

Abstract
Our knowledge of cell cycle events such as DNA replication and mitosis has been advanced significantly through the use of Xenopus egg extracts as a model system. More recently, Xenopus extracts have been used to investigate the cellular mechanisms that ensure accurate and complete duplication of the genome, processes otherwise known as the DNA damage and replication checkpoints. Here we describe several Xenopus extract methods that have advanced the study of the ATR-mediated checkpoints. These include a protocol for the preparation of nucleoplasmic extract (NPE), which is a soluble extract system useful for studying nuclear events such as DNA replication and checkpoints. In addition, we describe several key assays for studying checkpoint activation as well as methods for using small DNA structures to activate ATR.
Keywords: ATR, Chk1, Rad1, Hus1, Rad9, checkpoint, Xenopus, NPE, chromatin, chloroquine, replication
1. Introduction
The maintenance of genomic integrity is essential for the survival of all organisms. As a result, cells have evolved multiple processes to prevent genomic instability caused by normal DNA metabolism and insults from DNA damaging agents or replication inhibitors. These processes, which are coordinated by the DNA damage and replication checkpoints, include arrest of cell cycle progression, initiation of apoptosis, regulation of DNA repair, and maintenance of replication fork stability (1, 2). In eukaryotes, checkpoint activation is centrally mediated by the ATR (Ataxia Telangiectasia and Rad3-related) kinase and its homologs (3), which in combination with a number of other checkpoint proteins forms a signaling complex competent to phosphorylate and activate downstream targets (4).
The ATR-mediated checkpoint pathways are highly conserved, and similar mechanisms of checkpoint activation are found in organisms as divergent from humans as Saccharomyces cerevisae and Schizosacharomyces pombe (4). The evolutionarily conserved nature of this response has permitted the use of a number of systems for checkpoint analysis, ranging from yeast genetic systems to transgenic mice. These systems have provided valuable insights into the processes that lead to checkpoint activation, as have biochemical studies with purified proteins. Yet as activation of the checkpoint following many types of DNA damage has been linked to DNA replication (5-8), the Xenopus egg extract system has offered several advantages for analysis of checkpoint signaling and activation. This in vitro system involves the use of extracts prepared from the eggs of Xenopus laevis which can cycle through S and M phase in a highly synchronous manner, replicating chromatin in a semi-conservative fashion (9-13).
Indeed, various types of Xenopus egg extracts have proven to be quite effective for the study of DNA replication and these studies have revealed that replication in extracts occurs through mechanisms very similar to those in mammalian cells [reviewed in (14)]. The cell-free nature of the system allows extensive manipulation of the proteins present, through immunodepletion and protein addition experiments, as well as alteration of the DNA template, which can take the form of chromatin, plasmid or even simpler structures. For the cell cycle checkpoint field, this has allowed a level of biochemical analysis difficult to achieve in mammalian cells due to the essential nature of many proteins involved in checkpoint activation. Furthermore, these extracts have allowed the assessment of biochemical events reconstituted with purified proteins in a system that more closely resembles that of a cell.
Here we describe recently developed methods that are useful for the study of checkpoint activation in Xenopus extracts. Specifically, we outline the preparation of nucleoplasmic extracts (NPE), which is a relatively new and powerful addition to the repertoire of Xenopus extracts (15). These extracts have the ability to replicate double-stranded plasmid templates (15, 16), and have proven useful for understanding the connection between DNA replication and the nature of the DNA damage signal (17). Additionally, we discuss our experience using ATR-mediated phosphorylation events for studying activation of the checkpoint. Lastly, we discuss the use of plasmid-based DNA templates for activating ATR, as well as methods for assessing the replication of these templates in Xenopus extracts.
2. Extract preparation
Four different types of extracts derived from Xenopus eggs have been used for checkpoint studies. The first type is a low-speed (interphase) extract (LSE). To prepare this type of extract, eggs are harvested and centrifuged to separate the yolk, pigment, and lipid contents from the cytoplasm. The result is a cytoplasmic extract containing small amounts of lipids that is competent for nuclear formation and replication of chromatin (9, 10), and arrests in interphase following completion of replication. A second type of low speed extract, called cytostatic-factor arrested (CSF) extract, can be used to recapitulate a full cell cycle, including mitosis (18, 19). High speed centrifugation of LSE to remove membrane components produces the third type of extract, a high-speed extract (HSE) consisting of egg cytosol devoid of all lipid content (20, 21). HSE is capable of single-stranded M13 replication (21) and pre-RC formation (16); however, it cannot promote firing of origins of replication. The fourth type of extract is a nucleoplasmic extract (NPE), which is obtained by replicating sperm chromatin in LSE (15). The nuclei that form upon incubation of sperm chromatin with LSE are isolated and centrifuged to yield the highly concentrated, membrane-free NPE. Both chromatin and double-stranded plasmids can be replicated by sequential incubation in HSE followed by NPE (16).
2.1. Preparation of Xenopus interphase (LSE) and cytosolic (HSE) extracts
Xenopus cell-free extracts are obtained from the unfertilized eggs of female Xenopus laevis frogs. To induce egg production, female frogs are “primed” to lay eggs by injection of pregnant mare serum gonadotropin (PMSG), followed by an injection of human chorionic gonadotropin (HCG) several days later to induce the laying of eggs. Harvested eggs are then crushed by centrifugation and the cytoplasmic layer is isolated for use. For in-depth information on frog injection and egg collection techniques, we direct the reader to the excellent protocols previously described by Murray (22), Walter and Newport (23) and Tutter and Walter (24). These papers are also excellent references for the preparation of LSE, HSE, NPE and CSF extract, as well as the preparation of demembranated sperm chromatin from male Xenopus laevis frogs.
2.1.1. Special considerations for the preparation of HSE
When making LSE in preparation for HSE, care should be taken to remove only the light brown cytoplasmic layer while completely avoiding both the yolk and pigment below and the lipids at the top. We have also found that gently inverting the LSE 60-80 times prior to centrifugation increases the yield of HSE following ultracentrifugation.
2.2. Preparation of Xenopus nucleoplasmic extract (NPE)
First developed by Walter et al. (15), NPE is a highly concentrated nuclear fraction that can be used in combination with HSE to replicate both chromatin and double-stranded plasmids.
2.2.1. Special considerations for the preparation of NPE
2.2.1.1. LSE
When making LSE in preparation for NPE, it is helpful to completely remove the middle cytoplasmic layer until the lipid and pigment layers touch. During this process, invariably a small amount of the yellow lipid layer is also removed with the cytoplasmic layer. We find that these extra lipids removed with the cytoplasmic extract aid in nuclear formation following addition of chromatin.
