

Abstract
The essential enzyme thymidylate synthase-dihydrofolate reductase (TS-DHFR) is a validated drug target for many pathogens, but has been elusive in Cryptosporidium hominis, as active site inhibitors of the enzymes from related parasitic protozoa show decreased potency and lack of species specificity over the human enzymes. As a rational approach to discover novel inhibitors, we conducted a virtual screen of a non-active site pocket in the DHFR linker region. From this screen, we have identified and characterized a noncompetitive inhibitor, flavin mononucleotide (FMN), with micromolar potency that is selective for ChTS-DHFR versus the human enzymes. These results describe a novel allosteric pocket amenable to inhibitor targeting, and a lead compound with which to move towards potent, selective inhibitors of ChTS-DHFR.
Keywords: Cryptosporidium hominis, DHFR, Non-active site, Allosteric, Virtual screen, Glide
Cryptosporidium hominis is a water-borne, intestinal protozoan parasite, leading to the opportunistic infection cryptosporidiosis.1 The disease state is especially dangerous in children, the elderly, and immuno-compromised individuals, where it leads to the severe and life-threatening ‘wasting’ disease.1,2 Recently, C. hominis has also been classified as a Category B Biodefense Pathogen by the National Institutes of Health, due to the water safety threat to public health.3 As yet, there are few effective treatments for the parasite, and recent major outbreaks in Milwaukee and New York City have highlighted the need for novel therapeutic development.4,5
The essential enzymes thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are long-utilized, validated targets for anticancer, antimicrobial, and antiparasitic therapy.6-8 The most common strategy for targeting protozoan parasites has been through DHFR active site inhibitors.6 However, the drugs used for other species have proven ineffective against C. hominis DHFR, likely due to active site amino acid residues that are similar to resistant mutations in other protozoan species.9 Additionally, the development of ChDHFR active site inhibitors with greater potency has tended to compromise the specificity of those inhibitors versus the human enzyme.10,11 However, a detailed examination of the three-dimensional structure of the C. hominis protein has revealed unique features that may be exploited for species-specific inhibitor design. Whereas in humans the proteins are separate polypeptides, in parasitic protozoa the two exist on the same polypeptide chain as a bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) enzyme (Fig. 1).† As a consequence, many non-active site surfaces exist on this bifunctional enzyme that are absent in the human orthologs. Targeting a non-active site region of ChTS-DHFR would address both the DHFR active site resistance, as well as allow for the important therapeutic goal of species-specificity for the parasite versus human enzyme.
Figure 1.

Structure of TS-DHFR from Cryptosporidium hominis. DHFR (blue/cyan) and TS (red/pink) are shown with active site ligands colored in gray. The magnified inset shows active site Helix B (blue), the Crossover helix (cyan), and the novel inhibitor FMN (green) docked into the non-active site pocket in the DHFR linker region.
Recent work from our lab has shown that the ‘crossover helix’ from the DHFR linker region in ChTS-DHFR is necessary for optimal catalytic activity at the DHFR domain.12 The crossover helix from the linker region packs against the backside of the opposite DHFR active site (Fig. 1). Its main interaction is with Helix B of the active site, which is known to display mobile, coordinated motions during catalysis.13,14 Mutations of the crossover helix that disrupt the interaction with Helix B cause a severe decrease in DHFR enzyme activity, implying an important role for the crossover helix and flexible linker region. Inspection of the crystal structure of ChTS-DHFR,15 reveals a small pocket located directly beneath the crossover helix (Fig. 1). We hypothesized that designing a small molecule to bind in this pocket might alter the position of the crossover helix and thereby impart DHFR inhibition. As a means to explore the potential of this site for novel inhibitor development, we conducted a virtual screen of the pocket, and tested the top compound hits in our in vitro enzymatic assays.
To define the target pocket, we used a grid with outer box dimensions of 20 Å that encompassed residues on the DHFR domain, the TS domain, and the linker region including the crossover helix (Fig. 1). We used the docking program GLIDE (version 3.5) in standard precision (SP) mode to dock a library of 100,000 commercially available compounds, with molecular weight less than 500 g/mol, from the Comprehensive Medicinal Chemistry (CMC) Database.16 Before running the GLIDE screen, all compounds were treated with LigPrep (version 1.6) to generate lowest energy conformations. The resulting report from GLIDE SP ranked virtual hits by Glide-Score, which roughly approximates the free energy of binding.16
The resulting report from GLIDE ranked virtual hits by SP Glide-Score, and yielded a chemically diverse set of compounds. The top seven compounds, based on GLIDE Score, are shown in Table 1 (appended). We ordered and tested the first 50 available compounds for potency against ChTS-DHFR using a spectroscopic assay as previously described.17 The most potent of the compounds was GLIDE-ranked #13, flavin mononucleotide (FMN, Table 1), with an IC50 of 55 μM against ChTS-DHFR (Fig. 2A). This result was confirmed by an independent kinetic assay in which the substrate, methylene tetrahydrofolate, was radiolabeled, and the conversion to product was monitored by HPLC (data not shown).17 In separate experiments, the enzyme concentration was varied from 80 to 250 nM with no resulting change in IC50, signifying the inhibition is not due to aggregate formation. The major effect of the inhibitor is at the DHFR domain, as there was little inhibition of the TS reaction alone at 300 μM FMN. A few of the compounds were of comparable potency, including riboflavin (FMN without the terminal phosphate) and streptonigrin (Table 1), while the majority of the 50 compounds tested were much less effective.
