

Abstract
The RAG1/2 endonuclease initiates programmed DNA rearrangements in progenitor lymphocytes by generating double-strand breaks at specific recombination signal sequences. This process, known as V(D)J recombination, assembles the vastly diverse antigen receptor genes from numerous V, D, and J coding segments. In vitro biochemical and cellular transfection studies suggest that RAG1/2 may also play postcleavage roles by forming complexes with the recombining ends to facilitate DNA end processing and ligation. In the current study, we examine the in vivo consequences of a mutant form of RAG1, RAG1-S723C, that is proficient for DNA cleavage, yet exhibits defects in postcleavage complex formation and end joining in vitro. We generated a knockin mouse model harboring the RAG1-S723C hypomorphic mutation and examined the immune system in this fully in vivo setting. RAG1-S723C homozygous mice exhibit impaired lymphocyte development and decreased V(D)J rearrangements. Distinct from RAG nullizygosity, the RAG1-S723C hypomorph results in aberrant DNA double-strand breaks within rearranging loci. RAG1-S723C also predisposes to thymic lymphomas associated with chromosomal translocations in a p53 mutant background, and heterozygosity for the mutant allele accelerates age-associated immune system dysfunction. Thus, our study provides in vivo evidence that implicates aberrant RAG1/2 activity in lymphoid tumor development and premature immunosenescence.
Introduction
The immense diversity of genes encoding the variable regions of antigen receptors is generated through rearrangement of component V, D, and J segments via a cut-and-paste mechanism known as V(D)J recombination.1 These site-specific chromosomal rearrangements are initiated in progenitor lymphocytes by the recombination activating gene 1 (RAG1) and RAG2 proteins that comprise a lymphoid-specific endonuclease, RAG1/2.2,3 DNA double-strand breaks (DSBs) are generated by RAG1/2 at specific recombination signal sequences (RSSs) adjacent to the numerous rearranging V, D, and J coding exons.4 The broken ends are then processed and joined by ubiquitously expressed nonhomologous end-joining (NHEJ) DNA repair factors, including Ku70, Ku80, the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, DNA ligase IV, XRCC4 and Cernunnos/XLF.1,5,6
The RAG1/2 endonuclease initially binds to RSSs that are composed of a conserved heptamer and nonamer separated by relatively nonconserved spacer sequences of either 12 or 23 nucleotides.4 Efficient cleavage requires recombination between one 12 RSS and one 23 RSS in the context of a synaptic, paired complex mediated by RAG1/2. RAG1/2 nicks the duplex DNA at the junction between the RSS heptamer and coding sequences then uses the resulting 3′ OH in a direct transesterification reaction with the phosphodiester backbone of the opposing strand. The products of RAG1/2 cleavage are blunt, 5′ phosphorylated signal ends and covalently closed, hairpin coding ends. Sequence elements within the nonconserved 12 and 23 spacers7 and coding flanks8–11 contribute to RAG1/2 interaction with the recombining DNA and strongly influence coding segment utilization.7 Hence, the efficiency of binding and cleavage of individual RSSs is significantly affected by interactions of the RAG1/2 endonuclease with the heptamer, nonamer, 12 or 23 spacer, and coding sequences.
In addition to an essential role in initiating V(D)J rearrangements, RAG1/2 has been proposed to have important functions subsequent to the generation of DSBs. In vitro biochemical and cellular transfection assays have provided evidence that upon cleavage of the RSSs, the RAG proteins remain associated with the DNA ends in postcleavage complexes.12–21 The transient cleaved signal complex (CSC), composed of RAG1/2 and the 4 newly generated coding and signal ends, is formed upon generation of the DSBs.14–16 The hairpin coding ends are released from this complex and rapidly undergo nicking, processing, and ligation by the NHEJ proteins to form variable coding joints that can contain added or deleted nucleotides. The CSC likely serves to protect coding ends from inappropriate processing and specifically directs them to the NHEJ pathway. The blunt signal ends remain bound to RAG1/2 in a protective, stable signal end complex (SEC) and persist until precisely ligated by the NHEJ factors.14,17,18
In vitro studies of mutant RAG proteins provided evidence for the functional importance of RAG1/2 postcleavage complexes during the joining phase of V(D)J recombination.16,19–21 Of particular relevance here are RAG1 mutants in which serine 723 was replaced by alanine or cysteine.16 These mutants are proficient for inducing DNA breaks16,22 but exhibit defects in the stability of the CSC, which manifest as premature release of the coding ends in biochemical assays.16 Despite apparently normal interactions with signal ends in vitro,22,23 accumulation of signal ends and formation of both coding and signal joints were significantly reduced in transient transfection V(D)J recombination assays as a result of the RAG1-S723C mutation.16
In this study, we examine the impact of the S723C mutation in a fully in vivo setting through generation and characterization of a mouse model harboring this single amino acid substitution within the endogenous RAG1 locus. We demonstrate that the RAG1-S723C mutation significantly impairs lymphocyte development due to defective chromosomal V(D)J rearrangements and, in a p53 mutant background, results in predisposition to thymic lymphomas associated with chromosomal translocations. We also unexpectedly uncovered an accelerated age-associated immunodeficiency phenotype in RAG1-S723C heterozygous mice. These results have implications with regard to human immunodeficiencies and lymphoid malignancies.
Methods
RAG1-S723C knockin mice
Mice harboring the RAG1-S723C knockin mutation at the endogenous locus were generated via gene targeting. Approval for use of animals in this study was granted by the University of Michigan UCUCA office (protocol number 08758). See Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article) for a detailed description of the targeting strategy.
PCR analysis of IgH and TCR rearrangements
DJH and DJβ rearrangements in sorted pro- and pre-B cells and purified double-negative (DN) thymocytes, respectively, were analyzed by polymerase chain reaction (PCR) amplification as described24,25 (see Document S1 for details). PCR analyses were repeated 3 times on at least 3 independent samples of genomic DNA. Extrachromosomal Dδ2-Jδ1 signal joints were analyzed as described.26 PCR analyses were repeated 3 times on 3 independent sets of genomic DNA samples.
LM-PCR amplification of signal ends
Ligation-mediated (LM)–PCRs were carried out as previously described.27 Specific modifications to PCR amplification conditions and additional details are described in Document S1. For sequence analysis, the PCR-amplified products were either directly subcloned or first electrophoresed through a 1.2% agarose gel to eliminate contamination from nonspecific primer dimer products. PCR analyses were repeated 3 times on 3 independent samples of genomic DNA.
Characterization of tumors
Lymphoid tumors were analyzed by flow cytometry with antibodies against surface B-cell (CD43, B220, IgM) and T-cell (CD4, CD8, CD3, TCRβ, CD44, CD25) markers. Solid tumors were fixed in Bouin solution, paraffin embedded, and analyzed histologically by hematoxylin and eosin staining of sections. Spectral karyotyping was performed either on metaphases from cells derived from the primary tumor (tumors 135, S542) or early passage tumor cells cultured as previously described28 (all other tumors). Structural aberrations were considered clonal if present in 2 or more metaphases.29,30
Results
Generation of RAG1-S723C mice
To examine the in vivo consequences of the RAG1-S723C mutation on immune system development, we generated a mouse strain harboring this amino acid substitution via gene targeting (Figure S1). The homozygous RAG1-S723C mice, which were housed in a pathogen-free facility, were fertile, born in Mendelian numbers (Table S1), and survived into adulthood with no obvious developmental defects. Expression of the RAG1-S723C mutant allele was verified by subcloning and sequencing the wild-type and mutant cDNAs from RAG1+/S723C thymocytes (data not shown). In addition, we analyzed RAG1 protein levels in nuclear extracts from wild-type, RAG1+/S723C, and RAG1S723C/S723C thymocytes by Western blot analyses and observed that the RAG1-S723C protein is stable and present at levels comparable with the wild-type protein (Figure S1). Thus, the mutant RAG1 transcript is expressed in developing lymphocytes, and the amino acid substitution does not affect steady-state levels of the RAG1 protein.