2.2.1.2. Sperm chromatin
Optimal nuclei formation occurs around 4,000 nuclei/μL of extract. Typically, this is achieved by adding the appropriate amount of a 250,000 nuclei/μL chromatin stock to the extract. Unfortunately, the percentage of viable sperm heads (i.e., sperm that can form good nuclei) varies between preparations. To account for this variability, after a new chromatin preparation is made, a number of different dilutions of sperm (for example, 3000, 4000, and 5000 nuclei/μL) are tested to find the optimal concentration at which the largest nuclear layer is obtained. Each dilution of new sperm chromatin is tested in a 4.5 mL batch against a previously characterized, control sperm preparation at its optimal dilution. Note that if the volume of sperm to be added is under 100 μL, the sperm should be diluted 1:1 with sperm dilution buffer prior to addition to the LSE.
2.2.1.3. Nuclear layer
When pipetting the nuclear layer, the viscosity should be such that the nuclei are somewhat stringy (i.e., when the pipet tip is touched to the surface of the nuclei and pulled away, a thin string should follow it). We have noticed that these nuclei tend to yield higher quality NPE than layers that are elastic (i.e., instead of forming a thin string, the nuclei form a thick clump around the pipet tip when pulled away).
2.2.1.4. Notes on extract volumes
NPE should be prepared from 4.5 mL batches of LSE in 5 mL Falcon tubes. Volumes of extract below 4.5 mL result in smaller nuclear layers that are difficult to isolate, and volumes greater than 4.5 mL will prevent the cap from snapping onto the top of the 5 mL tube. Extract that is left over after the individual 4.5 mL batches are prepared should be used for other experiments or discarded. The expected yield from 18 mL of LSE (four 4.5 mL tubes) is anywhere from 100-400 μL of NPE, depending on the size of the nuclear layer.
2.2.2. Materials and reagents
10× ELB salt solution: 25 mM MgCl2; 500 mM KCl; 100 mM HEPES-KOH pH 7.7
10× MMR: 1 M NaCl, 20 mM KCl, 10 mM MgSO4, 20 mM CaCl2, 1 mM EDTA, 50 mM HEPES-KOH pH 7.8
ELB-sucrose: 1× ELB salts containing 250 mM sucrose
Nocodazole (5 mg/mL in DMSO) Sigma M1404
Leupeptin (10 mg/mL in dH2O) Chemicon EI8
Aprotinin (10 mg/mL in dH2O) Sigma A4529
Dithiothreitol (DTT, 1 M stock in dH2O) Fisher BP172
Cycloheximide (10 mg/mL in dH2O) Sigma C7698
Cytochalasin B (10 mg/mL in DMSO) Sigma C6762
Creatine phosphate (1 M stock in 10 mM KPO4, pH 7) Roche 621-714
Adenosine triphosphate (0.2 M stock in dH2O, pH adjusted to 7) Roche 519-987
Creatine kinase (5 mg/mL stock in 10 mM HEPES-KOH pH 7.5, 50% glycerol) Roche 126-969
Energy mix (for 1 mL extract): 20 μL 1 M creatine phosphate; 10 μL 0.2 M adenosine triphosphate; 1 μL 5 mg/mL creatine kinase
Sperm dilution buffer: 1 mM MgCl2; 100 mM KCl; 150 mM sucrose; 5 mM HEPES-KOH pH 7.7
Demembranated sperm chromatin (250,000 nuclei/μL), prepared as described in (22).
Fix: 0.3 volumes 37% formaldehyde; 0.6 volumes 80% glycerol; 0.1 volumes 10× MMR; 2 μg/mL DAPI
14 mL polypropylene Falcon tubes
5 mL polypropylene Falcon tubes
Polycarbonate ultracentrifuge tubes, 7×20 mm (Beckman 343775)
17×100 mm tube adaptors (Fisher 05-566-50A) for 14 mL Falcon tubes
13×100 mm tube adaptors (Sorvall 00366) for 5 mL Falcon tubes
Delrin adaptors (Beckman 358615) for 7×20 mm ultracentrifuge tubes
200 μL wide bore pipet tips
Fluorescent microscope (with 385-400 nm excitation filter for viewing DAPI-stained nuclei)
Refrigerated centrifuge (Beckman JS13.1 rotor)
Ultracentrifuge (Beckman TLS-55 rotor)
2.2.3. Preparation of NPE
Prime and induce 16-20 frogs and prepare LSE according to Walter et al. (23). On average, approximately 1-2 mL of LSE can be obtained from a single frog, and ideally at least 18 mL of LSE (four 4.5 mL tubes) should be used for each NPE preparation. Immediately after removing LSE from the tube after the crushing step, add cycloheximide (1:200), protease inhibitors (leupeptin and aprotinin, 1:1000), DTT (1:1000), and cytochalasin B (1:2000) and invert gently 40X. After mixing, add nocodazole (1:1500) and again gently invert 40X. Transfer the extract into 14 mL Falcon tubes (using multiple tubes if necessary) and spin in 17×100 mm adaptors at 16,000×g for 10 minutes at 4°C in a pre-cooled JS13.1 rotor to clarify the extract. Following the spin, aspirate off the top yellow lipid layer as well as any black residue along the side of the tube and pour the clarified LSE into a new 14 mL Falcon tube on ice, making sure to avoid transferring any of the black material at the bottom of the tube. Dilute the clarified extract 9% with room temperature ELB-sucrose and invert 20X. Make energy mix fresh (31 μL for each mL of clarified extract), add to the extract, invert 20X and let the extract warm to room temperature.
While the extract is warming, thaw an appropriate amount of demembranated sperm chromatin to give an approximate final concentration of 4,000 nuclei/μL. Slowly pipet each aliquot of chromatin to ensure that the sperm is fully resuspended, then pool the aliquots into one tube and gently mix by pipetting. Dilute the sperm chromatin 1:1 with sperm dilution buffer if necessary, and aliquot the pooled, diluted sperm into 1.7 mL eppendorf tubes (one tube for each 4.5 mL batch of LSE). Add 1 mL of the energized extract to the sperm and slowly pipet 15X, then add the extract-sperm mixture to a 5 mL Falcon tube already containing 3.5 mL of the energized extract. Cap and invert the tube 30X to mix the extract. Incubate the extract at room temperature for 100 minutes or until replication of chromatin is complete, inverting once every 15 minutes. Nuclei formation and replication can be monitored by adding 1 μL of the extract to 1 μL of fix and observing the DAPI-stained chromatin. Demembranated sperm initially appears as a thin blue curve, which then becomes fatter and elongated after chromatin decondensation. After nuclei formation (the nuclear envelope can seen using phase-contrast microscopy), the chromatin staining becomes hazy and mesh-like in appearance, and then recondenses to a solid blue mass upon completion of replication. The exact time to finish replication may vary slightly based on the extract or sperm chromatin, therefore incubation times may need to be adjusted. Replication should be complete before proceeding further in order to maximize the size of the nuclear layer.