Table 1.
Representative table of GLIDE virtual screen top hitsa
| Chemical formula (common name) | Structure | GLIDE Score (kcal/mol) | Experimental IC50 (μM) |
|---|---|---|---|
| C35NO11H33 (Neocarzinostatin) | 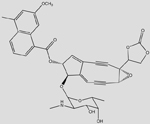 |
-11.71 | NA |
| C35N3O9H39 (Rubidazone) | 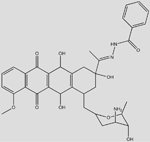 |
-9.75 | >500 |
| C31O7H48 (Digoxin) | 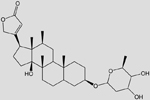 |
-9.18 | >500 |
| C16N2O11H28 (Chitodextrin) | 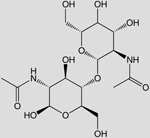 |
-8.91 | >500 |
| C19N2O3H24 (Disoxaril) | -8.42 | NA | |
| C26N4O4CL2H28 (Ketoconazole) | 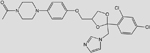 |
-8.41 | >500 |
| C25N4O8H22 (Streptonigrin) | 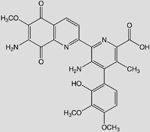 |
-8.41 | 80 |
| C17N4O9PH21 (Flavin mononucleotide, FMN) | 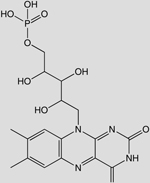 |
-8.08 | 55 |
Initial activity assays were conducted at 100 and 500 mM compounds; NA, compound not readily available to purchase.
Figure 2.

(A) Steady-state dose-response curve of FMN versus ChTS-DHFR. Line is a smooth fit. (B) Pre-steady-state burst amplitude dose response of FMN versus ChTS-DHFR. Line is a hyperbolic (binding curve) fit.
As a proof of principle, it is important to verify that the inhibitor is not binding at the active site. We conducted a steady-state rate profile for both DHFR substrates, dihydrofolate (H2F) and NADPH, in the presence and absence of FMN. The profiles show that the compound appears to be a noncompetitive (mixed) inhibitor for H2folate; in the presence of 200 μM FMN the Vmax decreased (2.35 ± 0.04 μmol/min versus 1.8 ± 0.1 μmol/min), but the Km was not significantly altered (5.8 ± 0.5 μM vs 7.2 ± 1.9 μM) (Fig. 3A). The compound appears to be an uncompetitive inhibitor for NADPH; in the presence of 200 μM FMN, both the Vmax (2.37 ± 0.03 μmol/min vs 1.77 ± 0.01 μmol/min) and the Km (4.2 ± 0.4 μM vs 3.0 ± 0.1 μM) decreased (Fig. 3A). Lineweaver-Burk plots better demonstrate the classical noncompetitive and uncompetitive character of the data (Fig. 3B).
Figure 3.

(A) Steady-state competition assay varying either dihydrofolate (left) or NADPH (right). Data fit to a hyperbolic equation. (B) Lineweaver-Burk plots for data in (A). Data fit to linear equation. For all graphs, circle (●) represents the reaction with no inhibitor, and square (■) represents the reaction with 200 μM FMN.
As a more thorough means to assess the mechanism of ChTS-DHFR inhibition by FMN, we conducted a pre-steady state kinetic analysis, observing steps of the catalytic cycle on the millisecond time scale. Single-enzyme-turnover experiments use enzyme concentration in excess of substrate, allowing for only one turnover and direct measurement of the rate of chemistry at the active site, as opposed to the overall rate-limiting step. We conducted DHFR single-enzyme turnover experiments, with enzyme in five-fold excess of the substrate H2F (data not shown). Using both a rapid chemical quench and stopped-flow fluorescence methodologies to monitor DHFR catalysis, we found that even at high concentrations of FMN (500 μM) there was no decrease in the rate of chemistry, signifying the effect of FMN is likely through a different step in the catalytic cycle. However, the amplitude of the single-turnover plot did show an FMN dose-dependent decrease. Kinetic modeling and negative controls with the known competitive inhibitor methotrexate showed this to be a characteristic of noncompetitive inhibitors.
