peptide (kcat, 2.45 s−1; Km, 1 mM; kcat/Km, 2,450 s−1⋅M−1) is a much better substrate.
To determine whether M2 possesses an APP β-secretase function in mammalian cells, these two cDNAs were transiently expressed in HeLa cells (8), were metabolically pulse-labeled with 35S-Met, and then were immunoprecipitated with anti-APP antibodies for visualization of APP-generated fragments after SDS/polyacrylamide electrophoresis and imaging. Cells expressing both APP and M2 produced the 97-kDa APP Nβ-fragment (from the N terminus to the β-secretase site) in the conditioned media (Fig. 4A, lane 2) and the 12-kDa βC-fragment (from the β-secretase site to the C terminus) in the cell lysate (Fig. 4B, lane 2). Controls that included transfection of APP alone produced little detectable Nβ-fragment (Fig. 4A, lane 1) nor the βC-fragment (Fig. 4B, lane 1). Bafilomycin A1, which is known to raise the intravesicle pH of lysosomes/endosomes and has been shown to inhibit APP cleavage by β-secretase (14), abolished the production of both APP fragments Nβ and βC (Fig. 4A, lane 3 and Fig. 4B, lane 3, respectively) in co-transfected cells. Transfection of cells with Swedish APP alone did not produce the βC-fragment band in the cell lysate (Fig. 4B, lane 4), but the co-transfection of Swedish APP and M2 did (Fig. 4B, lane 5). This Swedish βC-fragment band is more intense than that of the wild-type APP (Fig. 4B, lane 2). A 97-kDa Nβ-band is also seen in the conditioned media but is about the equal intensity as the wild-type APP transfection (result not shown). These results suggest that M2 processes the β-secretase site of APP in acidic compartments such as the endosomes. To establish the expression of transfected M2 gene, the pulse-labeled cells were lysed and immunoprecipitated by anti-M2 antibodies. A 70-kDa M2 band was seen in cells transfected with M2 gene (Fig. 4C, lane 1), which has the same mobility as the major band from HEK 293 cells (Fig. 4C, lane 3) known to express β-secretase (13). A very faint band of M2 is also seen, after a long film exposure, in untransfected HeLa cells (Fig. 4C, lane 2), indicating a very low level of endogenous M2, which was insufficient to produce Nβ- or βC-fragments without M2 transfection (Fig. 4A, lane 1 and Fig. 4B, lane 1). Antibody Aβ1–17, which specifically recognizes residues 1–17 in Aβ peptide, was used to confirm the correct β-secretase site cleavage. From immunoprecipitation of conditioned media from cells transfected with APP and M2, both Nα- and Nβ-fragments are visible using an antibody recognizing the N-terminal region of APP present in both fragments (Fig. 4C, lane 1). In Western blots, antibody Aβ1–17 recognized the immunoprecipitated Nα-fragment produced by endogenous α-secretase in the untransfected cells (Fig. 4C, lane 2). This antibody was, however, unable to recognize the Nβ-fragment (Fig. 4C, lane 3) known to be present in cells co-transfected with APP and M2 (compare the 95-kDa band in lanes 1 and 3). These observations confirmed that Nβ-fragment is the product of β-secretase site cut by M2, which abolished recognition of the epitope of Aβ1–17.
Figure 4.

Processing of APP by M2 in HeLa cells. (A) SDS/PAGE patterns of immunoprecipitated APP Nβ-fragment (97-kDa band) from the condition media (2 h) of pulse–chase experiments. Lanes: 1, transfection of APP alone; 2, co-transfection of APP and M2; 3, same as lane 2 except that bafilomycin A1 is included. (B) SDS/PAGE patterns of APP βC-fragment (12 kDa) immunoprecipitated from the conditioned media of the same experiment as in A. Lanes: 1, transfection of APP; 2, co-transfection of APP and M2; 3, as in lane 2 but with bafilomycin A1; 4, transfection of Swedish APP; 5, co-transfection of Swedish APP and M2. (C) SDS/PAGE patterns of immunoprecipitated M2 (70 kDa). Lanes: 1, M2 transfected cells; 2, untransfected HeLa cells after long time film exposure; 3, endogenous M2 from HEK 293 cells. (D) SDS/PAGE patterns of APP fragments (100-kDa Nα-fragment and 95-kDa Nβ-fragment) recovered from conditioned media after immunoprecipitation using antibodies specific for the N-terminal region of APP. Lanes: 1, autoradiogram of immunoprecipitation from co-transfection of APP and M2; 2 and 3, immunoblotted by using antibody Aβ1–17, specific for the Nα-fragment that does not recognize the Nβ-fragment; 2, transfection with APP alone; 3, co-transfection of APP and M2. Dots mark band positions.