RAG1-S723C homozygous mice exhibit impaired lymphocyte development
The impact of the RAG1-S723C mutation on lymphocyte development was initially assessed by examining the thymic cellularity of homozygous mutant mice. We observed a marked decrease (20- to 100-fold) in the number of thymocytes in 4- to 5-week-old RAG1S723C/S723C mice, indicating a substantial impairment in T-cell development in comparison with wild-type controls (Table 1). No significant decrease in thymocyte number was observed in RAG1+/S723C littermates at this age. Flow cytometric analyses of the RAG1-S723C mutant thymocytes were performed to determine the stage at which the developmental block occurs. During T-cell development, progenitors progress from the CD4−CD8− (DN) stage to the CD4+CD8+ (double positive, DP) stage upon successful V(D)J rearrangement of the T-cell receptor beta (TCRβ) locus. The majority of thymocytes in the RAG1-S723C homozygous mice were CD4−CD8− DN progenitors, indicating a severe impairment in T-cell development (Figure 1A; Table 1). DN thymocytes are divided into 4 subsets (DN1 through DN4) representing distinct developmental stages that can be identified based on CD44 and CD25 expression. Within the TCRβ locus, Dβ to Jβ rearrangements are initiated as thymocytes transition from the CD44+CD25+ DN2 to the CD44−CD25+ DN3 stage, and Vβ to DJβ rearrangements are completed at the DN3 stage. We observed a significant accumulation of CD44−CD25+ DN3 cells in the RAG1-S723C homozygous mice, consistent with a defect in TCRβ rearrangements due to impaired RAG endonuclease function (data not shown).
Table 1.
Impact of the RAG1-S723C mutation on lymphocyte development
| Genotype | Thymocyte no., ×106 | CD4+CD8+ thymocytes, % | Splenocyte no., ×106 | CD4+ splenic T cells, ×106 | CD8+ splenic T cells, ×106 | Pro-B cells, % of IgM− | Pro-B cells, ×105 | Pre-B cells, % of IgM− | Pre-B cells, ×105 |
|---|---|---|---|---|---|---|---|---|---|
| RAG1+/+, n = 5 | 200 ± 44 | 82.7 ± 3.6 | 71.6 ± 27 | 7.5 ± 3.8 | 3.9 ± 1.6 | 8.0 ± 0.4 | 0.4 ± 0.1 | 56.8 ± 4.9 | 2.9 ± 1.0 |
| RAG1+/S723C, n = 5 | 175.6 ± 68.9 | 81.7 ± 5.1 | 77.5 ± 23 | 8.2 ± 4.0 | 4.4 ± 1.6 | 9.8 ± 2.9 | 0.5 ± 0.2 | 52.4 ± 15.1 | 3.1 ± 1.8 |
| RAG1S723C/S723C, n = 8 | 4.6 ± 2.4 | 0.41 ± 0.89 | 18 ± 12.5 | 0.2 ± 0.1 | 0.07 ± 0.05 | 44.8 ± 5.5 | 1.2 ± 0.7 (P = .03) | 4.4 ± 0.6 | 0.18 ± 0.1 |
Figure 1.

Impaired lymphocyte development in RAG1-S723C homozygous mice. Flow cytometric analyses were performed on RAG1+/+, RAG1+/S723C, and RAG1S723C/S723C littermates, as indicated, at 5 weeks of age. (A) Thymocytes and lymph node cells were stained with αCD4 and αCD8 antibodies. (B) Bone marrow and splenocytes were stained with antibodies against the indicated cell-surface markers.
Despite the accumulation of progenitor T cells at the DN3 stage, we consistently observed a small population of thymocytes progress to the CD4+CD8+ DP as well as CD4+ and CD8+ single-positive (SP) stages in the majority of the RAG1S723C/S723C mice analyzed (Figure 1A; Table 1). The DP thymocytes in RAG1-S723C homozygous mice developed into mature CD4+ and CD8+ SP T cells that also expressed surface CD3 and TCRαβ in the peripheral lymphoid organs (ie, lymph nodes and spleen) (Figure 1A, data not shown).
In contrast to the partial T-cell immunodeficiency observed in the RAG1-S723C homozygous mutant mice, the impact on B-cell development was more severe. During B-cell development, the immunoglobulin heavy chain (IgH) locus initially undergoes DH to JH rearrangements during the B220+CD43+ pro-B stage of development. Subsequent productive VH to DJH rearrangements allow pro-B lymphocytes to progress to the pre-B stage in which cells lose expression of CD43. Flow cytometric analyses of bone marrow cells isolated from 4- to 5-week-old RAG1S723C/S723C mice revealed an impairment in B-cell development at the pro-B stage as indicated by an increase in the number and percentage of B220+CD43+ pro-B cells and a concomitant decrease in B220+CD43− pre-B cells (Figure 1B; Table 1). This block resulted in an absence of detectable numbers of B220+IgM+ immature B cells in bone marrow and mature B cells in peripheral lymphoid organs in the RAG1-S723C homozygous mutant mice (Figure 1B). No significant differences in B- or T-cell populations were observed in RAG1-S723C heterozygous mice compared with wild-type littermates at 4 to 5 weeks of age. Together, these results demonstrate that the RAG1-S723C mutation significantly impairs B- and T-cell development at the stages during which V(D)J recombination is initiated. However, low levels of productive rearrangements do occur in RAG1S723C/S723C mice as evidenced by the presence of more mature T-cell populations expressing TCRαβ and CD3.
Decreased levels of endogenous rearrangements in RAG1-S723C homozygous mice
We next investigated the effects of the RAG1-S723C mutation on endogenous D to J rearrangements to determine whether the observed impairment in lymphocyte development was due to decreased efficiency of chromosomal V(D)J recombination. To address this question, we used a PCR amplification strategy using DNA isolated from developing lymphocytes. We analyzed Dβ to Jβ rearrangements within the TCRβ locus in thymocytes using PCR primers located 5′ of Dβ1 or Dβ2 and 3′ of the most distal Jβ1 or Jβ2 segments, respectively. We observed very low levels of Dβ1 to Jβ1 rearrangements that were detectable only upon digestion of the genomic DNA with the restriction endonuclease, XbaI, which cleaves the locus between Dβ1 and the Jβ1 segments (Figure 2A). In addition, we detected only rare Dβ2 to Jβ2 rearrangements in RAG1S723C/S723C thymocytes (Figure 2A). Thus, the RAG1-S723C mutation severely impairs Dβ to Jβ rearrangements. Using the same PCR-based assay, we examined the levels of DH to JH rearrangements within the IgH locus in sorted pro-B- and pre-B-cell populations from homozygous RAG1-S723C mice and wild-type controls using a degenerate 5′ DH RSS primer and a primer 3′ of the most distal J segment, JH4. We observed that DH to JH rearrangements were substantially decreased in both the pro- and pre-B-cell populations in the RAG1-S723C homozygous mice (Figure 2B).
Figure 2.