Pre-steady-state burst experiments, using substrate in slight excess over enzyme, are also useful in assessing inhibitor mechanism. It has been shown that non-active site inhibitors, such as nonnucleoside reverse-transcriptase inhibitors (NNRTIs) of the enzyme HIV reverse transcriptase, cause a dose-dependent decrease in the burst amplitude, corresponding to a decrease in active enzyme concentration.18 This decrease can then be used to derive the dissociation constant. A noncompetitive inhibitor should be expected to have a Kd equivalent to the IC50. Indeed, FMN causes a dose-dependent decrease in DHFR burst amplitude, which fits well to a hyperbolic curve, yielding a Kd of 48 μM (Fig. 2B).
The docked pose of FMN is shown in Figure 4, along with the amino acids that are proposed to make hydrogen bonds to the inhibitor. Preliminary analysis of mutations to two of these residues (R190G, D201A) has indicated that the inhibitory effect of FMN on mutant proteins is approximately 20% and 10%, respectively, less than that against WT ChTS-DHFR. This provides additional evidence that the inhibitor is binding not only at a non-active site, but specifically in the intended pocket.
Figure 4.

The docked pose of FMN (green) and amino acids that make putative hydrogen bonds to the compound (gray).
A major goal of the virtual screen was to provide an initial candidate that could be further improved through structure guided optimization to a lead compound that was not only effective against ChTS-DHFR, but that was also species-specific. To provide an initial assessment for the species-specificity of FMN, we tested it against both human DHFR and human TS. The inhibitor showed selectivity for the parasite enzyme versus the human enzymes (Table 2). Additionally, FMN showed specificity within protozoan parasites, as it did not inhibit TS-DHFR from Toxoplasma gondii, which also contains a linker region (Table 2).
Table 2.
IC50 values of FMN against enzyme targetsa
| Enzyme | C. hominis TS-DHFR | Human DHFR | Human TS | T. gondii TS-DHFR |
|---|---|---|---|---|
| IC50 (μM) | 55 ± 7 | 325 ± 50 | >500 | >500 |
All reactions were run with identical enzyme and ligand concentrations. Values are the averages of at least three independent experiments.
It is important to note that while this compound with inhibitory activity in the micromolar range is primarily a first-generation candidate for a lead compound, it is in fact comparable to the few successes reported for Cryptosporidium inhibitors. The only species-selective inhibitor for C. hominis DHFR to date, trimethoprim, has an IC50 of 14 μM against the enzyme.19 Additionally, the only FDA-approved drug for cryptosporidial diarrhea, nitazoxanide, inhibits parasite growth in the range of 1-40 μM.20,21 Therefore, this virtual screen has produced inhibitor hits with real potential toward development of ChTS-DHFR species-specific inhibitors.
The putative position of the inhibitor (Fig. 1) has allowed us to further probe the function of the crossover helix in DHFR catalysis. Our mutational analysis of this helical region showed that the crossover helix modulates catalytic activity and is important for optimal DHFR function, yet the mechanism of this enhancement is not understood.12 It is known that Helix B of the DHFR active site moves in correlation with catalysis,13 and that the linker containing the crossover helix is somewhat flexible. We might hypothesize that a small molecule designed to occupy the pocket at the base of the crossover helix would appear to act like a wedge restricting the helix mobility. The fact that a compound directed to bind in this pocket inhibits the enzyme implies that the crossover helix flexibility, in addition to its interaction with Helix B, is necessary for maintaining optimal DHFR activity. Findings that Escherichia coli DHFR coupled-residue mutants that disrupt catalytic activity also cause a decrease in the movement of Helix B toward the active site are consistent with this mechanism.22
It initially seems curious as to why the inhibitor would display uncompetitive behavior with respect to NADPH, as the most commonly held pattern of allosteric inhibition is noncompetitive or mixed. This becomes clear when viewed in light of what is known about the catalytic movements in DHFR. Crystal structures of multiple points along the E. coli DHFR catalytic cycle have shown that the position of the M20 loop, which directly precedes Helix B, is dictated by the binding of NADPH. When NADPH is bound, the loop is in a ‘closed’ conformation; however, when NADPH is unbound the loop is in an ‘occluded’ conformation, which physically moves Helix B away from the active site.13 The movement analogous to this in C. hominis DHFR would effectively narrow the non-active site pocket discussed here. It is therefore not surprising that our compound, predicted to contact multiple residues impacted by the position of Helix B, would show altered binding. Our uncompetitive data imply that the conformational change in the absence of NADPH is enough to restrict binding of the inhibitor altogether.