After the incubation, tubes are spun in 13×100 mm adaptors at 16,000×g for 2 minutes at 4°C in a pre-cooled JS13.1 rotor. 1 mL of water should be added to the bottom of the adaptors to cushion the tube. At this point, the nuclei will form a thin, opaque white layer at the top of the tube. There is also a dark brown plug of extract below the nuclear layer. Tubes should be kept on ice following this spin. Using a wide bore 200 μL pipet tip, carefully pipet the nuclei into a 7×20 mm polycarbonate tube. The pipet tip should be touched just to the surface of the layer and approximately 20 μL of nuclei at a time should be drawn up slowly while rotating the tube. Avoid disturbing the lower plug, which can cause the LSE below to leak into and dilute the nuclear layer. Transfer the nuclei into a 7×20 mm ultracentrifuge tube. Place tubes in 7×20 mm Delrin adaptors and centrifuge the isolated nuclei at 55,000 rpm for 30 minutes at 2 °C in a pre-cooled TLS-55 rotor. Following the spin, there should be a tan pellet at the bottom, a clear middle layer (NPE) bracketed by hazy layers, and a thin white film on the top. Carefully aspirate off the film without taking the NPE below and pipet out the clear layer. NPE should be pooled if multiple tubes were spun. The hazy fractions can also be taken and kept as a separate batch; however, this NPE is typically less robust than the clear fraction. The NPE is mixed to ensure uniformity, divided into 10 μL aliquots, snap frozen in liquid nitrogen, and stored at −80°C.
2.3. Using geminin to inhibit replication in extracts
Recombinant Xenopus geminin can be used to efficiently inhibit DNA replication in Xenopus extracts (5, 17, 25, 26). For LSE, pre-incubate the extract with 250 nM geminin for 10 minutes at room temperature before beginning the experiment. For NPE experiments, pre-incubate HSE with 250 nM geminin for 10 minutes at room temperature before addition of DNA. Make sure to test the ability of the recombinant geminin to inhibit DNA replication by gel electrophoresis (see section 5).
3. Detection of phosphorylated checkpoint proteins in Xenopus extracts
DNA damage causes the recruitment of a number of checkpoint proteins to the site of damage, which in turn leads to assembly of the ATR kinase signaling complex (4). Once activated, ATR phosphorylates a number of proteins in order to regulate the cellular response to DNA damage (3). In order to investigate ATR activation, methods to assay the phosphorylation state of ATR targets have been developed.
3.1. ATR-mediated phosphorylation of Chk1
After genotoxic stress, ATR phosphorylates and activates its downstream effector kinase, Chk1 (27-30). Active Chk1 then phosphorylates and inactivates the Cdc25 phosphatase, leading to Cdc2 inhibition and interphase arrest [reviewed in (31)]. Because of the importance of ATR-mediated Chk1 activation in damage-induced cell cycle arrest, Chk1 phosphorylation has become a principal readout of ATR activity. In humans, Chk1 has been shown to be phosphorylated on two sites by ATR, S317 and S345, and phosphorylation of these sites has been shown to regulate Chk1 activation (29, 30). In Xenopus, only phosphorylation of the latter site (S344 in Xenopus Chk1) has been observed (17, 32, 33), although a uncharacterized TQ site at residue 314 in Xenopus Chk1 aligns with S317 in human Chk1.
3.1.1. Inducing Chk1 phosphorylation in Xenopus extracts
Chk1 phosphorylation can be induced in Xenopus extracts by many types of DNA damage, including ultraviolet (UV) irradiation, methylmethanesulfonate (MMS), and 4-nitroquinoline-1-oxide (4-NQO), as well as by the DNA polymerase inhibitor aphidicolin [(5, 34) and unpublished data]. For UV damage, the sperm or plasmid is aliquoted onto parafilm and treated with >250 J/m2 UV in a Stratalinker. For MMS treatment, MMS is added to a stock solution of sperm chromatin (∼250,000/μL) at 1% and incubated at room temperature for 30 minutes. Sperm chromatin is then diluted for use in extract 50-100 fold (2500-5000 nuclei/μL), a dilution at which MMS does not affect the ability of the extract to form nuclei. 4-NQO is dissolved in DMSO (50 mM stock) and added directly to the extract at a concentration of 500 μM. Aphidicolin is diluted in DMSO (30 mM stock) and added to the extract at 15-150 μM. Stock solutions of aphidicolin should be divided into one-time use aliquots, as we have seen loss of activity upon multiple freeze-thaw cycles.