PCR amplification of endogenous rearrangements in RAG1-S723C homozygous developing lymphocytes. (A) TCRβ rearrangements. Genomic DNA from purified DN thymocytes from RAG1+/+ (WT) and RAG1S723C/S723C (S723C) mice and a nonrearranging tissue (tail) was PCR amplified to detect Dβ1 to Jβ1 and Dβ2 to Jβ2 rearrangements. For Dβ1 to Jβ1 rearrangements, genomic DNA was first digested with the XbaI restriction endonuclease. Control PCR amplification of a nonrearranging locus was performed to normalize levels of input DNA. Rearrangements were detected by Southern blot hybridization using an oligonucleotide probe. Experiments (A-C) were repeated 3 times with genomic DNA samples from 3 different sets of mice; representative results are shown. (B) IgH rearrangements. Genomic DNA from sorted pro- and pre-B-cell populations from RAG1+/+ (WT) and RAG1S723C/S723C (S723C) mice and a nonrearranging tissue (kidney) was PCR amplified to detect DH to JH rearrangements. (C) Endogenous TCRδ signal joints. Levels of extrachromosomal signal joints formed between TCR Dδ2 and Jδ1 RSSs were detected by PCR amplification of genomic thymocyte DNA isolated from RAG+/+ (WT) and RAG1S723C/S723C (S723C) mice and a nonrearranging tissue (tail). Vertical lines have been inserted to indicate repositioned gel lanes.
We also examined levels of the reciprocal V(D)J recombination products that are generated upon ligation of the blunt, 5′ phosphorylated signal ends. We PCR amplified the signal joints formed between the Dδ2 and Jδ1 23 and 12 RSSs, respectively, located on stable extrachromosomal circular products that are excised upon rearrangement of the TCRδ locus. We observed that the RAG1-S723C homozygous mutation results in significantly reduced levels of signal joints (Figure 2C). Together, these results indicate that the impairment in B- and T-lymphocyte development observed in RAG1S723C/S723C mice is due to the diminished ability of the mutant RAG1/2 endonuclease to catalyze endogenous V(D)J recombination.
Qualitative analysis of RAG1-S723C–mediated V(D)J joints
To gain additional mechanistic insights into the consequences of the RAG1-S723C mutation on V(D)J recombination, the sequences of the rare endogenous junctions were analyzed. The hairpin coding ends generated by the RAG1/2 endonuclease undergo several processing steps prior to ligation that can result in gain or loss of nucleotides within the junctions.1 Endonucleolytic nicking of the hairpins by the Artemis-DNA-PKcs complex at sites away from the apex generates short, palindromic P nucleotides that are incorporated into the coding joints. Nucleotides can also be added via the template-independent DNA polymerase, TdT, or deleted by an undefined mechanism.
We used a nested PCR strategy to amplify D to J rearrangements from the endogenous IgH and TCRβ loci, then subcloned and sequenced the DJH and DJβ1 joints, respectively. We found that the coding junctions from both homozygous RAG1-S723C and wild-type mice exhibited the hallmark P and N nucleotide additions as well as small deletions of the coding flanks (Figure S2). In this regard, similar numbers of N nucleotides were observed in RAG1-S723C homozygous and wild-type mice within both the DJβ1 and DJH junctions. Likewise, the frequency and length of P nucleotide additions as well as the proportion of deleted coding flanks and extent of deletions in the RAG1-S723C homozygous mutant mice were comparable with those observed in wild-type controls (Figure S2; Document S1).
We also examined the fidelity of signal joining in wild-type and RAG-S723C mice. Precise end ligation of RSS heptamers generates the recognition sequence for the ApaLI restriction endonuclease. We amplified the signal joints formed between the Dδ2 and Jδ1 RSSs using a nested PCR strategy then digested the products with ApaLI. The PCR-amplified signal joints from wild-type and RAG1-S723C mutant thymocytes were digested to a similar extent (data not shown). Thus, the low-level endogenous coding and signal joints in the RAG1-S723C homozygous mice are structurally similar to those observed in wild-type mice.
Steady-state levels of signal ends in RAG1-S723C homozygous mice
To address whether the RAG1-S723C mutation had an impact on the generation and/or stability of V(D)J ends in vivo, we examined the steady-state levels of signal ends in developing lymphocytes using LM-PCR. Double-stranded DNA linkers were ligated to the blunt, 5′ phosphorylated ends present in genomic DNA isolated from progenitor lymphocytes. The ligation products were then PCR amplified using primers designed to detect signal ends at different rearranging segments, and specific LM-PCR products were identified by Southern blot analysis.
Signal ends located 5′ of Jβ2.7 and 3′ of Dβ1 within the TCRβ locus were readily detected in sorted CD4−CD8− DN thymocytes from RAG1S723C/S723C mice compared with controls (Figure 3A). However, a severe reduction in LM-PCR products corresponding to signal ends 3′ of Dβ2 (Figure 3A) as well as 5′ of Dδ2 and 5′ of the Jβ1.1-1.4 segments was observed (data not shown). We also performed LM-PCR analyses on genomic DNA from sorted B220+CD43+ pro-B cells and observed abundant products corresponding to cleavage events within the JH region of the IgH locus in the homozygous mutant mice. In contrast, signal ends 5′ of DFL16.1 were undetectable (Figure 3B). Together, these results demonstrated that the mutant RAG1/2 endonuclease possesses substantial cleavage activity at a subset of RSSs in vivo. However, the RAG1-S723C mutation also significantly reduced levels of cleaved signal end intermediates at other RSSs examined.
Figure 3.

LM-PCR analyses of signal ends in RAG1-S723C developing lymphocytes. Three-fold serially diluted linker ligated genomic DNA isolated from sorted (A) DN thymocytes and (B) sorted pro-B bone marrow cells from RAG1+/+, RAG1+/S723C, and RAG1S723C/S723C were used in PCR amplification reactions for detection of signal ends at the indicated loci. PCR amplification of a nonrearranging locus was performed as a normalization control. C indicates non–linker-ligated wild-type genomic DNA. (C) Location of LM-PCR products within the IgH JH locus. Δ bp indicates number of nucleotides deleted 5′ of the JH RSS depicted at the top of the columns; triangles, RSSs; and arrows, RAG1/2 cleavage sites. LM-PCR analyses were repeated 3 times on genomic DNA isolated from at least 3 independently sorted DN thymocyte and pro-B-cell samples. Representative results are shown.
During our analyses of signal ends within the JH locus, we consistently noted the presence of abundant LM-PCR products in RAG1S723C/S723C pro-B cells that were not of the predicted sizes (Figure 3B). We subcloned and sequenced the products to identify the positions of the DSBs, which presumably were blunt and 5′ phosphorylated as is required for successful linker ligation. In the RAG1S723C/S723C pro-B cells, we observed that only 4 of the 49 sequences represented intact JH signal ends (Figure 3C). The majority of DNA ends in the mutant pro-B cells were located at aberrant positions between 1 and 558 nucleotides away from the endogenous RSSs. In control pro-B cells, LM-PCR products emanating from DNA ends outside of the JH RSSs were also observed; however, the proportion of expected cleavage events was considerably higher compared with the RAG1-S723C homozygous mutant mice (14 of 32 sequences, P = .003) (Figure 3C). These findings provide evidence that dysfunctional RAG1/2 activity can generate aberrant DNA ends in vivo that do not participate in normal V(D)J recombination.
Examination of Omenn syndrome phenotypes
Inactivating mutations in RAG1 and RAG2 cause T−B− severe combined immunodeficiency (SCID) in human patients, whereas hypomorphic mutations are associated with combined immunodeficiency syndromes of varying severity, including Omenn syndrome (OS) and atypical or leaky SCID.31 Interestingly, the same missense, partial loss of function mutations can result in distinct clinical presentations, possibly due to genetic background and/or environmental factors.31 Whereas RAG-dependent immunodeficiencies lead to a broad range of phenotypes, OS patients exhibit a distinct combination of immunodeficiency and autoimmune features.32,33 OS patients are characterized by reduced or absent B cells and substantial, variable numbers of oligoclonal T cells, as well as autoimmune symptoms that manifest as early onset, generalized erythroderma and colitis. Additional OS phenotypes include enlarged lymphoid tissue, eosinophilia, low serum immunoglobulins, elevated serum IgE, hepatosplenomegaly, alopecia, chronic diarrhea, and failure to thrive. In atypical or leaky SCID, T cells are detectable in the periphery, although at reduced numbers, and the patients exhibit less severe immunopathological manifestations in the skin or gastrointestinal tract.31 As the RAG1-S723C hypomorphic mutation leads to a T+B−SCID phenotype similar to that observed in RAG-dependent human immunodeficiencies and OS mouse models,34,35 we assessed several key immunopathological phenotypes in RAG1S723C/S723C mice.
