

Introduction
The procedure for improving the quality and safety of any product or service, both directly related to the characteristics of the process through which the product or service is generated, involves a series of common, preliminary steps:
a description of the processes related to the life-cycle of the product,
identification of all the elements involved in the processes (informatics systems, procedures, human resources, infrastructures) with the creation of a detailed inventory of the materials and equipment,
the definition of measurable points of control for every element; the controls must be based on requisites identified a priori and on a mechanism in which the controller and the controlled person are not the same.
The information collected by quality management systems and risk analysis techniques is combined in order to determine the priorities for interventions aimed at making improvements.
Risk analysis techniques
The management of risks in complex processes, such as those present in most health care activities, must be conducted in a multidisciplinary manner. The main aim is to provide an efficacious service for patients, recognising and managing both human errors and system errors with the same level of detail as "good healthcare practices".
The need to prevent human error has led to the distinction of different types of faults: active errors, which cause immediate consequences, and latent errors, which are not manifested until the occurrence of a triggering event.
The aim of prevention is to identify every critical situation which could conceal a possible fault, active or not. The crucial aspect is the problem of exposing and managing latent errors, since it is particularly difficult to evaluate the direct and combined effects of such errors. The levels of safety for the patient must be improved by creating "protections" in which technology plays an important role and is connected with the managerial and organisational systems.
Various different methodological instruments can be used for the analysis of processes, depending on the level of detail required. All these techniques are aimed at an "early diagnosis" of possible faults, without neglecting the final client's satisfaction. Both qualitative and semi-quantitative approaches can be employed, these being based, respectively, on the use of purely descriptive terms or numerical indices assigned according to more objective definitions.
The concept of quality assurance rather than quality control is now widely accepted in quality management systems; that is, the tendency to guarantee a priori the quality of a product or a service, rather than ascertaining the quality a posteriori on the final output. In general, two broad approaches can be used:
- reactive analyses, in which a process is studied after an event, in order to identify all the causes which led to the occurrence of the event,
- proactive analyses, in which a process is reviewed with the aim of detecting critical points of the system in order to remove or markedly reduce them.
Simplified greatly, it can be said that a reactive analysis is based on an a posteriori examination of what has already happened in order to understand the causes of the failure; the strategies most widely used in this field are incident reporting, reviews, the techniques of "sign research" and root causes analysis. On the other hand, the proactive approach, typical of failure mode and effect analysis (FMEA) and failure mode, effect and criticality analysis (FMECA), is based on a preventive study of the process with the aim of eliminating critical elements of a system, before an incident occurs.
Given the complexity and the wide variability of their application, processes in a healthcare structure are excellent candidates for the use of these methods. The possibility of using an analytic instrument to collect and manage all the elements necessary to reveal the critical points as well as the capacity to define synthetic and objective indicators that can be applied to the process are fundamental factors when choosing the most appropriate method.
From a rapid overview of existing literature, it can be seen that, in the field of healthcare, these methods are used predominantly to:
- assess the potential advantage of introducing new processes, equipment, etc.;
- analyse processes in use, identifying their main critical points, monitoring the effect of any changes, whether minimal or substantial, and evaluating their impact on the process
- compare, both retrospectively and prospectively, old and new processes with various levels of criticity;
- guarantee the quality and safety of medical devices.
Clinical Risk Management and Quality Risk management
The techniques for analysing risks are risk management instruments, which represent the set of complex actions put in act to guarantee the safety of patients and healthcare staff and which constitute, together with quality management systems (QMS)1, one of the main applications of "clinical governance". Clinical risk management initially emerged as a response to financial aspects of healthcare related to the growing number of medico-legal actions and requests for compensation, but over the years is becoming part of the structured, planned activities of the system and of the interventions to improve the quality of healthcare services. In fact, safety derives from the capacity to plan and manage procedures able, on the one hand, to limit the effects of errors that do occur (protection) and, on the other hand, to reduce the probability that such errors occur (prevention). For a system to be effective and efficient there must be full integration between risk management and QMS, as experience from the industrial sector has already demonstrated (see ICH Q9 Quality risk management)2.
At the Transfusion Service of Meyer University Hospital in Florence, we had carried out various non-systematic risk analyses; subsequently, thanks to the initiatives and opportunities made available through collaboration with external companies (Pharma Quality Europe - PQE) and institutional activities (Prevention and Protection Service)3, we tested the structured use of a proactive technique of risk analysis (FMECA) in the real process of collecting, processing and distributing haematopoietic stem cells from related allogeneic donors in an attempt to identify risk mitigation interventions that could be applied in this context. We tried to analyse the risks in a systematic manner, in part spurred by institutional 4,5 and professional6 accreditation systems and by recent legislation, which has given great emphasis to the quality and safety of this process that is subject to regulations relating to both transfusion matters and tissue and cell banks5,7,8. The management of equipment and medical devices9 as well as the structure's informatics system have emerged from risk evaluations as critical points of great importance. Indeed, the informatics system has become of particular relevance since the introduction of Ministerial Decree 208/077. In paragraph (q) of article 1 of this decree, an "informatics system" is defined as a "system that incorporates data entry, electronic processing and the production of information to be used for the purposes of notification, automatic control and documentation". These systems, as indicated in various parts of the decree and, in particular, in article 5 "…must undergo regular controls of their reliability, be validated before use and be subjected to periodic servicing to ensure that they continue to fulfil the planned requisites."
FMEA/FMECA
From among the techniques mentioned above, we decided to test the FMECA one in this study. We will briefly describe the underlying principles of this technique, starting from its basis, FMEA.
FMEA is a method for analysing a system; its purpose is to identify possible modes of failures/errors, their causes and their effects on the performance of the system. It is a proactive method of analysis that closely follows the logical process used by a good planner. Its application makes it possible to identify and, therefore, mitigate, the risk of defects/errors in a product or in a service, at the same time as providing a reference against which to compare the real behaviour of the product. This technique does, however, have the intrinsic limitation of considering the types of failure as independent, which means that chain effects of different types of errors are not taken into account.