3.1.2. Phospho-specific antibody detection of endogenous Chk1
The most common method for assaying Chk1 phosphorylation is with a phospho-specific antibody raised against the ATR phosphorylation site S345 (Cell Signaling Technology Cat. # 2341, Figure 1A and 2). This antibody is raised against the human epitope, but works quite well for Xenopus Chk1 that is phosphorylated at S344. To visualize phosphorylated Chk1, run an 8% gel and follow the manufacturer's instructions closely for the western blotting procedure. As a loading control, total Chk1 levels can be analyzed with a commercially available Chk1 antibody (Santa Cruz Biotechnology Cat. # SC-8408).
Figure 1.
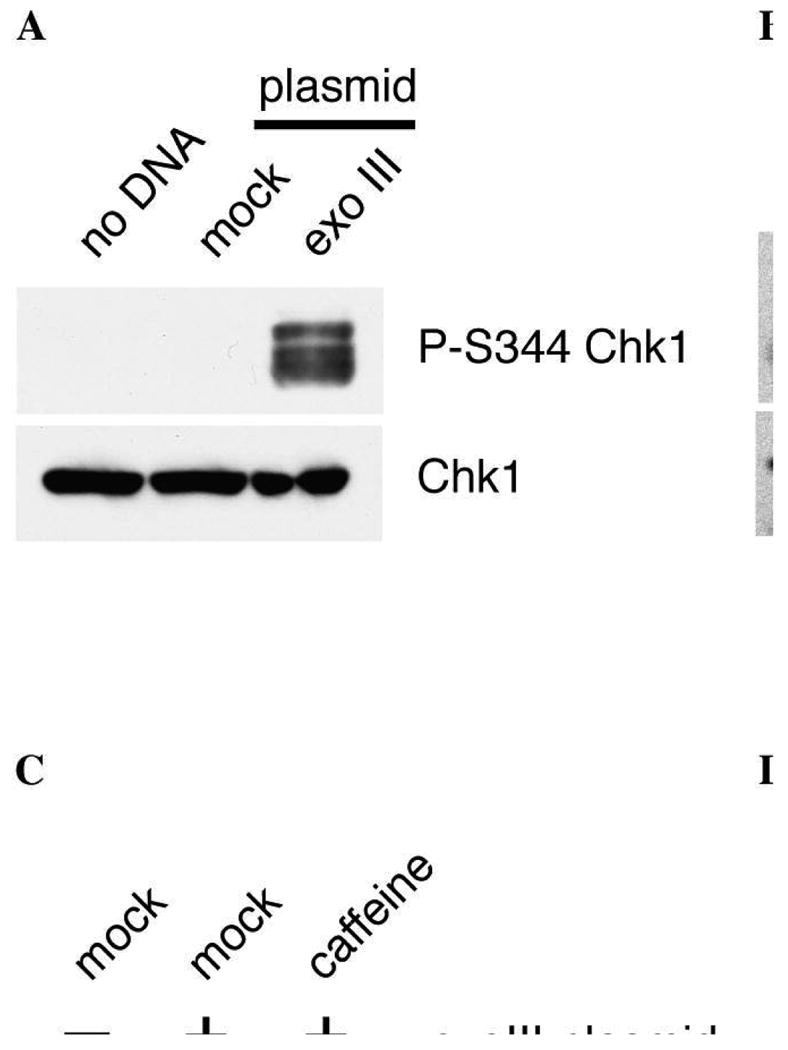
Exonuclease III-treated plasmid DNA can induce phosphorylation of checkpoint proteins. (A) ExoIII-treated DNA induces Chk1 phosphorylation at S344. Mock- or exoIII-treated plasmid (2.5 ng) was incubated with 5 μL Xenopus NPE for 30 minutes. 1 μL of each sample was run on an 8% SDS-PAGE gel and analyzed for phospho-Chk1 (P-S344) and total Chk1 by immunoblotting. (B) ExoIII-treated DNA induces RHR complex phosphorylation. ExoIII-treated plasmid DNA (5 ng) was incubated in 10 μL HSE for 30 minutes. 1 μL in vitro translated RHR complex (35S-Rad9, 35S-Hus1, cold Rad1) was added to the extract prior to plasmid addition to trace Rad9 and Hus1 phosphorylation. 1 μL extract was run out on a 10% SDS-PAGE gel. Rad9 and Hus1 were visualized by autoradiography, and Rad1 was analyzed by immunoblotting with anti-Rad1 antibodies. (C) Rad1 is phosphorylated on T5. ExoIII-treated plasmid DNA (2.5 ng) was incubated in 5 μL HSE for 30 minutes. To inhibit ATR activity, 5 mM caffeine was added to the extract prior to the addition of the plasmid. Rad1 was analyzed by immunoblotting with anti-Rad1 and anti-P-T5 phospho-specific antibodies. (D) ExoIII treatment of a nicked plasmid creates a heterogenous set of structures. Plasmid DNA (pEYFP) containing nicks was mock-treated or treated with 100 U exonuclease III for 2′ at 37 degrees. The mock- and exoIII-treated plasmid was electrophoresed along with an equivalent amount of single-stranded M13 DNA on an 0.8% agarose gel and stained with ethidium bromide. [Figures 1A and 1C reprinted with permission from Molecular Biology of the Cell, copyright 2006.]
Figure 2.
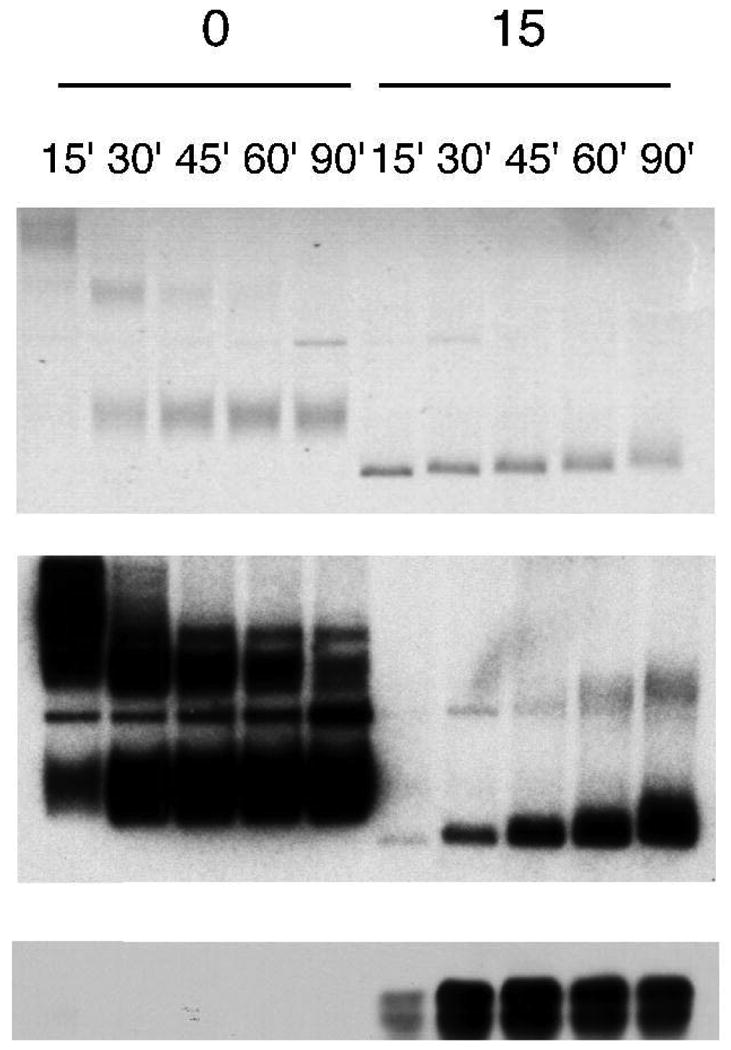
Aphidicolin induces hyperunwinding of plasmid DNA and Chk1 phosphorylation. Plasmid DNA was incubated in HSE for 30 minutes, and then transferred to an equal volume of NPE in the presence or absence of aphidicolin (final concentration 15 μM). 1 μL of each sample was analyzed for phospho-Chk1 (S344) or total Chk1 by immunoblotting at the indicated times (bottom panels). Replication was analyzed in parallel by incorporation of [α32P]-dCTP into plasmid DNA. DNA samples were analyzed via chloroquine agarose gel electrophoresis followed by SybrGold staining (top panel) and autoradiography (middle panel). [Reprinted with permission from Genes and Development, copyright April 15th, 2005.]
3.1.3. Using a Chk1 fragment for assaying ATR activity
In lieu of a phospho-specific antibody for Chk1, an in vitro-translated, 35S-labeled Chk1 fragment can be added to a Xenopus extract to assess the activity of ATR. This 218-amino acid C-terminal fragment (amino acids 258-473), originally identified by Michael et al. (25), undergoes a significant phosphorylation-induced mobility shift on SDS-PAGE in response to DNA damage. To use this assay, in vitro translate this fragment in the presence of 35S-methionine using a rabbit reticulocyte lysate system (TnT, Promega) and add 1 volume of 35S-labeled Chk1 to 20 volumes of extract. After the experiment is complete, it is necessary to isolate the nuclei from the extract to enrich for the phosphorylated form of the Chk1 fragment. To isolate nuclei from LSE, follow the first several steps of the protocol for chromatin isolation in Section 3.3.1.2. After spinning the LSE through the first ELB-sucrose cushion, aspirate the ELB-sucrose and resuspend the nuclei-enriched brown pellet in 1 mL ELB-sucrose and spin again at 5000×g for two minutes. Aspirate the ELB-sucrose and resuspend and boil the nuclear pellet in 40 μL sample buffer. Phosphorylation of the Chk1 fragment is analyzed by running 10 μL of the sample on an 8% SDS-PAGE gel and visualized by autoradiography.
