

Abstract
Background
In this study we compare outlier loci detected using a FST based method with those identified by a recently described method based on spatial analysis (SAM). We tested a panel of single nucleotide polymorphisms (SNPs) previously genotyped in individuals of goat breeds of southern areas of the Mediterranean basin (Italy, Greece and Albania). We evaluate how the SAM method performs with SNPs, which are increasingly employed due to their high number, low cost and easy of scoring.
Results
The combined use of the two outlier detection approaches, never tested before using SNP polymorphisms, resulted in the identification of the same three loci involved in milk and meat quality data by using the two methods, while the FST based method identified 3 more loci as under selection sweep in the breeds examined.
Conclusion
Data appear congruent by using the two methods for FST values exceeding the 99% confidence limits. The methods of FST and SAM can independently detect signatures of selection and therefore can reduce the probability of finding false positives if employed together. The outlier loci identified in this study could indicate adaptive variation in the analysed species, characterized by a large range of climatic conditions in the rearing areas and by a history of intense trade, that implies plasticity in adapting to new environments.
Background
Population genomics relies on the principle that loci across the genome are influenced by genome-wide evolutionary force, whereas selection is locus-specific and imprints a particular pattern of variability only on linked loci [1,2]. Increasing attention has been put in understanding what proportion of a genome or which genes are being shaped by selection. Directional (Darwinian) selection can leave a set of signatures in the genes under its influence, such as the rapid divergence of functional sites among species and the depression of polymorphism within species. On the basis of these signatures, it is possible to identify genes or chromosomal regions which are likely targets of positive selection [3,4].
This is achieved by comparing all the loci across the genome that respond similarly to demography and neutral history of populations with outlier loci that show patterns of variation that deviate from the rest of the genome. Identification of outliers is important for two main reasons: i. such loci are potentially under selection and could be a sign of adaptive variation; ii. they could also bias estimates of population genetic parameters such as gene flow, population size and structure, and therefore should be excluded from these analyses. However, selected loci could be used in studies to better understand adaptation or to plan conservation-management strategies [5].
The identification of genes that have undergone positive selection is an important step in understanding how populations have adapted to environmental changes. Such studies are increasingly widespread [6-10] and their application to livestock species can also reveal insights on their selection history.
Aberrant behaviour of a locus can range from having exceptionally high or low FST between populations, to having an excess or deficit of low frequency alleles in a population [5].
Several statistical methods, in which loci candidate for selection are identified in the extreme tails of empirical distributions, have become a widely used strategy in genome-wide scans for selection [11-17]. However, several studies pointed out important methodological conditions to ensure the correct application of these methods [18] or stressed their lack of power [19-21].
FST statistic can be used to assess if the variation of SNP allele frequencies among populations leads to signatures of selection [2,22]. If FST is determined only by genetic drift, all loci across the genome are affected in a similar way. In the presence of locus-specific selection pressure, deviation in FST values is observed in selected loci and in linked genetic markers. See Table 3 for SNPs showing simulated FST.
Table 3.
Environmental variables considered in SAM analysis
| Variable | Description |
| Altitude | DEM SRTM30, NASA |
| DTR | Diurnal Temperature Range in °C |
| FRS | Number of days with ground frost |
| PR | Precipitation in mm/month |
| PRCV | Coefficient of variation of monthly precipitation in % |
| REH | Relative humidity (%) |
| SUN | Percent of maximum possible sunshine (percent of daylength) |
| TMP | Mean temperature in °C |
| RDO | Number of days with > 0.1 mm rain per month |
| WIND | Windspeed in m/s 10 meters above the ground |
List of environmental variables considered in SAM analysis. Yearly means and monthly values were used for these climatic variables (a total of 118 environmental parameters).
In this study we want to compare outlier loci assessed using a FST based method [22] with those identified by a recently described method [8] based on spatial analysis (SAM). In the original paper Joost et al. tested the efficacy of SAM against FST based methods using AFLP and microsatellite data from two different species, showing a strong correspondence between the two approaches [8]. SAM is an implementation of logistic regression resembling the method applied by Jump et al. to plant populations with AFLP markers [23]; but it is used in an explorative way rather than for the confirmation of working hypotheses [8,24] and therefore the processing of all possible univariate association models between all environmental parameters and the presence or absence of all single alleles is carried out.