In summary, we have conducted a structure-guided virtual screen on a non-active site pocket of the therapeutically relevant target, ChTS-DHFR. Our screen yielded inhibitors with potency in the micromolar range, the most potent of which, FMN, has an IC50 of 55 μM. The inhibitor appears to display noncompetitive and uncompetitive behavior with respect to the natural substrates, and the binding is influenced by mutations at the docked site. This initial candidate compound displays selectivity, and potency in the range of known selective DHFR inhibitors, and therefore offers promise for obtaining lead compounds with future inhibitor design toward ChTS-DHFR. Perhaps more importantly, we have shown the inhibitory potential of an allosteric pocket previously unexplored in this enzyme. Work is currently underway to crystallize the enzyme in complex with the inhibitor identified here, to validate the computational modeling and allow for precise exploration of the binding pocket of this novel class of inhibitors.
Acknowledgments
This work was supported in part by NIH Grant AI 44630 (to K.S.A), GM 32136 (to W.L.J.), and NIH Grant 5T32-AI 07404 (to W.E.M.).
Footnotes
TS-DHFR, thymidylate synthase-dihydrofolate reductase is a functional designation as catalysis at TS precedes catalysis at DHFR; elsewhere the bifunctional enzyme is called DHFR-TS based on DHFR being N-terminal to TS.
References and notes
- 1.Mead JR. Drug Resist. Updates. 2002;5:47. doi: 10.1016/s1368-7646(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 2.Guarino A, Bruzzese E, De Marco G, Buccigrossi V. Paediatr. Drugs. 2004;6:347. doi: 10.2165/00148581-200406060-00003. [DOI] [PubMed] [Google Scholar]
- 3.Tzipori S, Widmer G. Trends Parasitol. 2008;24:184. doi: 10.1016/j.pt.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta M, Haas CN. J. Water Health. 2004;2:59. [PubMed] [Google Scholar]
- 5.O'Connor A. The New York Times. New York City: 2005. [Google Scholar]
- 6.Anderson AC. Drug Discov. Today. 2005;10:121. doi: 10.1016/S1359-6446(04)03308-2. [DOI] [PubMed] [Google Scholar]
- 7.Chu E, Callender MA, Farrell MP, Schmitz JC. Cancer Chemother. Pharmacol. 2003;52:S80. doi: 10.1007/s00280-003-0625-9. [DOI] [PubMed] [Google Scholar]
- 8.Then RL. J. Chemother. 2004;16:3. doi: 10.1179/joc.2004.16.1.3. [DOI] [PubMed] [Google Scholar]
- 9.Vasquez JR, Gooze L, Kim K, Gut J, Petersen C, Nelson RG. Mol. Biochem. Parasitol. 1996;79:153. doi: 10.1016/0166-6851(96)02647-3. [DOI] [PubMed] [Google Scholar]
- 10.Brophy VH, Vasquez J, Nelson RG, Forney JR, Rosowsky A, Sibley CH. Antimicrob. Agents Chemother. 2000;44:1019. doi: 10.1128/aac.44.4.1019-1028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson RG, Rosowsky A. Antimicrob. Agents Chemother. 2001;45:3293. doi: 10.1128/AAC.45.12.3293-3303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vargo MA, Martucci WE, Anderson KS. Biochem. J. in press. [Google Scholar]
- 13.Sawaya MR, Kraut J. Biochemistry. 1997;36:586. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Goodey NM, Benkovic SJ, Kohen A. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15753. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Neil RH, Lilien RH, Donald BR, Stroud RM, Anderson AC. J. Biol. Chem. 2003;278:52980. doi: 10.1074/jbc.M310328200. [DOI] [PubMed] [Google Scholar]
- 16.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J. Med. Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 17.Doan LT, Martucci WE, Vargo MA, Atreya CE, Anderson KS. Biochemistry. 2007;46:8379. doi: 10.1021/bi700531r. [DOI] [PubMed] [Google Scholar]
- 18.Spence RA, Kati WM, Anderson KS, Johnson KA. Science. 1995;267:988. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popov VM, Chan DC, Fillingham YA, Atom Yee W, Wright DL, Anderson AC. Bioorg. Med. Chem. Lett. 2006;16:4366. doi: 10.1016/j.bmcl.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 20.Gargala G, Delaunay A, Li X, Brasseur P, Favennec L, Ballet JJ. J. Antimicrob. Chemother. 2000;46:57. doi: 10.1093/jac/46.1.57. [DOI] [PubMed] [Google Scholar]
- 21.Theodos CM, Griffiths JK, D'Onfro J, Fairfield A, Tzipori S. Antimicrob. Agents Chemother. 1998;42:1959. doi: 10.1128/aac.42.8.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watney J, Agarwal P, Hammes-Schiffer S. J. Am. Chem. Soc. 2003;125:3745. doi: 10.1021/ja028487u. [DOI] [PubMed] [Google Scholar]