The processing of APP by M2 predicts the intracellular colocalization of the two proteins. Immunodetection observed by confocal microscopy of both APP (Fig. 5A) and M2 (Fig. 5B) revealed their colocalization in the superimposed scans (Fig. 5C). The distribution of both proteins is consistent with their residence in lysosomal/endosomal compartments.
Figure 5.
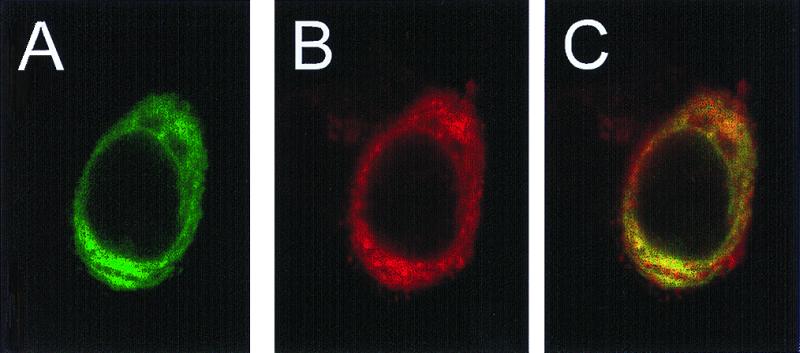
Intracellular localization of M2 and APP in a single cell. HeLa cells co-expressing APP and M2 were stained with antibodies directed toward APP (A, green image) and M2 (B, red image) and were visualized simultaneously by CSLM (see Materials and Methods) using a 100× objective. Areas of colocalization appear in yellow in overlay of two images in C.
In specificity studies, we found that M2pd cleaved its pro peptide (two sites) and the protease portion (two sites) during a 16-h incubation after activation (Table 1). Besides the three peptides discussed above, M2pd also cleaved oxidized bovine insulin B chain and a synthetic peptide Nch. Interestingly, we found that native proteins were not cleaved by M2pd (including β-amylase, alcohol dehydrogenase, carbonic anhydrase, cytochrome C, bovine and human serum albumin, hemoglobin, and chicken egg lysozyme; data not shown). The residues around all of these cleavage sites are aligned in Table 1. The specificity of M2 derived from these results will be further discussed below.
Discussion
The data presented herein strongly suggest that human M2 fulfills all of the criteria of a β-secretase that cleaves the β-amyloid precursor protein (APP): (i) M2 and APP are both membrane proteins present in human brain and co-localize in mammalian cells, (ii) M2 specifically cleaves the β-secretase site of synthetic peptides and of APP in cells, (iii) M2 preferentially cleaves the β-secretase site from the Swedish over the wild-type APP, and (iv) the acidic pH optimum for M2 activity and bafilomycin A1 inhibition of APP processing by M2 in the cells are consistent with the previous observations that β-secretase cleavage occurs in acidic vesicles (14). The spontaneous appearance of activity of recombinant pro-M2 in an acidic solutions is consistent with the idea that this zymogen can by itself generate activity in an acidic vesicle like an endosome. However, we note that the cleavage sites (Table 1) are not near the vicinity of the aligned pepsin N terminus (Fig. 1). It seems probable that the activation of proM2 in vivo may require another protease. While preparing this manuscript, the cloning of a human transmembrane aspartic protease BACE and its activity in the cleavage of APP β-secretase site were reported (15). The sequences of BACE and pro-memapsin 2 are identical. Although the approaches used for cloning, transfection, and expression were different in these two studies, both studies concluded that the new protease is β-secretase, the long-sought enzyme inexorably involved in Alzheimer's disease.
We have determined that the hydrolysis of the Swedish peptide by M2pd has a kcat/Km value ≈60× over that for the wild-type APP peptide, which explains the higher production of Aβ peptide and the early onset of Swedish familial Alzheimer's disease (12). The relative amount of β-secretase cleavage of APP and Swedish APP estimated from the cellular experiments is only about six-fold in favor of Swedish APP (13). We also observed only a few-fold increase of the βC-fragment from Swedish APP over that of native APP (Fig. 4B). Although β-secretase is known to be the rate-limiting step in the production of Aβ peptide from native APP (5), the cellular processing of Swedish APP, with 60× of catalytic efficiency, would likely make other steps in Aβ production to be rate limiting. The correct cleavage of a peptide from the processing site of presenilin 1 by M2pd (Table 1) could be taken to suggest that M2 may be the presenilin 1 processing protease, presenilinase. This seems to be improbable because the kinetics are not sufficiently robust for the required physiological efficiency. There may also be a question of accessibility of the presenilinase site by M2 that is located on the luminal side of the membrane. Evidence for the localization of the processing loop of presenilin 1 on the luminal (16) and cytosolic (17) sides of the membrane have both been reported.