The term FMECA is used when the analysis is extended to include the severity of the consequences of a non-conformity. FMECA incorporates a system for classifying the severity of the modes of failure, with the aim of enabling priorities to be made when planning any corrective measures to adopt. The general considerations on FMEA that we make below are, therefore, also valid for FMECA.
The decision to undertake a FMEA may be made because of the need to identify failures capable of causing unwanted effects on the system, to satisfy specifications and requisites imposed by a "client" (within or outside the system) or to improve the reliability, safety and ease of maintenance of the system itself. The aims of FMEA, therefore, include the identification and classification of functional failures of the system in relation to their relevant characteristics (measurability, ease of identification, actions that can be taken, etc.), an estimate of the severity and probability of the failure, the development of improvement plans to mitigate the types of faults and, finally, the possibility of supporting the development of maintenance projects designed to attenuate or reduce the probability of failures.
The following steps are essential parts of the FMEA methodology:
establish the fundamental underlying rules for the FMEA and the schedule guaranteeing the availability of skills and time necessary to start the analysis;
carry out the FMEA using appropriate spreadsheets or instruments, such as fault tree analysis, logical diagrams, etc.;
produce quantitative results and reports that include the conclusions and recommendations triggered by the analysis;
maintain the FMEA updated.
According to the FMEA, the process/product/service must be broken down into its elements, defining, for each of these:
- its function;
- a description;
- the mode of failure, that is, the way in which the product/process/service may not fulfil its function and thus satisfy the requisites, needs, expectations and requirements of the client (manifestation of the failure or of the non-conformity);
- the mechanism of the failure, that is, the chemical, physical or other process that gave rise to the failure;
- the cause of the failure, that is, the circumstances related to the project, to its realisation, to the use of the product/service that generated the failure;
- the effects, defined as "local" when they have repercussions on the element itself and "final" when, on the other hand, the failure has an impact at a higher level;
FMECA, applied in this study, has three additional parameters:
O (Occurrence): the probability of the occurrence of a non-conformity (intended in our case as the combination of modes of failure and effects on a patient, donor, or member of staff) in a predetermined period of time;
S (Severity): the severity/criticalness of the failure;
D (Detectability): the difficulty in detecting a nonconformity.
Each of these three non-dimensional parameters is arbitrarily assigned a value from 1 to 10 and the product of these three values provides a fourth index known as the Risk Priority Number (RPN)
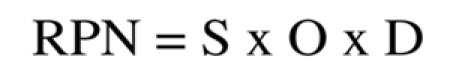 |
which must be as low as possible.
The value assigned to O is an increasing number on the scale of integers from 1 to 10 (as done for the S parameter), rather than the probability of the occurrence in a period of time; this approach, which is widely used in the literature, simplifies the whole evaluation process considerably.
The 'Detectability' parameter, D, is an estimate of the probability of identifying and eliminating a fault before this has negative effects on one of the three subjects considered (patient, donor, member of staff). For ease of calculation, this index is assigned values in an inverse order with respect to those used for S and O: high values of D (again expressed as an integer from 1 and 10) indicate greater difficulty in detection and, therefore, less probability of identification, thus contributing to increasing the final value of the RPN.
The range of whole numbers between 1 and 10 was chosen to maintain a certain conformity with the fields of application in which the use of FMECA has been consolidated for some time, such as analyses in the car industry of planning processes (DFMEA –design failure mode and effects analysis) and of production processes (PFMEA – production failure mode and effects analysis). In order to lower this index, project modifications are introduced to reduce the value of at least one of the indicators (the Severity is difficult to reduce). These strategies are known as design reviews (corrective actions).
The RPN can be useful for defining the scale of priorities for mitigation interventions to adopt for the various types of failure. However, as will be clarified later, the RPN must always be evaluated together with the three values that generate it. For example, for the same RPN, it would be wise to give precedence to resolving those modes of failure for which the severity values, S, are highest.
The choice of method to use to select items for risk reduction interventions is subjective and depends strictly on the type of application. According to the aim, intervention thresholds can be set based on the RPN (often corrective measures are taken for the items with the highest RPN in each phase, then the FMEA is updated and the procedure repeated), or on other parameters (typically the Severity factor, S) ignoring the RPN10–12.
Application of FMECA to the process of collecting, manipulating and distributing haematopoietic stem cells in the Transfusion Service of Meyer University Hospital
In this context the risk mitigation strategies should be developed to protect the product, the patient and the staff, without undervaluing the risks related to nonconformities with current legislation. The first task to perform with the purpose of managing risks is to identify them. In a first analysis, the risks can be grouped into the following general categories:
Adverse effects in the patient prior to transplantation
Adverse effects in the donor
Adverse effects in members of staff
Loss of product
"Insufficient" product
Contaminated product
Non-conformity with legislation.
These risks can, for example, be caused or fostered by the use of "open processes", the use of complex instruments (e.g. apheresis equipment, immunomagnetic separators, freezing systems, cyrocontainers), the lack of standardised quality control tests, the use of potentially infective material, and the clinical status of the recipient.
The FMECA was planned by the head of the Transfusion Service's QMS and lecturers at the Department of Electronics and Telecommunication at the University of Florence, who also compiled the tables and subsequent processed the information. The occasion was taken to review the description of the processes carried out in the Service, in order to a have a complete, updated overview of the micro- and macro-activities, staff, equipment and informatics systems (software/hardware – SW/HW) involved. The collaboration of Pharma Quality Europe (PQE) was useful in the evaluation of the SW/HW systems, which were given particular attention. The head of the hospital's Protection and Prevention Service contributed to evaluating the risks to staff. Besides the professional experience of the staff, the data recorded by the structure's QMS (non-conformities, adverse events in patients and donors, etc.) were of help in evaluating the probabilities of certain occurrences. Indeed, one of the greatest difficulties was evaluating these probabilities, particularly for events that are so rare that they have never occurred in the Service and for which the scientific literature does not provide useful data.