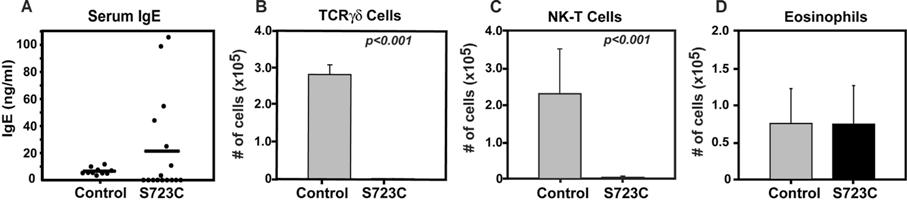
We observed that RAG1-S723C mutant mice did not develop the characteristic OS clinical manifestations, including alopecia, erythroderma, hepatosplenomegaly, lymphoadenopathy, failure to thrive, diarrhea, and colitis (Table S2). We found that the majority of RAG1-S723C mutant mice exhibited lower serum IgE levels compared with controls; however, a subset of mice did exhibit 5- to 10-fold higher levels (Figure S3A). The RAG1-S723C mutation also resulted in dramatically reduced levels of thymic and peripheral γδ T cells, which have been reported at normal or elevated levels in a subset of patients harboring RAG1 mutations36,37 (Figure S3B; Table S3). In addition, we observed significantly decreased, yet detectable, numbers of natural killer T (NKT) cells in RAG1S723C/S723C mice (Figure S3C; Table S3). RAG1-S723C homozygous mice had similar numbers of eosinophils compared with controls as determined by flow cytometry, indicating the absence of the characteristic eosinophilia associated with OS (Figure S3D; Table S3). Thus, overall, this phenotype is not suggestive of OS and rather resembles leaky SCID that has been reported in a subset of human subjects with hypomorphic RAG mutations.
RAG1-S723C heterozygosity causes accelerated age-dependent depletion of B and T lymphocytes
The lymphocyte development phenotypes of younger (3-5 weeks old) RAG1+/S723C mice were indistinguishable from wild-type controls; however, we hypothesized that the RAG1-S723C mutant protein may act in a dominant-negative manner since RAG1 functions in the context of a multimeric complex with RAG2.38 It is well established that aging results in diminished immune system function associated with reduced lymphocyte production in the bone marrow and thymus, decreased levels of V(D)J rearrangements, and accumulation of oligoclonal mature B and T cells.39,40 Thus, we explored the possibility that aged RAG1-S723C heterozygous mice would exhibit immune system defects.
We observed that older (12 months) RAG1+/S723C mice exhibit a dramatic impairment in T-cell development as evidenced by a significant decrease in number and percentage of CD4+CD8+ DP thymocytes compared with age-matched wild-type controls (Figure 4A; Table S4). In the bone marrow, significantly fewer B220+CD43+ pro-B cells progressed to the B220+CD43− pre-B and B220+IgM+ stages of development (Figure 4B; Table S4). The impairment in B and T lymphopoiesis in aged RAG1+/S723C mice was associated with significantly reduced levels of D to J and V to DJ rearrangements in thymocytes and bone marrow cells compared with age-matched controls (data not shown). In addition, consistent with the established phenotypes of aging mice, RAG1-S723C heterozygosity did not reduce the number of SP T cells or IgM+ B cells in the periphery39,40 (Figure 4; Table S4). Older RAG1S723C/S723C mice exhibited a similar impairment in B- and T-cell development as observed in younger mice (Figure 4).
Figure 4.

Significant impairment in B- and T-cell development in older RAG1-S723C heterozygous mice. Flow cytometric analyses were performed on RAG1+/+, RAG1+/S723C, and RAG1S723C/S723C mice at 12 months of age. (A) Thymocytes and splenocytes were stained with αCD4 and αCD8 antibodies. (B) Bone marrow cells and splenocytes were stained with antibodies against the indicated cell-surface markers.
The RAG1-S723C mutation leads to interlocus trans-rearrangements and thymic lymphomas
Our results indicating aberrant generation of broken DNA ends in RAG1-S723C homozygous mice led us to hypothesize that the DSBs may engage in chromosomal translocations. To address this question, we determined whether interchromosomal V(D)J rearrangements between the TCRγ and TCRβ loci, which are located on chromosomes 13 and 6, respectively, were present in RAG1-S723C mutant thymocytes using a nested PCR approach.41 Interchromosomal trans-rearrangement is a predictor of global chromosomal translocations41–44 and occurs at elevated frequencies in thymocytes that harbor mutations in genes that predispose to lymphoid neoplasia, including ATM,44–47 DNA-PKcs,41 and Nbs1.43,44 We detected PCR products corresponding to trans-rearrangements between TCRγV3S1 and TCRβJ2 in RAG1-S723C mutant thymocytes as well as ATM-null thymocytes (Figure 5A). In contrast, we did not readily observe interchromosomal rearrangements in wild-type, p53−/−, or RAG2−/− thymocytes, as previously reported.41,43,44 The PCR products from RAG1-S723C mutant thymocytes were subcloned and sequenced to verify that they represented the predicted interchromosomal events. We observed several unique clones containing flanking sequence from TCRγV3S1 and TCRβJ2 (Figure 5A). The junctions contained N and P nucleotides and small deletions, similar to the coding joints analyzed from intrachromosomal V(D)J recombination events in the RAG1S723C/S723C mice (Figure 5A).
Figure 5.

Rearrangements in RAG1-S723C thymocytes and RAG1-S723C/p53 double-mutant thymic lymphomas. (A) TCRγV3S1 (chr 13) to TCRβJ2S7 (chr 6) interchromosomal trans-rearrangements were PCR amplified from genomic thymocyte DNA isolated from mice of the indicated genotypes (top panel). S723C indicates RAG1S723C/S723C; Kid, kidney. Products corresponding to trans-rearrangements were detected by Southern blot analysis. Levels of rearrangements were normalized to PCR amplification of a nonrearranging locus (middle panel). Representative results are shown. Vertical lines have been inserted to indicate repositioned gel lanes. PCR products corresponding to TCRγV3-TCRβJ2 rearrangements in RAG1-S723C thymocytes were subcloned and sequenced (bottom panel). Coding sequences for TCRγV3 and TCRβJ2 are shown in boxes (bold); nucleotides added (center column) include P nucleotides (underlined) and N nucleotides; number of clones of each sequence are indicated. (B) Genomic DNA isolated from RAG1-S723C/p53 double-mutant lymphomas was digested with EcoRI then analyzed by Southern blot. Dβ1 to Jβ1 rearrangements were detected using a probe located 5′ of the Dβ1 segment (diagram). Individual tumors are indicated, top. C indicates wild-type thymus DNA; Kid, nonrearranging tissues; GL, unrearranged, germline band; and R, D-Jβ1 rearrangements. Amounts of input DNA were normalized to a nonrearranging locus (LR8). (C) PCR amplification of Dβ1 to Jβ1, Dβ2 to Jβ2, and Vβ8 to DJβ1 rearrangements were performed as described in Figure 2A using genomic DNA (250 ng) isolated from RAG1-S723C/p53 double-mutant lymphomas (tumor numbers indicated, top), wild-type thymocytes (C), and a nonrearranging tissue (tail). Input DNA levels were normalized to a nonrearranging locus (ATM). Vertical lines have been inserted to indicate repositioned gel lanes.