3.1.4. Considerations for phospho-Chk1 assays
It is important to note that in LSE, good nuclei formation is necessary for robust Chk1 phosphorylation. Additionally, Chk1 phosphorylation in response to non-chromatin templates is difficult to assay in LSE or HSE, presumably due to the presence of cytoplasmic phosphatase activities. The phosphatase inhibitor tautomycin has been used with some success to help enrich the phosphorylated fraction of Chk1 (33). Alternatively, Chk1 phosphorylation can be induced by small DNA templates or plasmids in the NPE system (Figure 1A and 2) (17, 35).
3.2. Phosphorylation of the Rad9-Hus1-Rad1 (RHR) complex
Phosphorylation of all three subunits of the Xenopus RHR complex occurs in response to DNA damage in Xenopus extracts, and these phosphorylation events can be monitored by the analyzing electrophoretic mobility shift in each protein on an SDS-PAGE gel (Figure 1B) (35, 36).
3.2.1. Phospho-specific antibody detection of Rad1
Rad1 is robustly phosphorylated in an ATR-dependent manner on T5 in response to DNA damage, and phospho-specific antibodies against this residue have been generated (Figure 1C). Phosphorylation of this residue is dependent on ATR activation and the TopBP1 protein, yet it is independent of both Claspin and the carboxy-terminal region of Rad9 (35). Phosphorylation of Chk1 on S345 requires all of these proteins (17, 35), indicating that the requirements for Rad1 T5 phosphorylation are distinct for those for Chk1 S345 phosphorylation. Therefore phosphorylation of Rad1 on T5 can be used as a complementary readout for ATR activation.
3.2.2. Monitoring the phosphorylation state of 35S-labeled RHR complex
In lieu of antibodies capable of western detection of the three RHR complex subunits, in vitro-translated, 35S-labeled RHR complex can be added as a tracer to the extract and be used to assess phosphorylation induced by DNA damage (35). In order to prepare the 35S-labeled RHR complexes, each subunit is in vitro translated individually, mixed in a 1:1:1 ratio and incubated for 30 minutes to assemble the heterotrimeric complex. The assembled RHR complex is then added to the extract at 5-10% by volume.
Importantly, Rad1 and Hus1 are of almost identical size (31 kD) so only one of those two subunits can be 35S-labeled at a time. Rad9 runs at approximately 45-55 kD and does not interfere with assessment of Rad1 or Hus1 phosphorylation by SDS-PAGE. The phosphorylation state of the radiolableled RHR complex subunits is best visualized on a 10% gel followed by autoradiography.
3.3. Isolating chromatin-bound checkpoint proteins from LSE and NPE
One of the more powerful tools developed in the Xenopus extract system has been a straightforward and reproducible assay for the isolation of chromatin-associated proteins. This simple assay has been very useful for understanding binding requirements for replication and checkpoint proteins, and has led to the development of a number of models for how these processes work (5, 16, 25, 27, 32).
3.3.1 Isolating chromatin from LSE
3.3.1.1 Materials and reagents
10X ELB salt solution: 25 mM MgCl2; 500 mM KCl; 100 mM HEPES-KOH pH 7.7
ELB-sucrose cushion: 1X ELB salts containing 0.8 M sucrose
Chromatin extraction buffer (CEB): 50 mM HEPES-KOH pH 7.7; 50 mM KCl; 5 mM MgCl2; 5 mM EGTA; 0.1% NP-40; 500 μM spermidine; 150 μM spermine; 0.057% β-mercaptoethanol
Spermine (0.5 M stock in dH2O) Sigma S2876
Spermidine (1 M stock in dH2O) Sigma S2626
Swinging bucket microfuge (capable of 11,700×g, similar to Eppendorf 5417C)
1.7 mL eppendorf tubes
Sample buffer
3.3.1.2. Procedure for isolation of chromatin from LSE
To isolate sperm chromatin from LSE, it is generally preferable to work with a minimum of 50 μL extract containing >4000 sperm/μL. This ensures a robust isolation of chromatin-bound proteins. After the experiment is completed, the extract is carefully layered on top of a 1 mL cushion of room temperature ELB-sucrose in a 1.7 mL eppendorf tube. The extract will sit on top of the dense sucrose-containing ELB. The extract is then centrifuged through the sucrose cushion at 5000×g in the swinging bucket microfuge for 2 minutes. In order to get the best nuclear enrichment, care should be taken not to disturb the layer of extract on top of the cushion while moving the tubes to the centrifuge. This step isolates the nuclei from the cytoplasmic fraction of the extract. After centrifugation, a loose pellet of tan nuclei should be visible at the bottom of the tube. Aspirate the ELB-sucrose from the top of the nuclei, being careful not to get too close to the pellet (leave ∼50 μL of liquid above the pellet).
At this point, it is necessary to lyse the nuclei and extract the chromatin with a detergent and poly-amine containing buffer. Add 250 μL CEB and mix by pipet action 10-15 times. Incubate on ice for 15 minutes. After the incubation with CEB, slowly pipet the CEB-chromatin mix on top of another 1 mL ELB-sucrose cushion in an eppendorf tube. The CEB-chromatin mix will layer on top of the dense sucrose-containing ELB. Again, it is important not to disturb the layer at this point in order to get maximum enrichment of chromatin-bound versus nuclear proteins. Spin the tubes at 11,700×g for 2 minutes in the swinging bucket microfuge. The chromatin will form a near-invisible pellet at the bottom of the tube. Carefully aspirate the ELB-sucrose supernatant leaving 10-20 μL in the tube, add 30 μL of sample buffer, and boil for 5-10 minutes. If the isolation worked, there will be a significant amount of chromatin in the pellet. The presence of chromatin will make the sample buffer extremely viscous, and necessitates shearing of the chromatin with a pipet or syringe.
3.3.2. Isolating chromatin from NPE
3.3.2.1. Materials and reagents
10X ELB salt solution: 25 mM MgCl2; 500 mM KCl; 100 mM HEPES-KOH pH 7.7
NPE resuspension buffer: 1X ELB salts containing 0.25 M sucrose and 0.2% Triton X-100
ELB-sucrose cushion: 1X ELB salts containing 0.5 M sucrose
ELB-sucrose wash buffer: 1X ELB salts containing 0.25 M sucrose
Beckman microfuge B (horizontal rotor, 11,000 RPM fixed-speed)
1.7 mL eppendorf tubes
2X Sample buffer
3.3.2.2. Procedure for isolation of chromatin from NPE
To isolate chromatin from NPE, a 10 μL reaction containing >4000 nuclei/μL is sufficient. Begin by resuspending the 10 μL NPE reaction in 60 μL cold NPE resuspension buffer. Slowly pipet the resupended NPE on top of a 180 μL cushion of ELB-0.5 M sucrose in a 1.7 mL eppendorf tube. The diluted NPE will layer on top of the more dense ELB-0.5 M sucrose. Gently place the eppendorf tubes in the horizontal rotor of the microfuge and spin the tubes for 30 seconds at the fixed speed of 11,000 RPM. The chromatin will form a near-invisible pellet at the bottom of the tube. Aspirate the supernatant down to ∼1 μL, and add 200 μL of cold ELB wash buffer. Spin the tubes again at 11,000 RPM for 30 seconds. Aspirate the wash buffer down to ∼1 μL and add 10-20 μL of 2X sample buffer. Boil the samples for 5-10 minutes and, if necessary, shear the DNA in the samples with a pipet or syringe before loading on an SDS-PAGE gel.