The level of detail to use must be established in order that each phase can, in its turn, be analysed in more depth, by breaking it down yet further.
The critical points and description of the process of collecting, processing and distributing haematopoietic stem cells
Given its characteristics (treatment of "non-repeatable" products, type of diseases treated, involvement of healthy donors, use of complex equipment, etc.) the process under study is recognised as having numerous critical points that can be examined from two points of view: that of the people involved (donors, recipients and members of staff)13,14 and that of the process.
"Flow charts" representing the whole process of collecting, manipulating and distributing haematopoietic stem cells were used to highlight all the relative critical points which could be identified either theoretically, according to the literature, or through the professional experience of the staff15–27. The overall process was divided into six macrophases, each identified by a number indicating its chronological position:
Selection and suitability of the donor
Stem cell mobilisation
Collection
Manipulation and validation
Freezing and cryopreservation
Thawing and distribution
For each of these macrophases, a table divided into four parts was used (Figure 1).
Figure 1.

As an example, part of the table used to analyse macrophase 4 – Manipulation and validation
The tables consisted of a:
-
Schematic description of the activity, including: (a) a list of the phases, (b) staff, (c) equipment, (d) software, (e) documents, (f) materials, and (g) premises.
Each entry is repeated for each type of fault, in order to enable its unique identification.
Description of the failure and its relative effects on patients, donors and staff
Quantitative evaluation of the failure
Any corrective or preventive actions taken.
The mechanisms and causes of faults were not recorded, nor were the mild side effects inextricably related to the process (e.g. headache or bone pain during stem cell mobilisation); furthermore, a choice was made not to describe and consider phases of the transplant programme managed by others or not directly interfaced with the structure.
The choice of combining an analysis of the process with an evaluation of the effects of the modes of failure was found to be useful for a better identification and classification of the risks. In fact, the attribution of O, S and D values can differ substantially depending on the consequences for patients, donors and members of staff. For this reason, the process breakdown structure (PBS) codes reported in the tables, identified by progressive numbers, take into account the different effects on the people involved in the process. The PBS, in analogy to work breakdown structure (WBS) techniques used in the field of project management, is structured hierarchically in three levels whose numerical values identify, respectively, macrophases, phases and types of faults with their effects.
Criteria for attributing the O, S and D values
One of the major limitations of the technique is the lack of standardised scales of values for the O, S and D parameters and the consequent difficulty in identifying criteria with which to assign the scores in such a way as to reduce the subjective component related to direct, personal experience.
The Occurrence and Detectability scores were divided into three classes (low, medium, high), whereas for the Severity score, the values between 6 and 10 were left independent as representative of well identifiable and particularly severe (sometimes catastrophic) adverse events. Indeed, the most severe adverse events were used as the end of the scale for the S values, attributing maximum values8,9,13 to the most severe events conceivable, for example, the death of a healthy donor, distribution of an unsuitable product, loss of the product.
Those factors with consequences on subsequent phases were considered worsening factors of the Severity parameter (see tables I, II, III).
Table I.
Criteria for attributing the score for the Severity paramet er
| Score | Class | Description (examples) |
|---|---|---|
| 1–3 | Mild | Repetition of the organisational inconvenience/activity causing loss of efficiency/efficacy |
| 4–5 | Moderate | Medico-legal problems/loss of information/interruption of the process |
| 6 | Severe | Distribution of a quantitatively insufficient product |
| 7 | Loss of the product (breakage, tampering) | |
| 8 | Distribution of a qualitatively inadequate product (contaminated) | |
| 9 | Distribution of an unsuitable product, in that it is destined for another recipient | |
| 10 | Adverse events in healthy donor/staff member leading to irreversible lesions up to death |
Table II.
Criteria for attributing the score for the Occurrence parame ter
| Score | Class | Description (examples) |
|---|---|---|
| 1–3 | Low | Event that has never occurred and/or is rarely reported in the literature or in benchmarking analyses |
| 4–6 | Medium | Event that has occurred occasionally since starting the activity |
| 7–10 | High | Event that has occurred many times since starting the activity |
Table III.
Criteria for attributing the score for the Detectability par ameter.
| Score | Class | Description (examples) |
|---|---|---|
| 1–3 | High | Event detectable during the performance of the activity |
| 4–6 | Medium | Event not detectable during the performance of the activity, but for which there are reliable instruments for detection in the subsequent phase |
| 7–10 | Low | Event not detectable or detectable only at the end of the process |
Methodology and results of the data analysis
The data were analysed, independently for each of the six macrophases identified, as follows: the various items of the PBS – which represent, as said, the possible combinations between subphases, types of faults and effects on patients, donors, and staff –were re-ordered by decreasing RPN in order to highlight the points with the highest priority for intervention.
During this operation, all the items with a RPN=0 were excluded since they did not represent any type of failure. This first operation led to a list of items without any chronological consequences, although these could easily be identified since the above-described PBS code had been adopted.
The mean RPN, represented in the graph by a dashed horizontal line, was then calculated for each macrophase. The mean RPN is useful for both an analysis of the distribution of the priority of interventions within the lifecycle of the whole process and because possible threshold levels for risk mitigation interventions can be related to it.
The results of the FMECA analysis carried out are summarised in tables IV, V and VI.
Table IV.