The occurrence of TCRγ-TCRβ trans-rearrangements in RAG1S723C/S723C thymocytes suggested that DSBs generated by the mutant RAG1/2 endonuclease could engage in aberrant chromosomal translocations; thus, we examined the RAG1-S723C mutant mice for tumor predisposition. We observed that the RAG1-S723C homozygous mutation alone does not predispose to tumorigenesis, as the mutant mice survive tumor free beyond 12 months of age. To enhance the oncogenic potential of unrepaired DNA DSBs, we introduced p53 deficiency into the RAG1-S723C mutant background through mouse breedings. We observed that RAG1S723C/S723Cp53−/− mice exhibit a significant decrease in survival in comparison with the p53−/− cohort (RAG1+/S723Cp53−/− and RAG1+/+p53−/− mice; P = .017, log-rank test) and controls (P < .001) (Figure 6A). All of the RAG1S723C/S723Cp53−/− mice succumbed to tumorigenesis, and the majority of tumors observed were immature TCRβ− T-cell lymphomas (90%, Table S5). In comparison, although the control p53−/− cohort was also predominantly predisposed to T-cell lymphomas (66%, Table S548,49), the majority of these tumors (16 of 23 thymic lymphomas) were surface TCRβ+, indicating they emanated from later stages of T-cell development compared with the RAG1-S723C/p53 mutant tumors (P < .001, 2-tailed Fisher exact test). Solid tumors and surface IgM+ B-cell lymphomas were frequently observed in the p53−/− cohort (12 of 35 tumors), whereas 3 of the 31 mice in the RAG1S723C/S723Cp53−/− cohort succumbed to solid tumors (P = .02, 2-tailed Fisher exact test), and no lymphoid tumors other than TCRβ− T-cell lymphomas were observed (Table S5).
Figure 6.

RAG1-S723C/p53 double-mutant mice are predisposed to thymic lymphomas with chromosomal translocations. (A) Survival of a cohort of control (RAG1+/+, RAG1+/S723C, RAG1S723C/S723C, n = 39), p53−/− (RAG1+/+p53−/−, RAG1+/S723Cp53−/−, n = 35), and RAG1S723C/S723Cp53−/− (n = 31) mice was observed for a period of 35 weeks. Kaplan-Meier survival curves were compared using the 2-tailed log-rank test with a 95% confidence interval (RAG1S723C/S723Cp53−/− vs p53−/− cohorts, P = .017; RAG1S723C/S723Cp53−/− vs control cohorts, P < .001). (B) RAG1S723C/S723Cp53−/− thymic lymphomas harbor chromosomal translocations and fusions. DAPI staining (left panels) and SKY analysis (right panels) of tumors S542 and S922 containing clonal chr 15;15 and 12;12 short-arm fusions and t(14;8) translocations, respectively. Arrows indicate translocated chromosomes.
We analyzed the rearrangement status of the TCRβ locus in the RAG1S723C/S723Cp53−/− lymphomas to assess the molecular events that occurred in the tumors. We performed Southern blot analyses of EcoRI-digested genomic DNA isolated from the RAG1-S723C/p53 double-mutant tumors using a probe located 5′ of Dβ1 (Figure 5B). The majority of tumors exhibited D-Jβ1 rearrangements on one of the 2 alleles. As Dβ to Jβ rearrangements are severely reduced in RAG1S723C/S723C thymocytes (Figure 2A), the relative intensities of the germline and rearranged bands observed in DNA isolated from tumor cells suggest that the rearrangements are clonal. We also examined the Dβ1 to Jβ1, Dβ2, and Jβ2 and Vβ8 to DJβ1 rearrangements via PCR amplification and observed several tumors harboring specific D-Jβ or V-DJβ rearrangements (Figure 5C). Together, these results indicate that the mutant RAG1-S723C/RAG2 endonuclease was active in generating DSBs in progenitor lymphocytes from which the tumors emanated, and the lymphomas are of a clonal origin.
To determine whether the RAG1S723C/S723Cp53−/− lymphomas were distinct from those caused by p53 deficiency alone, we analyzed the karyotypes of metaphases from primary and early passage cultured tumor cells using spectral karyotyping (SKY), a multicolor chromosome painting technique. Thymic lymphomas that arise in p53-deficient mice usually feature aneuploidy without clonal translocations,28,30,50–54 although clonal chromosomal aberrations have been observed in a small subset of p53−/− lymphomas in a mixed C57BL/6;129/Sv genetic background (M. Gostissa and F.W.A., unpublished, August 2007). RAG-null mice in a p53 mutant background are also predisposed to thymic lymphomas51,55; however, similar to the p53-null tumors, they are characterized by aneuploidy and do not harbor clonal translocations involving rearranging loci.51 Thus, in the absence of RAG-induced DSBs, clonal translocations are not a common feature of the lymphomas in p53 deficiency. Furthermore, inactivation of NHEJ genes, which are required for joining of V(D)J ends, does not increase the frequency of oncogenic translocations in the RAG/p53 double-null background. In this regard, thymic lymphomas that arise in Ku80−/−RAG2−/−p53−/− mutant mice lack clonal translocations,50 and XRCC4−/−RAG2−/−p53−/− mice die without any sign of lymphoma.56 Interestingly, DNA-PKcs mutation in a RAG2−/−p53−/− background leads primarily to progenitor B-cell malignancies associated with translocations involving chromosomes without V(D)J loci,57 thereby leading to the notion that DNA-PKcs deficiency leads to accumulation of unrepaired random DSBs that engage in oncogenic events.
We found that 6 of the 7 RAG1S723C/S723Cp53−/− lymphomas contained chromosomal translocations and/or short-arm fusions in a clonal population of tumor cells (Figure 6B; Table 2). We observed 2 tumors with translocations involving chromosomes that harbor rearranging TCR loci (chr 6 and 14), suggesting that the clonal events were initiated by the mutant RAG1-S723C/RAG2 endonuclease. Intriguingly, we also observed clonal translocations involving chromosomes lacking V(D)J loci. We also performed SKY analyses on several thymic lymphomas arising in RAG1+/S723Cp53−/− mice, as our analyses of lymphocyte development in older mice indicated that heterozygosity for the S723C mutation may confer additional phenotypes. We found that RAG1+/S723Cp53−/− lymphomas also harbored clonal chromosomal translocations (Table 2). The thymic lymphomas arising in our p53-null cohort were largely characterized by aneuploidy with one harboring a clonal translocation, consistent with previous studies.28,30,50–54 Thus, the RAG1-S723C/p53 thymic lymphomas featuring frequent clonal translocations and fusions are karyotypically distinct from the p53−/− lymphomas.
Table 2.