3.3.3. Considerations for analyzing chromatin-bound proteins
It is difficult to keep the volume of individual chromatin isolation reactions constant, therefore we generally load the entire sample in order to ensure even loading between samples. For a loading control, a member of the ORC complex (Orc1-6) is commonly used. The levels of the DNA binding protein RPA and many checkpoint proteins are generally increased after genotoxic stress (5, 32, 36), and therefore are not good readouts for visualizing the amount of chromatin loaded.
4. Plasmid-based methods for activating ATR in Xenopus extracts
4.1. Using a gapped plasmid structure to activate ATR
Activation of ATR in Xenopus extracts using the simple DNA structure poly dA(70)/poly dT(70) was first described by Kumagai and Dunphy (33). Later, Costanzo et al. (37) showed that sperm chromatin treated with exonuclease III (exoIII) could also activate the ATR-mediated checkpoint. Merging these two findings, we found that exoIII treatment of a nicked plasmid activates ATR in a number of types of extract, including LSE, HSE, and NPE. We have found the exoIII-treated plasmid to be the most robust method for inducing RHR complex phosphorylation (35).
4.1.1. Preparation of exoIII-treated DNA
4.1.1.1. Materials and Reagents
0.5 mL 5-6 kB plasmid (e.g. pEGFP) at 1 mg/mL
Phenol/chloroform/isoamyl alcohol (25:24:1)
100% Ethanol
3 M Sodium Acetate pH 5.2
Exonuclease III (NEB M0206)
NEB Buffer 1 (10 mM Bis Tris Propane-HCl; 10 mM MgCl2; 1 mM DTT; pH 7.0)
37°C water bath
4.1.1.2. Procedure
A 5-6 kB plasmid (e.g. pEGFP) at a concentration of 1 mg/mL is first phenol/chloroform extracted to strip all bacterial DNA binding proteins then ethanol precipitated to remove all traces of phenol. The plasmid is re-dissolved in the original volume of dH2O, then snap frozen and thawed ten times to create nicks in the DNA. 1 μg of this plasmid is then treated with 5-10 U exoIII in 20 μL Buffer 1 for two minutes at 37°. The plasmid is kept on ice, and should be used within several hours. To ensure that the exoIII has not completely degraded the plasmid, an aliquot is run on an 0.8% agarose gel containing ethidium bromide to visualize the DNA (Figure 1D). The plasmid band should be fuzzy, indicating a heterogenous population of single- and double-stranded DNA structures.
4.1.2. Using an exoIII-treated plasmid to activate the checkpoint
ExoIII-treated DNA will activate ATR in several types of Xenopus extracts, including LSE, HSE, and NPE (35, 37). Energy mix, 2 mM DTT, and 20 μM nocodazole (see Section 2.2.2) are added to the extract prior to addition of the DNA. ExoIII-treated DNA is an extremely efficient activator of ATR, and incubation of 500 pg DNA/μL extract for 15-30 minutes will induce Chk1 phosphorylation in NPE or RHR complex phosphorylation in LSE, HSE, or NPE.
4.2. Using a double-stranded plasmid to activate the checkpoint in NPE
One of the major advantages of the HSE/NPE system is the ability to replicate double-stranded plasmids, shown by Walter et al. (15). Addition of a double-stranded plasmid into HSE leads to formation of a pre-replication complex on the plasmid, which is followed by single round of semi-conservative replication after subsequent addition of NPE (15, 16). Our lab has used this system to monitor activation of ATR in response to varying types of replication stress and damaging agents, including aphidicolin and UV irradiation (17). Use of a plasmid in the NPE system also allows us to study structural intermediates that occur during replication and checkpoint activation through gel electrophoresis (see section 5).
4.2.1. Considerations for plasmid-induced checkpoint activation in NPE
4.2.1.1. Concentrations
Typical plasmid concentrations used in replication experiments are 1-10 ng/μL, but as much as 50 ng/μL can be replicated successfully. For checkpoint activation, at least 10 ng/μL should be used. If plasmids are stored in TE, MgCl2 is added to a final concentration of 20 mM prior to addition into extract.
4.2.1.2. Radiolabeling
In order to monitor replication, trace amounts of α32P-dCTP (0.1 μCi/μL HSE) can be added to HSE prior to addition of the plasmid. Incorporation can be monitored by gel electrophoresis and autoradiography (see section 5).
4.2.1.3. UV and aphidicolin treatment
UV and aphidicolin dosages used to activate Chk1 phosphorylation are typically 250-1500 J/m2 and 15-150 μM, respectively (Figure 2) (17). If necessary, dilute aphidicolin in PIPES buffer prior to use. It should be noted that high concentrations of aphidicolin (>300 μM) have been shown to prevent Chk1 phosphorylation by inhibiting the polymerase activity of DNA Polymerase α, thereby preventing loading of the RHR complex (17).
4.2.1.4. HSE and NPE dilution
Reagents can be added to HSE or NPE up to 30% of the starting volume without loss of activity.
4.2.2. Materials and reagents
Creatine phosphate (1 M stock in 10 mM KPO4, pH 7) Roche 621-714
Adenosine triphosphate (0.2 M stock in dH2O, pH adjusted to 7) Roche 519-987
Creatine kinase (5 mg/mL stock in 10 mM HEPES-KOH pH 7.5, 50% glycerol) Roche 126-969
Energy mix (for 1 mL extract): 20 μL 1 M creatine phosphate; 10 μL 0.2 M adenosine triphosphate; 1 μL 5 mg/mL creatine kinase
Nocodazole (0.5 mg/mL in DMSO) Sigma M1404
10 mM PIPES, pH 7.4
Aphidicolin (30 mM in DMSO) Sigma A0781
α32P-dCTP (1 μCi/μL, 3000 Ci/mmol)
UV Stratalinker (Stratagene)
Replication stop buffer: 0.5% SDS; 20 mM EDTA pH 8.0
Proteinase K (10 mg/mL in water) Sigma P6556
DNA loading buffer with bromophenol blue
SDS-PAGE loading buffer
4.2.3. Procedure
Quickly thaw aliquots of HSE and NPE and put on ice. To HSE, add 0.31 μL energy mix (made fresh) and 0.133 μL nocodazole per 10 μL extract and pipet to mix. If the replicating plasmid is to be radiolabeled, add α32P-dCTP to a final concentration of 0.1 μCi/μL HSE. Add plasmid to the HSE, pipet to mix, and incubate at room temperature. If UV irradiation is being used, the plasmid must be irradiated prior to addition to the extract (see 3.1.1 for procedure). For aphidicolin treatment, add the desired concentration to the HSE 15 minutes after addition of the plasmid and pipet to mix. Add an equal volume of energized NPE to the HSE/plasmid reaction 30 minutes after plasmid addition and pipet to mix. NPE should be energized (0.31 μL energy mix per 10 μL NPE) 5 minutes before addition and warmed to room temperature. For faster replication, up to 2 volumes of NPE can be added to HSE (15). Samples can then be taken at various time points, added to SDS loading buffer and boiled, and then run on SDS-PAGE gels and analyzed for Chk1 phosphorylation by western blotting.