Summary of the mean, mode, median, range and standard deviati on of the RPN values for each macrophase and for the whole process
| Macrophases | RPN Mean | Mode | Median | Range | Standard Deviation |
|---|---|---|---|---|---|
| 1. Donor selection and suitability | 62.55 | 70 | 70 | 120–10 | 29.72 |
| 2. Stem Cell mobilisation | 51.86 | 63 | 56 | 90–15 | 22.21 |
| 3. Collection | 57.50 | 72 | 56 | 120–10 | 28.16 |
| 4. Manipulation and validation | 47.61 | 40 | 40 | 135–5 | 28.14 |
| 5. Freezing and cryopreservation | 61.06 | 126 | 52 | 126–8 | 39.57 |
| 6. Thawing and distribution | 49.46 | 72 | 48 | 108–6 | 24.45 |
| Overall value | 55.49 | 48 | 54 | 135–5 | 30.05 |
Table V.
List of items with an RPN greater than 100, in decreasing or der. S scores of 8 or above are shown in grey
| PBS | Phase | O | S | D | RPN |
|---|---|---|---|---|---|
| 4.02.01 | Control of the level of contamination prior to entry | 3 | 9 | 5 | 135 |
| 5.03.01 | Validation of the bag | 2 | 9 | 7 | 126 |
| 5.23.03 | Definitive storage in liquid nitrogen (liquid or vapour phase) | 2 | 9 | 7 | 126 |
| 5.25.02 | Evaluation of the report on the sterility controls | 2 | 9 | 7 | 126 |
| 5.15.01 | Labelling of daughter bags | 2 | 9 | 7 | 126 |
| 5.15.03 | Labelling of daughter bags | 2 | 9 | 7 | 126 |
| 1.05.02 | Information to the donor and release of informed consent | 5 | 6 | 4 | 120 |
| 3.03.01 | Performance of the medical visit | 3 | 8 | 5 | 120 |
| 5.06.01 | Entry of data into the SW form/number of aliquots/calculation of volume | 4 | 6 | 5 | 120 |
| 5.06.02 | Entry of data into the SW form/number of aliquots/calculation of volume | 3 | 8 | 5 | 120 |
| 3.26.01 | Post-donation information | 4 | 7 | 4 | 112 |
| 5.05.01 | Registration of donor’s and patient’s data on the freezing form | 4 | 7 | 4 | 112 |
| 5.19.01 | Freezing at −80°C | 2 | 8 | 7 | 112 |
| 5.15.02 | Labelling of daughter bags | 2 | 8 | 7 | 112 |
| 5.15.04 | Labelling of daughter bags | 2 | 8 | 7 | 112 |
| 5.23.02 | Definitive storage in liquid nitrogen (liquid or vapour phase) | 2 | 8 | 7 | 112 |
| 3.06.01 | Labelling of bags | 3 | 9 | 4 | 108 |
| 3.11.04 | Cleaning and disinfection of the skin - venipuncture – connecti on to the cell separator | 3 | 9 | 4 | 108 |
| 3.25.05 | Blood sampling and sending test-tubes for analysis | 3 | 9 | 4 | 108 |
| 6.06.01 | Verification of the correct storage of the bags | 3 | 9 | 4 | 108 |
| 3.07.01 | Evaluation of venous accesses: unsuitable accesses | 6 | 6 | 3 | 108 |
| 1.06.01 | Performance of the medical visit | 3 | 7 | 5 | 105 |
| 1.10.05 | Blood sampling and sending test-tubes for tests of suitability | 3 | 7 | 5 | 105 |
| 1.10.07 | Blood sampling and sending test-tubes for tests of suitability | 3 | 7 | 5 | 105 |
| 1.14.01 | Updating of the donor’s healthcare records | 4 | 5 | 5 | 100 |
Table VI.
Means and respective ranges of the O, S and D values
| Macrophases | O:Mean | Range | S:Mean | Range | D:Mean | Range |
|---|---|---|---|---|---|---|
| 1. Donor selection and suitability | 3.36 | 2–8 | 5.73 | 3–7 | 3.39 | 1–5 |
| 2. Stem Cell Mobilisation | 3.00 | 1–5 | 6.14 | 4–10 | 3.24 | 1–9 |
| 3. Collection | 3.19 | 2–6 | 6.33 | 3–9 | 2.88 | 1–*9 |
| 4. Manipulation and validation | 2.45 | 1–5 | 5.66 | 2–9 | 3.39 | 1–5 |
| 5. Freezing and cryopreservation | 2.50 | 1–4 | 6.98 | 3–10 | 2.58 | 1–7 |
| 6. Thawing and distribution | 2.62 | 1–5 | 6.89 | 5–10 | 2.78 | 1–5 |
| Overall score | 2.86 | 1–8 | 6.33 | 2–10 | 3.21 | 1–9 |
The analysis of the mean RPN and standard deviation for each of the single macrophases shows that these values remain similar to those calculated for the whole "population", that is, all the individual phases of the whole process (Table IV).
These findings, although far from representing a particular statistical result, do support the choice to apply risk mitigation interventions by analysing the individual macrophases separately and then repeating the procedure on the whole "population".
The phases taken into particular consideration were those with an RPN of 100 or more and those with a Severity score greater than 8, subdivided by macrophase (Table V). The results are discussed below phase by phase.
Selection and suitability of the donor
The risks requiring particular attention in this macrophase are those related to carrying out the medical visit, the correctness of the donors' records, the release of informed consent and the management of examinations of suitability for donation. The first two points are particularly important in the management of foreign donors: the inability to find a cultural mediator and/or information documents not translated into the potential donor's language can lead to a delay or even lack of harvesting of haematopoietic progenitor cells for transplantation; a donor can be induced to undergo procedures at risk during the stem cell mobilisation with severe clinical complications (e.g. rupture of the spleen) and legal consequences for the donor and staff, respectively. The RPN analysis also confirmed the importance of both a careful medical assessment, in that a less than meticulous evaluation can lead to the acceptance of an unsuitable donor, again with possible clinical consequences (for the donor) and legal implications (for the staff), and of the correct compilation of clinical records, since incomplete records imply a lack of registration of relevant clinical information and will have severe legal consequences for the staff.