Spectral karyotyping of thymic lymphomas
| Tumor | Genotype | Translocations | Frequency | Flow cytometry |
|---|---|---|---|---|
| 83 | RAG1S723C/S723Cp53−/− | t(8;17) | 20/20 | CD4+CD8+TCRβ− |
| [2 translocated chromosomes per metaphase] | CD8SP | |||
| 135 | RAG1S723C/S723Cp53−/− | t(15;12) | 2/18 | CD4+CD8+TCRβ− |
| t(10;5) | 1/18 | |||
| S542 | RAG1S723C/S723Cp53−/− | Chr 15;15 SAFs | 16/18 | CD4+CD8+TCRβ− |
| Chr 12;12 SAFs | 5/18 | |||
| t(5;19) | 1/18 | |||
| S642 | RAG1S723C/S723Cp53−/− | Chr 3;6 SAFs | 5/24 | CD4−CD8− |
| t(14;1) | 2/24 | |||
| S779 | RAG1S723C/S723Cp53−/− | t(15;12) | 1/18 | CD4+CD8+TCRβ− |
| S878 | RAG1S723C/S723Cp53−/− | t(15;9) | 17/18 | CD4−CD8+TCRβ− |
| S922 | RAG1S723C/S723Cp53−/− | t(14;8) | 11/22 | CD4+CD8+TCRβ− |
| S786 | RAG1+/S723Cp53−/− | t(9;11) | 2/9 | CD4+CD8+TCRβ+ |
| S801 | RAG1+/S723Cp53−/− | Chr 15;15 SAFs | 28/33 | CD4+CD8+TCRβ− |
| Chr 11;11 SAFs | 22/33 | |||
| Chr X;X SAFs | 5/33 | |||
| Chr 16;16 SAFs | 4/33 | |||
| Chr 6;6 SAFs | 4/33 | |||
| Chr 3;3 SAFs | 2/33 | |||
| t(16;12) | 1/33 | |||
| S900 | RAG1+/S723Cp53−/− | Chr 5;5 SAFs | 15/24 | CD4+CD8+TCRβ+ |
| t(11;16) | 13/24 | |||
| S771 | RAG1+/+p53−/− | None | 0/19 | CD4−CD8+TCRβ+ |
| S839 | RAG1+/+p53−/− | None | 0/20 | CD4−CD8+TCRβ+ |
| S908 | RAG1+/+p53−/− | None | 0/27 | CD4+CD8+TCRβ+ |
| S919 | RAG1+/+p53−/− | None | 0/12 | CD4−CD8+TCRβ+ |
| S926 | RAG1+/+p53−/− | None | 0/20 | CD4−CD8+TCRβ+ |
| S1324 | RAG1+/+p53−/− | t(15;14) | 22/25 | CD4+CD8+TCRβ− |
SAFs indicates short-arm fusions.
Discussion
Leaky severe combined immunodeficiency due to the RAG1-S723C hypomorphic mutation
In this study, we examined the in vivo phenotypic impact of a RAG1 mutation, S723C, shown previously to have a minimal effect on RAG1/2 cleavage activity, but a significant impact on postcleavage complex formation and end joining in vitro.16 We found that mice harboring the gene-targeted RAG1-S723C homozygous mutation exhibit a significant impairment in lymphocyte development at the stages when V(D)J rearrangements occur. In contrast to RAG1- and RAG2-null mice, we observed “leaky” T-cell development in RAG1-S723C mutant mice, as evidenced by small populations of CD4+CD8+ DP thymocytes and mature peripheral CD4+ and CD8+ SP T cells expressing surface TCRαβ. In addition, D to J rearrangements were observed by PCR amplification in RAG1-S723C mutant developing lymphocytes. Hence, mice harboring the RAG1-S723C hypomorphic allele can be clearly distinguished from RAG nullizygosity by the ability to catalyze a low level of productive V(D)J recombination events in vivo. The phenotypes of RAG1-S723C mice more closely parallel those of human leaky SCID in which patients exhibit low or absent mature B cells, decreased thymic output, residual circulating T cells, and a mild manifestation of additional immunopathological phenotypes.31
Aberrant DNA DSB formation in RAG1-S723C developing lymphocytes
We detected substantial levels of signal ends at a subset of endogenous RSSs, indicating that the RAG1-S723C mutant enzyme was capable of efficiently binding and cleaving these RSSs. However, accumulation of signal ends at other RSSs was significantly reduced or undetectable. We also observed numerous aberrant DNA ends within the rearranging JH locus in developing mutant B lymphocytes. This phenotype is unique to the RAG1-S723C mice as RAG1/2 cleavage products of unpredicted sizes have not been reported in NHEJ deficiencies26,58–62 nor in truncated, “core” RAG163 or core RAG2 knockin mice.64 Together, these findings indicate that the RAG1-S723C mutation results in aberrant DNA DSB formation during lymphocyte development.
We propose the following, not mutually exclusive, explanations to account for the observed in vivo signal end phenotypes. The RAG1-S723C mutation may destabilize binding to the cleaved DNA ends, thereby leading to their deprotection and subsequent degradation. This molecular defect would result in the severe reduction or absence of signal ends observed in RAG1-S723C developing lymphocytes. Likewise, the LM-PCR products of unexpected sizes within the JH locus could result from nucleolytic resection resulting in blunt end structures, consistent with the notion that the signal ends are prematurely released from postcleavage complexes and rendered susceptible to degradation. Although the RAG1-S723C mutation did not significantly destabilize SECs in biochemical23 and transient transfection homologous recombination assays using core RAG1-S723C and core RAG-2 proteins,22 this mutation may have a greater impact in our studies that examined the full-length RAG1/2 proteins and V(D)J rearrangements in the normal context of a chromosome. In this regard, in cellular transfection V(D)J recombination assays, full-length RAG1-S723C/RAG2 severely reduced accumulation of signal ends, whereas core RAG1-S723C/core RAG2 had a significantly milder effect,16 thereby indicating that the truncated portions of the proteins may play a role in modulating the stability of signal end complexes. Thus, our in vivo findings of reduced accumulation of intact signal ends in developing lymphocytes parallel the in vitro studies using the full-length RAG1-S723C protein.16
Alternatively, the severely reduced levels of signal ends at particular RSSs in RAG1-S723C developing lymphocytes could be due to decreased cleavage activity in the context of chromosomal V(D)J recombination. The sequence composition of the heptamer, nonamer, 12 and 23 spacers as well as coding flanks influences the RSS cleavage efficiency by RAG1/2.4,7–11 Thus, some RSSs may represent poor substrates for binding or cleavage by the mutant RAG1-S723C/RAG2 endonuclease. Although a computational model65 developed to assess the recombinogenic potential of RSSs did not identify specific sequence elements that correspond to the different levels of intact signal ends observed in our study, additional factors, such as proximity to adjacent sequence elements and/or chromatin context of individual RSSs,66–68 may influence cleavage efficiency by the mutant endonuclease. Our observations also raise the intriguing possibility that the S723C mutation alters or loosens the cleavage specificity of the RAG1/2 endonuclease. The S723 residue resides within a central domain of RAG1 (amino acids 528-760 of 1040) that binds with sequence specificity to RSSs.38 Biochemical studies of purified RAG1-S723C indicate that this amino acid is important for proper interactions with flanking coding region DNA during both precleavage and postcleavage events.23 Thus, perturbations in specific contacts between RAG1/2 and the recombining DNA may alter cleavage activity.
What are the potential consequences of aberrant DNA cleavage by a dysfunctional RAG1/2 endonuclease? Cleavage of cryptic RSSs or non-B conformation DNA structures by RAG1/2 has been hypothesized as one mechanism by which oncogenic chromosomal translocations arise in some human lymphoid malignancies.69,70 In addition, unbiased scans of human and murine genomes revealed cryptic RSSs that can be efficiently cleaved by RAG1/2,71,72 indicating that abundant cleavage sites outside of the antigen receptor loci exist in mammalian genomes. A mutant RAG1/2 endonuclease with altered or loosened cleavage specificity could more frequently generate DNA breaks at the numerous cryptic RSS sequences or non-B form structures present in the genome. Furthermore, premature release of V(D)J ends in developing lymphocytes would result in accumulation of unprotected, unrepaired DSBs. Aberrant DNA ends generated by a dysfunctional RAG1/2 endonuclease via either mechanism possess significant potential to engage in oncogenic chromosomal translocations.
In this regard, we found that RAG1-S723C/p53 double-mutant mice are predisposed to thymic lymphomas that harbor frequent chromosomal translocations. Two of the 7 RAG1S723C/S723Cp53−/− tumors harbored chromosome 6 or 14 translocations, consistent with the notion that RAG1-S723C/RAG2-induced DNA DSBs at the rearranging TCR loci may engage in aberrant repair events. Intriguingly, 4 of the 7 RAG1S723C/S723Cp53−/− lymphomas contained translocations and short-arm fusions that involve the chromosomes lacking rearranging TCR loci. We speculate that the S723C mutation may permit more frequent endonucleolytic cleavage of cryptic RSSs or non-B form DNA structures within the genome, thereby generating substrates for aberrant DSB repair events, including chromosomal translocations.