5. Monitoring DNA replication in Xenopus extracts
Agarose gel electrophoresis offers a simple method to track plasmid replication as well as observe structural intermediates during replication and checkpoint activation. Native gel electrophoresis is commonly used to track plasmid or chromatin replication either visually (through staining with dyes such as ethidium bromide or SybrGold) or quantitatively (by measuring incorporation of radionucleotides) (5, 16, 17). However, native gels yield a limited amount of structural information and therefore alkaline denaturing gels and chloroquine gels are also useful for plasmid and chromatin analysis. Alkaline gels denature the DNA sample and can be used to analyze single-stranded intermediates formed during replication (38-40). Agarose gels containing chloroquine, an intercalating agent, have been previously utilized in studying plasmid unwinding at replication forks during checkpoint activation (16, 17).
5.1. Use of native agarose gels to analyze replication products
5.1.1. Materials and reagents
Agarose
50× TAE: 2 M Tris; 50 mM EDTA pH 8.0; 5.7% (v/v) glacial acetic acid
Replication stop buffer: 0.5% SDS; 20 mM EDTA pH 8.0
Proteinase K (10 mg/mL in water) Sigma P6556
Phenol/chloroform/isoamyl alcohol (25:24:1)
100% Ethanol
3 M Sodium Acetate pH 5.2
DNA loading buffer with bromophenol blue
SybrGold (Invitrogen S-11494)
Vacuum pump and gel dryer
Phosphorimager and Phosphorimager screen
5.1.2. Sample preparation
Typical plasmid concentrations used in replication experiments are 1-10 ng/μL extract. Typical chromatin concentrations used are 4000 nuclei/μL extract. Samples containing sperm chromatin or plasmids are first treated with a minimum of one volume of replication stop buffer. This is followed by treatment of the extract with 500 μg/mL of proteinase K at 37°C for >30 minutes. At this point, DNA loading buffer can be added and the samples can be electrophoresed, or alternatively they can be phenol/chloroform extracted, ethanol precipitated, and dried in a speed vac in order to isolate the DNA and reduce the volume.
5.1.3. Native gel procedure
Add 0.8% agarose (w/v) to 1× TAE, and heat until TAE is boiling and agarose is fully dissolved. Cool for 10-15 minutes and then pour into a gel casting set-up. Allow the agarose to solidify. Load samples (>2 ng DNA or 1×104 nuclei) and run at 100-140 V in 1× TAE (approximately 30 minutes for a 7.5 cm gel and 90 minutes for a 13 cm gel). Upon completion, the gel can be stained by incubating in 1× TAE containing SybrGold (1:1000 dilution) for 30-60 minutes on an orbital shaker. The gel should be covered with foil during incubation due to the light sensitivity of the SybrGold dye. Overnight incubations can also be done for increased sensitivity. When running radiolabeled samples, the area of the gel below the leading dye front should be cut off to prevent background from unincorporated radionucleotides. The gel can then be dried down onto filter paper using a gel dryer and vacuum pump and exposed to a phosphorimager screen for quantification.
5.2. Use of alkaline gel conditions to analyze replication products
DNA samples can be denatured under alkaline conditions and analyzed using alkaline gel electrophoresis. This method has been useful for monitoring nascent replicating strands of DNA normally complexed with the parental DNA in Xenopus extracts (38-40). These nascent DNA strands are seen as a broad band that moves up in molecular weight as replication progresses.
5.2.1 Materials and Reagents
Agarose
10 M NaOH stock
0.5 M EDTA pH 8.0 stock
Alkaline Running Buffer (1X): 30mM NaOH; 1mM EDTA
Alkaline Gel Loading Buffer (5X): 250mM NaOH; 5mM EDTA; 2.5% Ficoll-400, 0.025% bromocreosol green
Trichloroacetic acid
5.2.2. Sample preparation
See 5.1.2. for information on sample preparation. It is important to note that samples prepared for alkaline gel electrophoresis require phenol/chloroform extraction and ethanol precipitation, followed by resuspension in alkaline gel loading buffer.
5.2.3. Alkaline gel procedure
Make a 1% agarose-dH2O solution and boil to dissolve the agarose. When the melted agarose solution cools to approximately 55°C, add 10 M NaOH stock to 50 mM and 0.5 M EDTA stock to 1 mM. Pour gel and let solidify. After the gel solidifies, add running buffer to the gel running unit and load samples (>2 ng plasmid or 1×104 nuclei) that have been resuspended in 1X loading buffer. Run the gel at 110 volts until the dye front runs three-quarters the length of the gel. Fix the gel in 7% trichloroacetic acid for 30 minutes, dry gel and visualize by autoradiography.
5.3. Use of chloroquine gels to analyze replication products
During checkpoint activation with replication inhibitors such as UV irradiation or aphidicolin, the MCM helicase functionally uncouples from the polymerase and continues to unwind the plasmid template DNA (16, 17, 41). Upon digestion and extraction of bound proteins, the plasmid strands reanneal and the now hyperunwound plasmid takes on a high degree of negative supercoiling. This unwound plasmid form, known as U-form, can be detected using an agarose gel containing chloroquine. On agarose gels, supercoiled plasmids run with a higher mobility than normal plasmids. Chloroquine is an intercalating agent that unwinds DNA and causes compensatory positive supercoiling, which causes a negatively supercoiled plasmid to exhibit a decrease in mobility. However, U-form plasmids, being hyperunwound, still retain a high degree of negative supercoiling and continue to run as a high mobility band, which is then distinguishable from normal replicating plasmid forms that have been retarded due to chloroquine (Figure 2). For further information on U-form mobility and appearance and protocols on U-form detection, we direct the reader to several papers in the primary literature (16, 17, 24, 41).
5.3.1. Considerations for chloroquine gels
5.3.1.1. Chloroquine
The concentration of chloroquine can be adjusted depending on the type of resolution desired (16). Chloroquine should be made fresh prior to use.
5.3.2. Materials and reagents
Agarose
10× TBE: 0.89 M Tris, 0.89 M Borate, 0.02 M EDTA pH 8
Chloroquine, 100 mM in water (Sigma C6628)
DNA loading buffer with bromophenol blue
Ribonuclease A (Qiagen 19101)
5.3.3. Sample preparation
See 5.1.2. for information on sample preparation. As with alkaline gels, samples require phenol/chloroform extraction (pipet to mix, do not vortex) and ethanol precipitation followed by resuspension in DNA loading buffer containing 100 ng/mL ribonuclease A.