During the phase of blood collection and sending samples for controls of suitability, errors in labelling or malfunction of the SW/HW systems involved can lead to an incorrect evaluation of the donor making it necessary to repeat the investigations (with possible legal consequences and/or organisational inconvenience for the staff). The validation of SW/HW systems could limit the frequency and consequences of their malfunction.
Stem Cell Mobilisation
There were no RPN of 100 or more for this macrophase, because although severe adverse effects, death and delayed onset severe adverse events in the donor have very high S values, the RPN values are medium or medium-high given that the probability of occurrence of such events is very low. The only point to highlight is the risk connected to errors in evaluation of the conformity of the drug used in the mobilisation.
Collection
The problems related to providing foreign donors with correct information recur in this macrophase. The aim is to prevent donors from carrying out potentially harmful activities after the stem cell collection. Likewise, risks associated with careless medical assessments (acceptance of an unsuitable donor, with possible clinical consequences for the donor and legal implications for the staff) must be avoided. In this macrophase, faults in labelling blood samples or a malfunction of SW/HW can cause errors in the critical phase of validation, with effects ranging from the need to repeat the investigations of the donor to the far more serious erroneous validation of the product, while a fault in labelling the bags can lead to the re-infusion of a wrong bag or contribute to the disposal of the unit.
Another delicate problem is the management of invasively obtained vascular accesses, whose possible contamination can lead to the collection of a contaminated product and, therefore, of a risk of infection for the patient, with legal consequences for the staff. Incorrect performance of the procedure can lead to severe adverse effects in the donor (e.g. convulsions, hypovolaemic shock, gas emboli). The phase of removing the product is also critical in that it involves risks related to the seal of the connecting tubes and to the removal of the bag.
Manipulation and validation
The most critical point of the whole process (with an RPN of 135) occurs in this macrophase: the control of contamination (of the laminar flow hood and premises) before their use. In the case of malfunction or inappropriate use, not only can non-sterile cells be reinfused, but staff may also become contaminated. Another delicate step is the determination of the volume of the collection, because an error in weighing and in calculating the volume can lead to an erroneous evaluation of the product and to the product being processed using the wrong parameters. Centrifugation to reduce the volume, during which the bag can break with consequent loss of the product, is another critical point. Finally, the step of verifying the conformity of the bags should not be overlooked, given that the acceptance of unsuitable bags could prejudice the outcome of the manipulation process.
Freezing and cryopreservation
The control of contamination (of the laminar flow hood and premises) is a critical step also in this macrophase, being important in various phases, such as the fractioning of the collection bag into aliquots to freeze and the addition of the cryopreservation solution.
The donors' and patients' data are recorded on the freezing form through the use of a calculation spreadsheet which, if set out poorly (overwriting of data, wrong formulae, lack of protection) can lead to a product with unsuitable characteristics, with possible legal consequences for the staff. The number of aliquots and their volumes are also calculated using an electronic spreadsheet: in this case a damaged datasheet or underestimate or overestimate of the number of CD34+ cells can lead to an error in the subdivision of the cells collected; in particular, an overestimate of the number of cells in the sample will lead to the final product being insufficient and the type of fault is, therefore, more serious, even for the same RPN.
As far as concerns the management of the bags, an error in labelling can lead to the wrong bag being infused or contribute to the disposal of the unit; during the transport of the bags to the cryopreservation area, a fall by the person carrying the units can lead to loss of the product, particularly if the containers used are not suitable, and to injury to the member of staff; during the phase of freezing to −80°C, a malfunction of the freezer can lead to a defective freezing process (e.g. too slow) with consequent damage to the product; finally, during the definitive storage in liquid nitrogen (liquid or vapour phase) an error in the placement of the bag can lead to the risk of it being lost. The placement of a serologically positive unit (e.g. positive for hepatitis B or C virus) in a liquid phase can lead to contamination of the container and a risk for the other units, while malfunctioning of the equipment can expose staff to severe adverse events.
The validation of the product involves transfusion managment software but, unlike the same phase for the blood components, is not automated. There is a risk of erroneous validation of products with unsuitable characteristics.
The evaluation of the sterility of the sample is important because if this is not carried out, there is a risk of releasing uncontrolled products: if the product is not sterile and cannot be replaced, there are risks inherent in the management of a contaminated transplant, while mistaken interpretation of results can lead to the infusion of a contaminated product without the possibility of taking the appropriate precautions.
Thawing and distribution
Before thawing the samples, the request itself must be evaluated and the presence and correct storage of the bags must be verified; furthermore, the bags stored in liquid phase must be transferred to the vapour phase before thawing. If the request is evaluated by untrained staff, errors may occur in the subsequent phases; the consequences of missing, incorrectly stored or damaged bags are varied (delay or lack of transplant, infusion of an unsuitable product, need for the donor to undergo repeat apheresis), but all have obvious clinical consequences for the recipient and legal consequences for the staff. Breakage and/or contamination of the bag during thawing or transport to the ward can make it necessary to repeat the apheresis or infuse a contaminated product. To avoid infusion of a wrong bag, care must be taken during the phases of identifying and assigning the bag, printing the labels and making the delivery report, which depend on the good function of the SW/HW systems. The viability of the product and the residue of the infusion must be controlled to determine their efficacy. Finally, all the risks related to the use of liquid nitrogen in the cryopreservation process (burns, adhesion, inhalation, contact with the eyes, freezing, death) must be taken into due consideration.
Conclusions
The first finding to report is the compression of the scale of RPN values which occurs when applying FMECA in this context: against a theoretical range of RPN values from 0–1000, the actual range was compressed to between 5 and 135. Since the RPN is the product of the values for three factors parameters (O, S and D), its increase is not linear and it is particularly sensitive to non-parallel increases in the three parameters. In fact, in our case the most severe events (with high S values) were also usually the rarest ones and the ones most easily detected (i.e. with low O and D values). The mean values of O, S and D (Om=2.86; Dm= 3.21; Sm=6.33) bear witness to this tendency.