Data presented in Table 1 suggest that M2 has an overall broad substrate specificity. Among the eight subsites, present in all aspartic proteases, the most stringent specificity for M2 occurs at S1′, which prefers small or negatively charged side chains, such as Ala, Asp, and Ser, or no side chain, Gly. Our data on the P1 preference are in general agreement but somewhat broader than the results of a cellular mutagenesis study (18) in which Tyr, Leu, Phe, and Met were found at P1. This is expected from a higher sensitivity for product detection in our experiments. Other subsites show relatively high tolerance for side chains of different sizes and charges. From the sequences of the peptide substrates that were cleaved, there appears to be a trend of requiring at least two large hydrophobic residues in these eight subsites. These specificity findings suggest that the P1/P1′ residues at the α-secretase site (Lys/Leu) and the γ-secretase site (Val/Ile and Ala/Thr) are not favored by M2 and would predict that they are not substrates. The failure of M2 to cleave native proteins is consistent with the requirement in a substrate of a stretch of peptide in an extended conformation. The hydrolysis of Nch peptide between residues 1–2 and 3–4 (Table 1) suggests that the hydrolysis by M2 does not require a substrate to fill all eight substrate binding subsites. This may indicate that a relatively short peptidic transition-state analogue can be a good inhibitor for M2.
Even though the molecular architecture of the two memapsins are similar, because M1 is absent in the brain (Fig. 2A), the question as to whether it can cleave the APP β-secretase site may be physiologically irrelevant. However, the presence of both M1 and M2 in many other tissues argues for important roles of this new class of aspartic proteases in other physiological processes. The poor kinetic parameters for the hydrolysis of β-secretase site of native APP by M2 suggest that this cleavage may be a tolerated aberration in normal human brain.
Acknowledgments
The authors thank Dr. J. Donald Capra (Oklahoma Medical Research Foundation) for critical reading of the manuscript, Dr. Ken Jackson, Jihua Wang, and Jim Henthorn of the Warren Medical Research Foundation, University of Oklahoma Health Sciences Center, for assistance in mass spectrometry and confocal microscopy, and Dr. David Powell (SmithKline Beecham Pharmaceuticals) for helpful discussion. G.K. is a Scientist Development Awardee of the American Heart Association (9930115N). J.T. is holder of the J. G. Puterbaugh Chair in Biomedical Research at the Oklahoma Medical Research Foundation.
Abbreviations
- APP
β-amyloid precursor protein
- M1
memapsin 1
- M2
memapsin 2
- EST
expressed sequence tag
Footnotes
References
- 1.Selkoe D J. Nature (London) 1999;399A:23–31. [Google Scholar]
- 2.Wolfe M S, Xia W, Ostaszewski B L, Diehl T S, Kimberly W T, Selkoe D J. Nature (London) 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 3.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 4.Podlisny M B, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, et al. Neurobiol Dis. 1997;3:325–337. doi: 10.1006/nbdi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 5.Sinha S, Lieberburg I. Proc Natl Acad Sci USA. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin X, Lin Y, Tang J. Methods Enzymol. 1994;241:195–224. doi: 10.1016/0076-6879(94)41066-6. [DOI] [PubMed] [Google Scholar]
- 7.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1985. [Google Scholar]
- 8.Lin X, Dashti A, Schinazi R F, Tang J. FASEB J. 1993;7:1070–1080. doi: 10.1096/fasebj.7.11.8370478. [DOI] [PubMed] [Google Scholar]
- 9.Perez R G, Squazzo S L, Koo E H. J Biol Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- 10.Tang J, Wong R N S. J Cell Biochem. 1987;33:53–63. doi: 10.1002/jcb.240330106. [DOI] [PubMed] [Google Scholar]
- 11.Koelsch G, Loy J, Lin X, Tang J. Adv Exp Med Biol. 1998;436:245–252. doi: 10.1007/978-1-4615-5373-1_34. [DOI] [PubMed] [Google Scholar]
- 12.Mullan M, Houlden H, Windelspecht M, Fidani L, Lombardi C, Diaz P, Rossor M, Crook R, Hardy J, Duff K, et al. Nat Genet. 1992;2:340–342. doi: 10.1038/ng1292-340. [DOI] [PubMed] [Google Scholar]
- 13.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung A Y, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe D J. Nature (London) 1992;260:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 14.Knops J, Suomensaari S, Lee M, McConlogue L, Serbert P, Sinha S. J Biol Chem. 1995;270:2419–2422. doi: 10.1074/jbc.270.6.2419. [DOI] [PubMed] [Google Scholar]
- 15.Vassar R, Bennett B D, Babu-Khan S, Kahn S, Mendiaz E A, Denis P, Teplow D B, Ross S, Amarante P, Loeloff R, et al. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 16.Dewji N N, Singer S J. Proc Natl Acad Sci USA. 1997;94:1425–1430. [Google Scholar]
- 17.De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop P S, et al. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- 18.Citron M, Taplow D B, Selkoe D J. Neuron. 1995;14:661–670. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]