5.3.4. Chloroquine gel procedure
Add 0.8% agarose (w/v) to 100 mL 1× TBE and heat until TBE boils and agarose is fully dissolved. Cool until TBE is warm to touch, then add chloroquine to a final concentration of 4 μM. Swirl to mix and then pour into a 12×14 mm gel tray. Allow gel to solidify for 2-3 hours. Load samples (20-30 ng) and run at 100 V in 1× TBE containing 4 μM chloroquine until the bromophenol blue dye front runs off (∼3.5 hrs). The gel can be stained with SybrGold and dried down for quantification as detailed in Section 5.1.3.
6. Concluding Remarks
The essential nature of many proteins involved in checkpoint signaling have made it difficult to study certain aspects of these pathways in intact cellular systems. Xenopus egg extracts provide a cell-free system that can recapitulate many of the events that take place in the nucleoplasm. The in vitro nature of this system allows for extensive manipulation of protein and DNA content, yet the checkpoint pathways can be completely reconstituted in a manner not possible with recombinant in vitro systems. These advantages guarantee that the Xenopus model system will continue to provide powerful methods for studying DNA damage checkpoint signaling in higher eukaryotes.
Acknowledgments
We thank Tony Byun, Christina MacDougall, Debbie Chang, and the Walter lab for helpful discussions concerning extract techniques. This research is supported by a fellowship from the CA BCRP (8GB-0091) to P.J.L., from the DOD Breast Cancer Research Program (W81XWH-04-1-0311) to C.V., and by grants to K.A.C. from the NIH (GM062193) and American Cancer Society (RSG-05-028-01-CCG). K.A.C. is a Leukemia and Lymphoma Society Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou BB, Elledge SJ. Nature. 2000;408:433–39. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Cimprich KA. Curr Biol. 2003;18:R231–R33. doi: 10.1016/s0960-9822(03)00158-1. [DOI] [PubMed] [Google Scholar]
- 3.Abraham RT. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 4.Melo J, Toczyski D. Curr Opin Cell Biol. 2002;14:237–45. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 5.Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA. Genes Dev. 2002;16:2327–32. doi: 10.1101/gad.1013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stokes MP, Van Hatten R, Lindsay HD, Michael WM. J Cell Biol. 2002;158:863–72. doi: 10.1083/jcb.200204127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ward IM, Minn K, Chen J. J Biol Chem. 2004;279:9677–80. doi: 10.1074/jbc.C300554200. [DOI] [PubMed] [Google Scholar]
- 8.Tercero JA, Longhese MP, Diffley JF. Mol Cell. 2003;11:1323–36. doi: 10.1016/s1097-2765(03)00169-2. [DOI] [PubMed] [Google Scholar]
- 9.Lohka MJ, Masui Y. Science. 1983;220:719–21. doi: 10.1126/science.6601299. [DOI] [PubMed] [Google Scholar]
- 10.Blow JJ, Laskey RA. Cell. 1986;47:577–87. doi: 10.1016/0092-8674(86)90622-7. [DOI] [PubMed] [Google Scholar]
- 11.Hutchison CJ, Cox R, Drepaul RS, Gomperts M, Ford CC. EMBO J. 1987;6:2003–10. doi: 10.1002/j.1460-2075.1987.tb02464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray AW, Kirschner MW. Nature. 1989;339:275–80. doi: 10.1038/339275a0. [DOI] [PubMed] [Google Scholar]
- 13.Felix MA, Pines J, Hunt T, Karsenti E. EMBO J. 1989;8:3059–69. doi: 10.1002/j.1460-2075.1989.tb08457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arias EE, Walter JC. Front Biosci. 2004;9:3029–45. doi: 10.2741/1457. [DOI] [PubMed] [Google Scholar]
- 15.Walter J, Sun L, Newport J. Mol Cell. 1998;1:519–29. doi: 10.1016/s1097-2765(00)80052-0. [DOI] [PubMed] [Google Scholar]
- 16.Walter J, Newport J. Mol Cell. 2000;3:617–27. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- 17.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohka MJ, Maller JL. J Cell Bio. 1985;101:518–23. doi: 10.1083/jcb.101.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray AW, Solomon MJ, Kirschner MW. Nature. 1989;339:280–86. doi: 10.1038/339280a0. [DOI] [PubMed] [Google Scholar]
- 20.Newport J. Cell. 1987;48:205–17. doi: 10.1016/0092-8674(87)90424-7. [DOI] [PubMed] [Google Scholar]
- 21.Mechali M, Harland RM. Cell. 1982;30:93–101. doi: 10.1016/0092-8674(82)90015-0. [DOI] [PubMed] [Google Scholar]
- 22.Murray AW. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- 23.Walter J, Newport J. In: Eukaryotic DNA Replication. Cotterill S, editor. Oxford University Press; Oxford: 1999. pp. 201–22. [Google Scholar]
- 24.Tutter AV, Walter JC. Methods Mol Biol (Clifton, NJ. 2006;322:121–37. doi: 10.1007/978-1-59745-000-3_9. [DOI] [PubMed] [Google Scholar]
- 25.Michael WM, Ott R, Fanning E, Newport J. Science. 2000;289:2133–37. doi: 10.1126/science.289.5487.2133. [DOI] [PubMed] [Google Scholar]
- 26.McGarry TJ, Kirschner MW. Cell. 1998;93:1043–53. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- 27.Hekmat-Nejad M, You Z, Yee MC, Newport J, Cimprich KA. Curr Biol. 2000;10:1565–73. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- 28.Guo Z, Kumagai A, Wang SX, Dunphy WG. Genes Dev. 2000;14:2745–56. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Piwnica-Worms H. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Sanchez Y. DNA Repair (Amst) 2004;3:1025–32. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Kumagai A, Dunphy WG. Mol Cell. 2003;11:329–40. doi: 10.1016/s1097-2765(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 33.Kumagai A, Dunphy WG. Mol Cell. 2000;6:836–49. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 34.Kumagai A, Guo ZJ, Emami KH, Wang SX, Dunphy WG. J Cell Biol. 1998;142:1559–69. doi: 10.1083/jcb.142.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupardus PJ, Cimprich KA. Mol Biol Cell. 2006;17:1559–69. doi: 10.1091/mbc.E05-09-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones RE, Chapman JR, Puligilla C, Murray JM, Car AM, Ford CC, Lindsay HD. Mol Biol Cell. 2003;14:3898–910. doi: 10.1091/mbc.E03-03-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. Mol Cell. 2003;11:203–13. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 38.Shechter D, Costanzo V, Gautier J. Nat Cell Biol. 2004;6:648–55. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- 39.Luciani MG, Oehlmann M, Blow JJ. J Cell Sci. 2004;117:6019–30. doi: 10.1242/jcs.01400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yanow SK, Gold DA, Yoo HY, Dunphy WG. J Biol Chem. 2003;278:41083–92. doi: 10.1074/jbc.M307144200. [DOI] [PubMed] [Google Scholar]
- 41.Pacek M, Walter JC. EMBO J. 2004;23:3667–76. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]