As can be deduced from the data presented in table VII, the highest scores (5–10) were assigned more frequently to the S parameter: this fact limits the possibility of improvement, because the severity of an event, even if it can be reduced by protective-type interventions, is the most difficult parameter to control.
Table VII.
Relative frequencies of the scores attributed to the O, S an d D parameters, calculated for the whole process. The highest frequencies for each score are shown in grey
| Score | Occurrence (O) | Relative frequency (%)Severity (S) | Detectability (D) |
|---|---|---|---|
| 1 | 22.0 | 0.0 | 78.0 |
| 2 | 70.9 | 0.7 | 28.4 |
| 3 | 53.7 | 3.7 | 42.5 |
| 4 | 33.1 | 7.4 | 59.5 |
| 5 | 12.0 | 63.2 | 24.8 |
| 6 | 8.9 | 91.1 | 0.0 |
| 7 | 1.7 | 83.3 | 15.0 |
| 8 | 2.7 | 97.3 | 0.0 |
| 9 | 0.0 | 95.2 | 4.8 |
| 10 | 0.0 | 100.0 | 0.0 |
When deciding the priority of interventions, it is clearly important to use a critical approach rather than relying on a purely numerical method of selection: for example, the most catastrophic types of faults (S=10) do not appear in the list of PBS with the highest RPN. Furthermore, the same type of fault can be associated with different RPN depending on the effects caused: an "error of labelling" during the activity "blood sampling for analysis" in the mobilisation phase (item 2.08.05) has a RPN score of 90, while the same item at the end of the collection phase (item 03.25.05) reaches a RPN of 108. It can also occur that an item with a lower RPN, for example, item 1.05.01 ("lack of request of informed consent from the donor", RPN=63) but a higher S score (S=7) warrants the same or greater attention than an item with a higher RPN, for example, item 1.14.02, (a "malfunction of software" which prevents updating of the donor's clinical records, RPN=75), but that has a lower S score (S=5).
Organisational inconvenience and legal consequences are among the most frequent effects for staff members. The former occurs in part because of the difficulties in managing a process included in the complex pathway of haematopoietic transplantation and, therefore, opportune interventions to improve organisational aspects are essential features of risk mitigation. The latter effects, also related to staff errors, are explained by the criticality of the process and the recent proliferation of overlapping regulations which make conformity of the systems ever more difficult. Although the FMECA technique was not conceived to prevent intrinsic staff errors, it can help to identify, as in our analysis, fundamental preventive actions, such as staff training, and thus contribute indirectly to the management of this type of error (Figure 2).
Figure 2.

The frequency of two of the main preventive/corrective actions (staff training and SW/HW validation) in the various phases
Table V, which reports the items with a RPN above 100, highlights the importance of "traditional" critical points in the process, such as controlling contamination of the product, obtaining informed consent from the donor and managing cryogenic equipment, whose great importance could have been hypothesised a priori.
However, the table also highlights the particular attention that must be given to risks3 related to the SW/HW systems which, also in this systematic analysis, are shown to warrant a high priority. As already emphasised, the introduction of automation leads to more efficient, but not necessarily safer, systems, because these are associated with new, important critical points, whose treatment requires sophisticated and expensive instruments. Although there is less automation and integration of SW/HW systems in the process of collecting and manipulating haematopoietic stem cells than in the "standard" transfusion process, the role of such integrated systems is determinant and increasing in many critical decisional steps. Indeed, the results of the FMECA show that possible corrective actions for risk reduction include the development of plans to test and validate these systems (Figure 2).
The recent ministerial decree 208/077, which makes this type of intervention unavoidable, does not, however, report the immediately applicable models or standards. In the absence of these, the only possible option seems to be to introduce, also into health care processes, the instruments used to guarantee the safety of SW/HW systems foreseen by pharmaceutical Good Manufacturing Practice (GMP)28,29 according to the fourth edition of Good Automated Manufacturing Practice (GAMP4)30. The definition of "validation" reported in the decree7, "the preparation of documented and objective evidence demonstrating that the predefined requisites of a procedure or of a specific process can be systematically met", is the same as that reported in the GMP since its earliest versions (1987). The GAMP4 calls for the planning of a "life-cycle" of an informatics system, which goes from the initial version of the User Requirement Specifications through to the phase of final validation (Performance Qualification) according to a "V model" (Figure 3).
Figure 3.

The life-cycle of the planning, realisation and validation of a SW/HW system
The V model is based on the principle that validation documentation must be integrated synergistically with the planning documentation, paralleling this latter's development, although always giving due attention to the separation of the roles between clients and provider, so that there is no overlapping or conflicts of interests.
In summary, in sectors in which the safety of people is at great risk (e.g. the aeronautic, nuclear, pharmaceutical and health care industries) reactive type risk analyses and mitigation techniques based on adverse events and non-conformities are not sufficient. What is needed is proactive instruments that are able to provide the management system with information useful for introducing appropriate preventive actions and evaluating the priority of interventions. It is clear that the QMS and risk management information and instruments must be integrated in order to function effectively and efficiently. Many operations involved in both approaches are the same or so similar as to be able to be carried out in a coordinated manner. The integrated use of FMECA was found to be very useful in this particular context.
The underlying concepts of FMEA/FMECA10 are, in brief, the breakdown of the system with the definition of the relative functional diagrams and the indication of modes of failure and critical points. Some considerations can be made regarding these elements, which are the real contribution to the problem of risk management in a process such as that under evaluation:
The methodology can really be applied also to this type of process, with the advantage of making the various phases of the process "objective", thus decreasing the probability of overlooking some of them in the risk analysis.