Here we evaluate how FST and SAM perform with a different kind of marker, single nucleotide polymorphisms (SNPs), which are increasingly employed due to their high number, low cost and easy of scoring. Besides, SNP can be found inside gene candidates for artificial or natural selection and therefore they might be more informative for this purpose rather than neutral or random markers, like microsatellites and AFLPs.
We analysed goat breeds of southern areas of the Mediterranean basin (Italy, Greece and Albania) sampled in the context of the European ECONOGENE Project http://www.econogene.eu using a set of SNPs previously described [25]. See Table 2 for Breeds analysed, country of origin and their sample sizes (N).
Table 2.
Breeds analysed, country of origin and their sample sizes (N).
| Breed | Origin | N |
| Argentata dell'Etna | Italy | 31 |
| Bionda dell'Adamello | Italy | 31 |
| Camosciata | Italy | 31 |
| Capore | Albania | 31 |
| Dukati | Albania | 31 |
| Girgentana | Italy | 32 |
| Greek goat | Greece | 31 |
| Grigia molisana | Italy | 31 |
| Hasi | Albania | 31 |
| Liquenasi | Albania | 31 |
| Mati | Albania | 30 |
| Muzhake | Albania | 31 |
| Orobica | Italy | 31 |
| Sarda | Italy | 31 |
| Skopelos | Greece | 31 |
| Valdostana | Italy | 31 |
List of environmental variables considered in SAM analysis. Yearly means and monthly values were used for these climatic variables (a total of 118 environmental parameters).
Results
Six alleles at five loci (Lipase, Casein Alpha S1, Interleukin 2, Integrin Beta-1 and Growth Hormone Receptor) out of 27 were found to lie outside the 95% confidence region of the conditional joint distribution of FST and mean heterozygosity (Figure. 1) by FDIST2 analysis. They are potentially under selection (P < 0.05) (Tab. 3).
Figure 1.

Plot of FST against heterozygosity for the 27 SNPs analysed. Distribution of FST values as a function of the within-population heterozygosity (HS) based on the 27 goat SNPs analysed. The envelope of values corresponds to neutral expectations (with FST = 0.078) in the infinite-allele model constructed according to the method of Beaumont and Nichols [21], with a confidence level set to 95%.
With a significance threshold (ST) set to 95% (corresponding to P < 5.04E-06), SAM detected 16 loci associated with at least one environmental parameter, among which 3 are also identified by FDIST2 (LIPE, CSN1S1-5 and IL2_ln2). Conversely, Integrin beta-1 and Growth Hormone Receptor, identified by FDIST2, are not detected by SAM at any confidence level, and are not associated with any environmental parameter. With a ST of 1.01E-13 (confidence level of 99.99999999%, Bonferroni correction included,), SAM identified 3 alleles at 2 loci (CSN1S1 and LIPE) to be significantly associated with at least one environmental variable. Of the two SNPs analysed in CSN1S1 gene, one (CSN1S1_ex9) is involved in 38 significant models and is associated with 7 among 10 "families" of environmental parameters (duration of sunshine between may and September; relative humidity in January, May to September, and December; number of days with > 0.1 mm rain per month from may to September, and the yearly mean; temperature from April to September; precipitation from June to September; diurnal temperature range from march to December, and the yearly mean; number of days with ground frost per month from March to November, and the yearly mean). The second SNP in CSN1S1 (CSN1S1-5) is associated with 3 environmental parameters which are relative humidity in May, the number of days with ground frost per month in April, and diurnal temperature range in October. Finally, one LIPE allele is associated with relative humidity from May to September, the coefficient of variation of monthly precipitation in March, diurnal temperature range from March to December, as well as with the yearly mean, and with wind speed from April to May (see Figure 2).
Figure 2.