RAG1-S723C heterozygosity results in accelerated age-associated immune system decline
We observed an accelerated onset of the hallmark signs of age-associated immune system dysfunction in RAG1-S723C heterozygous mice compared with age-matched wild-type controls. Aging in humans and mice causes a reduction in lymphocyte production in the thymus and bone marrow leading to an accumulation of oligoclonal populations of mature peripheral lymphocytes.39,40 This, in turn, results in a decline in antigen-specific immune responses in aged individuals. Twelve-month-old RAG1-S723C heterozygous mice exhibit significant defects in B- and T-cell development as evidenced by fewer CD4+CD8+ DP thymocytes as well as decreased numbers of B220+CD43− pre-B and B220+IgM+ B cells in the bone marrow. Thus, although the molecular mechanisms underlying age-associated immunosenescence remain unclear, our results suggest that defective RAG1/2 activity can influence the rate of decline of immune system function. These findings have potential implications for human disease as heterozygous carriers of inherited RAG1 or RAG2 hypomorphic alleles may be predisposed to early onset immune system dysfunction.
In summary, the RAG1-S723C knockin mutant mouse represents a unique model of leaky SCID. We have demonstrated that the RAG1-S723C hypomorphic mutation leads to impaired lymphocyte development, decreased V(D)J rearrangements, aberrant DNA cleavage and/or premature end release, and potentially oncogenic rearrangements in vivo. Our studies also revealed that older RAG1-S723C heterozygous mice exhibit accelerated onset of age-associated immune system dysfunction. Together, these findings suggest that hypomorphic mutations in the RAG1 or RAG2 genes in the human population not only cause combined immunodeficiencies but may also predispose to tumorigenesis and premature immunosenescence.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH, Bethesda, MD) through the University of Michigan's Cancer Center Support Grant (5 P30 CA46592); NIH grant AI063058 (NIAID), Pew Scholars Award (Pew Charitable Trusts, San Francisco, CA), Munn IDEA award (UM Cancer Center, Ann Arbor, MI) to J.S.; NIH grant AI32524 (NIAID) to D.G.S.; and NIH grant AI35714 (NIAID) to F.W.A. T.M. is a Fulbright Scholar. D.G.S. and F.W.A. are Investigators of the Howard Hughes Medical Institute.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.G. and M.C. performed experiments and analyzed the results; G.W., B.T., T.M., and H.-L.C. performed experiments; J.D. generated vital reagents in the laboratory of F.W.A.; D.G.S., D.O.F., and J.S. conceptualized and designed experiments and interpreted the results; J.S. wrote the paper; and all authors read and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: JoAnn Sekiguchi, Department of Internal Medicine, University of Michigan, Ann Arbor, MI 48109; e-mail: sekiguch@med.umich.edu.
References
- 1.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–131. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 2.Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 3.Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 4.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 5.Buck D, Malivert L, de Chasseval R, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 6.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Feeney AJ, Goebel P, Espinoza CR. Many levels of control of V gene rearrangement frequency. Immunol Rev. 2004;200:44–56. doi: 10.1111/j.0105-2896.2004.00163.x. [DOI] [PubMed] [Google Scholar]
- 8.Jung D, Bassing CH, Fugmann SD, Cheng HL, Schatz DG, Alt FW. Extrachromosomal recombination substrates recapitulate beyond 12/23 restricted VDJ recombination in nonlymphoid cells. Immunity. 2003;18:65–74. doi: 10.1016/s1074-7613(02)00507-1. [DOI] [PubMed] [Google Scholar]
- 9.Gerstein RM, Lieber MR. Coding end sequence can markedly affect the initiation of V(D)J recombination. Genes Dev. 1993;7:1459–1469. doi: 10.1101/gad.7.7b.1459. [DOI] [PubMed] [Google Scholar]
- 10.Yu K, Lieber MR. Mechanistic basis for coding end sequence effects in the initiation of V(D)J recombination. Mol Cell Biol. 1999;19:8094–8102. doi: 10.1128/mcb.19.12.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuomo CA, Mundy CL, Oettinger MA. DNA sequence and structure requirements for cleavage of V(D)J recombination signal sequences. Mol Cell Biol. 1996;16:5683–5690. doi: 10.1128/mcb.16.10.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leu TM, Eastman QM, Schatz DG. Coding joint formation in a cell-free V(D)J recombination system. Immunity. 1997;7:303–314. doi: 10.1016/s1074-7613(00)80532-4. [DOI] [PubMed] [Google Scholar]
- 13.Ramsden DA, Paull TT, Gellert M. Cell-free V(D)J recombination. Nature. 1997;388:488–491. doi: 10.1038/41351. [DOI] [PubMed] [Google Scholar]
- 14.Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- 15.Bailin T, Mo X, Sadofsky MJ. A RAG1 and RAG2 tetramer complex is active in cleavage in V(D)J recombination. Mol Cell Biol. 1999;19:4664–4671. doi: 10.1128/mcb.19.7.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai CL, Drejer AH, Schatz DG. Evidence of a critical architectural function for the RAG proteins in end processing, protection, and joining in V(D)J recombination. Genes Dev. 2002;16:1934–1949. doi: 10.1101/gad.984502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agrawal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- 18.Jones JM, Gellert M. Intermediates in V(D)J recombination: a stable RAG1/2 complex sequesters cleaved RSS ends. Proc Natl Acad Sci U S A. 2001;98:12926–12931. doi: 10.1073/pnas.221471198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu JX, Kale SB, Yarnell Schultz H, Roth DB. Separation-of-function mutants reveal critical roles for RAG2 in both the cleavage and joining steps of V(D)J recombination. Mol Cell. 2001;7:77–87. doi: 10.1016/s1097-2765(01)00156-3. [DOI] [PubMed] [Google Scholar]
- 20.Huye LE, Purugganan MM, Jiang MM, Roth DB. Mutational analysis of all conserved basic amino acids in RAG-1 reveals catalytic, step arrest, and joining-deficient mutants in the V(D)J recombinase. Mol Cell Biol. 2002;22:3460–3473. doi: 10.1128/MCB.22.10.3460-3473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarnell Schultz H, Landree MA, Qiu JX, Kale SB, Roth DB. Joining-deficient RAG1 mutants block V(D)J recombination in vivo and hairpin opening in vitro. Mol Cell. 2001;7:65–75. doi: 10.1016/s1097-2765(01)00155-1. [DOI] [PubMed] [Google Scholar]
- 22.Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- 23.Nagawa F, Hirose S, Nishizumi H, Nishihara T, Sakano H. Joining mutants of RAG1 and RAG2 that demonstrate impaired interactions with the coding-end DNA. J Biol Chem. 2004;279:38360–38368. doi: 10.1074/jbc.M405485200. [DOI] [PubMed] [Google Scholar]
- 24.Schlissel MS, Corcoran LM, Baltimore D. Virus-transformed pre-B cells show ordered activation but not inactivation of immunoglobulin gene rearrangement and transcription. J Exp Med. 1991;173:711–720. doi: 10.1084/jem.173.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gärtner F, Alt FW, Monroe R, et al. Immature thymocytes employ distinct signaling pathways for allelic exclusion versus differentiation and expansion. Immunity. 1999;10:537–546. doi: 10.1016/s1074-7613(00)80053-9. [DOI] [PubMed] [Google Scholar]
- 26.Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 27.Hempel WM, Stanhope-Baker P, Mathieu N, Huang F, Schlissel MS, Ferrier P. Enhancer control of V(D)J recombination at the TCRbeta locus: differential effects on DNA cleavage and joining. Genes Dev. 1998;12:2305–2317. doi: 10.1101/gad.12.15.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rooney S, Sekiguchi J, Whitlow S, et al. Artemis and p53 cooperate to suppress oncogenic N-myc amplification in progenitor B cells. Proc Natl Acad Sci U S A. 2004;101:2410–2415. doi: 10.1073/pnas.0308757101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petiniot LK, Weaver Z, Barlow C, et al. Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proc Natl Acad Sci U S A. 2000;97:6664–6669. doi: 10.1073/pnas.97.12.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Celeste A, Difilippantonio S, Difilippantonio MJ, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Hum Mutat. 2006;27:1174–1184. doi: 10.1002/humu.20408. [DOI] [PubMed] [Google Scholar]