The multidisciplinary team (staff who are expert in health care, technical and organisational aspects) is involved in every stage of the work, with the advantage of providing a global perspective both during the application of the method and identification of the types of fault, and during the analysis of the critical points and possible solutions; this ensures less loss of time, given that the various aspects of each problem are immediately discussed by everyone, and, overall, better results in terms of the efficacy of the solutions proposed.
The methodology used does not provide solutions, but focuses attention on problems. The breakdown of the process highlights the possible faults of the system, attributing them a semi-quantitative weight and indicating the priorities for both more detailed analysis and subsequent improvement plans, once again demonstrating how the integration between "quality systems" and risk management improves the efficacy and efficiency of the management of processes and rationalises the choice of the priority of interventions.
There are situations in which critical points are related to activities conducted externally (e.g. in other laboratories) for which the lack of direct control constitutes the main "weak" factor, highlighting the need to broaden the evaluation of suppliers.
FMECA, effective in relation to system errors due to technical-organisational faults, is ineffective in preventing intrinsic staff errors (e.g., a mistaken clinical evaluation). It can, however, help to identify, as in our analysis, fundamental preventive actions, such as staff training, thus contributing indirectly to the management of this type of fault. FMECA, by facilitating the creation of an integrated system between technical, health care and organisational aspects, moves out of line or eliminates some of the "holes" in the Swiss cheese described by Reason31–33.
It is customary that the last step of the FMECA procedure is a periodic update of the analysis to "measure" the progress of the improvement plans. This partially corrects one of the limitations of the method, the way the various indices are assigned; indeed, the repetition of the analysis reveals the trend, making it less important to establish the starting point exactly.
One of the assumptions of the technique, which partially limits its field of action, is that of considering the faults as independent (an all the more critical aspect when considering processes that involve both software and hardware). This prevents a chain effect of different errors from being taken into account. In these cases it is worth supporting the FMECA with other analytic instruments, such as fault tree analysis11,12.
The RPN index has some limitations that should be taken into consideration, such as excessive sensitivity to small changes in one of its three constituent parameters when the other two parameters have high values and vice versa. Thus, although it is a useful, synthetic index, it must be analysed in context with its three components O, S and D. In some cases it could be worth modifying the effects of the single parameters on the RPN product by using factors of correction.
Legislation and references
- 1.Sistemi di Gestione per la Qualità – Requisiti. UNI EN ISO 9001:2000. 2000. [Google Scholar]
- 2.ICH Harmonized Tripartite Guideline Quality Risk Management Q9. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2005. [Google Scholar]
- 3.Bambi F, Spitaleri I, Gianassi S, et al. Raccolta e manipolazione delle cellule staminali emopoietiche -Comunicazione al Convegno dei servizi trasfusionali. Stresa. 2007;(RE13):37–40. [Google Scholar]
- 4.Conferenza Stato Regioni, Accordo 10 luglio 2003, Linee-guida in tema di raccolta, manipolazione e impiego clinico delle cellule staminali emopoietiche CSE. [24th July 2008];Ministero della Salute. Available at http://www.normativasanitaria.it/jsp/dettaglio.jsp?aggiornamenti=&attoCompleto=si&id=5924&page=&anno=null,doc.
- 5.Decreto Legislativo 6 novembre 2007, n.191 Attuazione della direttiva 2004/23/CE sulla definizione delle norme di qualità e di sicurezza per la donazione, l'approvvigionamento, il controllo, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umani. [24th July 2008];Ministero della Salute. Available at http://www.normativasanitaria.it/jsp/dettaglio.jsp?aggiornamenti=&attoCompleto=si&id=26011&page=&anno=null,doc.
- 6.International Standards for Cellular Therapy Product Collection, Processing, and Administration. 3rd ed. Joint Accreditation Committee-ISCT & EBMT (JACIE); 2007. [Google Scholar]
- 7.Decreto Legislativo 9 novembre 2007, n.208 Attuazione della direttiva 2005/62/CE che applica la direttiva 2002/98/CE per quanto riguarda le norme e le specifiche comunitarie relative ad un sistema di qualità per i servizi trasfusionali. [24th July 2008];Ministero della Salute. Available at http://www.normativasanitaria.it/jsp/dettaglio.jsp?aggiornamenti=&attoCompleto=si&id=23571&page=&anno=null,doc.
- 8.Decreto Legislativo 19 agosto 2005, n. 191 Attuazione della direttiva 2002/98/CE che stabilisce norme di qualità e di sicurezza per la raccolta, il controllo, la lavorazione, la conservazione e la distribuzione del sangue umano e dei suoi componenti. [24th July 2008];Ministero della Salute. Available at http://www.normativasanitaria.it/jsp/dettaglio.jsp?aggiornamenti=&attoCompleto=si&id=20989&page=&anno=null,doc.
- 9.Application of Risk Management to Medical Devices. 2007 ISO 14971: 2007. [Google Scholar]
- 10.Metodi di Analisi per l'Affidabilità dei Sistemi. Procedura di Analisi dei Modi e degli Effetti di Guasto (FMEA) 2nd ed. 2006. CEI EN 60812. [Google Scholar]
- 11.Analisi ad Albero delle Avarie. 2nd ed. 2007. CEI EN 61025. [Google Scholar]
- 12.Gestione della Fidatezza. Parte 3-1: Guida Applicativa -Tecniche di Analisi per la Fidatezza - Guida ai Metodi. 2nd ed. 2005. CEI EN 60300-3-1. [Google Scholar]
- 13.Guide to Safety and Quality Assurance for the Transplantation of Organs, Tissues and Cells. 3rd ed. Council of Europe; 2006. [Google Scholar]
- 14.Decreto Legislativo 19 settembre 1994, n. 626 Attuazione delle direttive 89/391/CEE, 89/654/CEE, 89/655/CEE, 89/656/CEE, 90/269/CEE, 90/270/CEE, 90/394/CEE, 90/679/CEE, 93/88/CEE, 95/63/CE, 97/42, 98/24, 99/38 e 2001/45/CE riguardanti il miglioramento della sicurezza e della salute dei lavoratori durante il lavoro. [28th July 2008];Ministero della giustizia. http://www.giustizia.it/cassazione/leggi/dlgs626_94.html,doc.