- 32.Marrella V, Poliani PL, Sobacchi C, Grassi F, Villa A. Of Omenn and mice. Trends Immunol. 2008;29:133–140. doi: 10.1016/j.it.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: Inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122:1082–1086. doi: 10.1016/j.jaci.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 34.Marrella V, Poliani PL, Casati A, et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J Clin Invest. 2007;117:1260–1269. doi: 10.1172/JCI30928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khiong K, Murakami M, Kitabayashi C, et al. Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J Clin Invest. 2007;117:1270–1281. doi: 10.1172/JCI30513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehl S, Schwarz K, Enders A, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. 2005;115:3140–3148. doi: 10.1172/JCI25221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Villartay JP, Lim A, Al-Mousa H, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115:3291–3299. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De P, Rodgers KK. Putting the pieces together: identification and characterization of structural domains in the V(D)J recombination protein RAG1. Immunol Rev. 2004;200:70–82. doi: 10.1111/j.0105-2896.2004.00154.x. [DOI] [PubMed] [Google Scholar]
- 39.Min H, Montecino-Rodriguez E, Dorshkind K. Effects of aging on early B- and T-cell development. Immunol Rev. 2005;205:7–17. doi: 10.1111/j.0105-2896.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 40.Labrie JE, 3rd, Borghesi L, Gerstein RM. Bone marrow microenvironmental changes in aged mice compromise V(D)J recombinase activity and B cell generation. Semin Immunol. 2005;17:347–355. doi: 10.1016/j.smim.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 41.Lista F, Bertness V, Guidos CJ, Danska JS, Kirsch IR. The absolute number of trans-rearrangements between the TCRG and TCRB loci is predictive of lymphoma risk: a severe combined immune deficiency (SCID) murine model. Cancer Res. 1997;57:4408–4413. [PubMed] [Google Scholar]
- 42.Allam A, Kabelitz D. TCR trans-rearrangements: biological significance in antigen recognition vs the role as lymphoma biomarker. J Immunol. 2006;176:5707–5712. doi: 10.4049/jimmunol.176.10.5707. [DOI] [PubMed] [Google Scholar]
- 43.Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J. 2002;21:1447–1455. doi: 10.1093/emboj/21.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang J, Ferguson D, Song H, et al. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol. 2005;25:661–670. doi: 10.1128/MCB.25.2.661-670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipkowitz S, Stern MH, Kirsch IR. Hybrid T cell receptor genes formed by interlocus recombination in normal and ataxia-telangiectasis lymphocytes. J Exp Med. 1990;172:409–418. doi: 10.1084/jem.172.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stern MH, Lipkowitz S, Aurias A, Griscelli C, Thomas G, Kirsch IR. Inversion of chromosome 7 in ataxia telangiectasia is generated by a rearrangement between T-cell receptor beta and T-cell receptor gamma genes. Blood. 1989;74:2076–2080. [PubMed] [Google Scholar]
- 47.Kobayashi Y, Tycko B, Soreng AL, Sklar J. Transrearrangements between antigen receptor genes in normal human lymphoid tissues and in ataxia telangiectasia. J Immunol. 1991;147:3201–3209. [PubMed] [Google Scholar]
- 48.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 49.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 50.Difilippantonio MJ, Petersen S, Chen HT, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao MJ, Zhang XX, Hill R, et al. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 1998;18:3495–3501. doi: 10.1128/mcb.18.6.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haines BB, Ryu CJ, Chang S, et al. Block of T cell development in P53-deficient mice accelerates development of lymphomas with characteristic RAG-dependent cytogenetic alterations. Cancer Cell. 2006;9:109–120. doi: 10.1016/j.ccr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Bassing CH, Suh H, Ferguson DO, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 54.Vanasse GJ, Halbrook J, Thomas S, et al. Genetic pathway to recurrent chromosome translocations in murine lymphoma involves V(D)J recombinase. J Clin Invest. 1999;103:1669–1675. doi: 10.1172/JCI6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nacht M, Jacks T. V(D)J recombination is not required for the development of lymphoma in p53-deficient mice. Cell Growth Differ. 1998;9:131–138. [PubMed] [Google Scholar]
- 56.Zhu C, Mills KD, Ferguson DO, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 57.Gladdy RA, Taylor MD, Williams CJ, et al. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell. 2003;3:37–50. doi: 10.1016/s1535-6108(02)00236-2. [DOI] [PubMed] [Google Scholar]
- 58.Gao Y, Sun Y, Frank KM, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 59.Gu Y, Seidl KJ, Rathbun GA, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–665. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 60.Gao Y, Chaudhuri J, Zhu C, Davidson L, Weaver DT, Alt FW. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity. 1998;9:367–376. doi: 10.1016/s1074-7613(00)80619-6. [DOI] [PubMed] [Google Scholar]
- 61.Taccioli GE, Amatucci AG, Beamish HJ, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- 62.Rooney S, Sekiguchi J, Zhu C, et al. Leaky scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol Cell. 2002;10:1379–1390. doi: 10.1016/s1097-2765(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 63.Dudley DD, Sekiguchi J, Zhu C, et al. Impaired V(D)J recombination and lymphocyte development in core RAG1-expressing mice. J Exp Med. 2003;198:1439–1450. doi: 10.1084/jem.20030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang HE, Hsu LY, Cado D, Cowell LG, Kelsoe G, Schlissel MS. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 2002;17:639–651. doi: 10.1016/s1074-7613(02)00448-x. [DOI] [PubMed] [Google Scholar]
- 65.Cowell LG, Davila M, Kepler TB, Kelsoe G. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 2002;3:0072.1–0072.20. doi: 10.1186/gb-2002-3-12-research0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krangel MS. T cell development: better living through chromatin. Nat Immunol. 2007;8:687–694. doi: 10.1038/ni1484. [DOI] [PubMed] [Google Scholar]
- 67.Abarrategui I, Krangel MS. Noncoding transcription controls downstream promoters to regulate T-cell receptor alpha recombination. EMBO J. 2007;26:4380–4390. doi: 10.1038/sj.emboj.7601866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sen R, Oltz E. Genetic and epigenetic regulation of IgH gene assembly. Curr Opin Immunol. 2006;18:237–242. doi: 10.1016/j.coi.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 69.Raghavan SC, Lieber MR. DNA structures at chromosomal translocation sites. Bioessays. 2006;28:480–494. doi: 10.1002/bies.20353. [DOI] [PubMed] [Google Scholar]
- 70.Raghavan SC, Gu J, Swanson PC, Lieber MR. The structure-specific nicking of small heteroduplexes by the RAG complex: implications for lymphoid chromosomal translocations. DNA Repair (Amst) 2007;6:751–759. doi: 10.1016/j.dnarep.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marculescu R, Vanura K, Montpellier B, et al. Recombinase, chromosomal translocations and lymphoid neoplasia: targeting mistakes and repair failures. DNA Repair (Amst) 2006;5:1246–1258. doi: 10.1016/j.dnarep.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 72.Cowell LG, Davila M, Yang K, Kepler TB, Kelsoe G. Prospective estimation of recombination signal efficiency and identification of functional cryptic signals in the genome by statistical modeling. J Exp Med. 2003;197:207–220. doi: 10.1084/jem.20020250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}