- 15.Wiltbank T, Giordano GF. The safety profile of automated collections: an analysis of more than 1 million collections. Transfusion. 2007;47:1002–5. doi: 10.1111/j.1537-2995.2007.01224.x. [DOI] [PubMed] [Google Scholar]
- 16.Anderlini P, Donato M, Chan KW, et al. Allogeneic blood progenitor cell collection in normal donors after mobilization with filgrastim: the M.D. Anderson Cancer Center experience. Transfusion. 1999;39:555–60. doi: 10.1046/j.1537-2995.1999.39060555.x. [DOI] [PubMed] [Google Scholar]
- 17.Chen-Plotkin AS, Vossel KA, Samuels MA, Chen MH. Encephalopathy, stroke, and myocardial infarction with DMSO use in stem cell transplantation. Neurology. 2007;68:859–61. doi: 10.1212/01.wnl.0000256716.04218.5b. [DOI] [PubMed] [Google Scholar]
- 18.Faulkner LB, Tucci F, Tamburini A, et al. G-CSF serum pharmacokinetics during peripheral blood progenitor cell mobilization: neutrophil count-adjusted dosage might potentially improve mobilization and be more cost-effective. Bone Marrow Transplant. 1998;21:1091–5. doi: 10.1038/sj.bmt.1701241. [DOI] [PubMed] [Google Scholar]
- 19.Bambi F, Faulkner LB, Azzari C, et al. Pediatric peripheral blood progenitor cell collection: haemonetics MCS 3P versus COBE Spectra versus Fresenius AS104. Transfusion. 1998;38:70–4. doi: 10.1046/j.1537-2995.1998.38198141501.x. [DOI] [PubMed] [Google Scholar]
- 20.Bambi F, Fontanazza S, Messeri A, et al. Use of percutaneous radial artery catheter for peripheral blood progenitor cell collection in pediatric patients. Transfusion. 2003;43:254–8. doi: 10.1046/j.1537-2995.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 21.Iannalfi A, Bambi F, Tintori V, et al. Peripheral blood progenitor uncontrolled-rate freezing: a single pediatric center experience. Transfusion. 2007;47:2202–6. doi: 10.1111/j.1537-2995.2007.01447.x. [DOI] [PubMed] [Google Scholar]
- 22.Basini V, Cinotti R, Di Denia P. FMEA/FMECA Analisi dei Modi di Errore/Guasto e dei Loro Effetti nelle Organizzazioni Sanitarie - Sussidi per la Gestione del Rischio 1. ISSN 1591-223X 2002. Dossier. 75 [Google Scholar]
- 23.DeRosier J, Stalhandske E, Bagian JP, Nudell T. Using Health Care Failure Mode and Effect Analysis: the VA National Center for patient safety's prospective risk analysis system. The Joint Commission Journal on Quality Improvement. 2002;28:248–67. doi: 10.1016/s1070-3241(02)28025-6. [DOI] [PubMed] [Google Scholar]
- 24.Saif MW, Leitman SF, Cusack G, et al. Thromboembolism following removal of femoral venous apheresis catheters in patients with breast cancer. Ann Oncol. 2004;15:1366–72. doi: 10.1093/annonc/mdh347. [DOI] [PubMed] [Google Scholar]
- 25.Eder AF, Kennedy JM, Dy BA, et al. Bacterial screening of apheresis platelets and the residual risk of septic transfusion reactions: the American Red Cross experience (2004–2006) Transfusion. 2007;47:1134–42. doi: 10.1111/j.1537-2995.2007.01248.x. [DOI] [PubMed] [Google Scholar]
- 26.Pulsipher MA, Levine JE, Hayashi RJ, et al. Safety and efficacy of allogeneic PBSC collection in normal pediatric donors: the Pediatric Blood and Marrow Transplant Consortium Experience (PBMTC) 1996–2003. Bone Marrow Transplant. 2005;35:361–7. doi: 10.1038/sj.bmt.1704743. [DOI] [PubMed] [Google Scholar]
- 27.Michon B, Moghrabi A, Winikoff R, et al. Complications of apheresis in children. Transfusion. 2007;47:1837–42. doi: 10.1111/j.1537-2995.2007.01405.x. [DOI] [PubMed] [Google Scholar]
- 28.Decreto Legislativo 24 aprile 2006, n. 219 Attuazione della direttiva 2001/83/CE (e successive direttive di modifica) relativa ad un codice comunitario concernente i medicinali per uso umano, nonché della direttiva 2003/94/CE. [28th July 2008];Ministero della Salute. http://www.normativasanitaria.it/jsp/dettaglio.jsp?aggiornamenti=&attoCompleto=si&id=23926&page=&anno=null,doc.
- 29.Medical Products for Human and Veterinary Use: Good Manufacturing Practice. Eudralex [Google Scholar]
- 30.GAMP 4, Good Automated Manufacturing Practice (GAMP) Guide for Validation of Automated Systems. 4th ed. International Society for Pharmaceutical Engineering; 2001. [Google Scholar]
- 31.Reason J. Human Error: Cambridge University Press; 1990. [Google Scholar]
- 32.Reason J. Human errors: models and management. Br Med J. 2000;320:768–70. doi: 10.1136/bmj.320.7237.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reason J, Carthey J, de Leval MR. Diagnosing "vulnerable system syndrome": an essential prerequisite to effective risk management. Qual Health Care. 2001;10 (Suppl 2):21–5. doi: 10.1136/qhc.0100021... [DOI] [PMC free article] [PubMed] [Google Scholar]