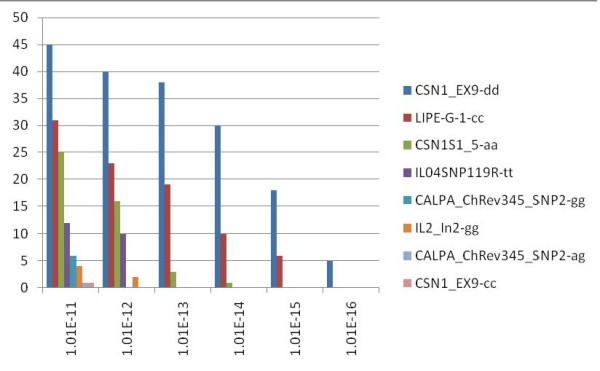
Plot of significant association models between alleles and environmental parameters. Histogram of the number of significant association models between genotypes and environmental parameters according to the 6 highest significance levels (Bonferroni correction included) in the analysis. From 1.01E-13 or a confidence level of 99.999999% only CSN1S1 and LIPE are significantly present. Names of environmental variables are listed in the text. Names of loci in the legend are followed by the genotype [25] associated to the models.
One CSN1S1_ex9 allele is the unique involved in the 5 most significant models (1.01E-16 Bonferroni correction included). At this level of significance, it is associated with relative humidity in July and August, with the number of days with > 0.1 mm rain per month in August, with precipitation in August, and with the number of days with ground frost in April.
Discussion
Rapid adaptation to environments is expected to have shaped at least some of the genetic diversity in marginal European goat breeds [8]. We thus expected that evidence for divergent selection should be detected in goat breeds, as a response to human and environment mediated selection and to migration history [26].
Outlier approaches, in which genes potentially under selection are identified in the extreme tails of empirical distributions, have become a widely used strategy in genome-wide scans for ascertaining selection signatures [11,14,16,17].
Most studies have been conducted using neutral or random markers, like microsatellites or AFLP, however the increasing use of SNPs as a genetic marker due to their rapid discovery rate provides the opportunity to identify genome-wide signatures of selection, as shown in humans [27,28]. There is potentially little power to detect outlier loci when examining populations in pairwise comparisons with biallelic markers [29]. On the contrary, in multi-population analysis the variability in FST among loci is reduced facilitating the detection of outlier loci [22]. Lao et al. [30] have shown that the frequency distributions of SNPs with ability to detect geographical population structure are most likely shaped by local positive selection rather than by genetic drift. The set of SNPs used for this study have been proven very efficient in detecting geographical patterning (Pariset et al, Population analysis of goat breeds from Italy, Albania and Greece reveals precise geographical patterning. Manuscript in preparation).
By calculating FST and mean heterozygosis as a measure of genetic differentiation for each locus, we have identified six loci at five genes in goat populations, which have been potentially target of selection according to the distribution of their genetic variation. One of them is related to body size and can be easily selected for by man, two are related to nutritional property of milk (CSN1S1 and LIPE) and they can be both selected by man or by adaptive advantage of the offspring. Interleukin-2 is a gene showing evidence for positive selection in mammals [31] as involved in immune response. The last gene, ITGB1, affects several traits, many of which potentially under selection.
Moreover, by using univariate logistic regression analysis with geographical variables, we could identify 3 of the 5 genes ascertained using FDIST2. The combined approach reveals that these three genes are under a selection pressure driven by the environment. One allele at locus CSN1S1 is very significantly associated with relative humidity during warm months (July and August), with the frequency of humidity and of precipitation during a warm summer month as well (number of days with > 0.1 mm rain per month and precipitation in August), and with the number of days with ground frost in April. These conditions (warm, wet) are favourable for parasite number to increase over time.
The last association with ground frost is more difficult to interpret, but April could correspond – grosso modo – to the moment when the animals come out of the goatshed and this could explain why the association doesn't exist during the other months in winter. Integrin beta-1 and Growth Hormone Receptor, identified by FDIST2, are not detected by SAM at any confidence level, and are not associated with any environmental parameter. Indeed, such markers can only be detected by their low variance in frequency among populations, and this action requires a dedicated model. Thus, because of their nature they cannot be detected using a regression approach like SAM does. Outlier loci, possibly under selection, can also be driven by other kinds of selection than natural selection. SAM proposes an environment-centered analysis, but there is room left for non-selected environmental effects, and FDIST or BayeScan are able to identify these outliers. This is a reason why Sam can not be used independently, without comparing results with specialized software packages based on theoretical approaches in population genetics, and focused on the analysis of genetic data
While directional selection tends to reduce within-population genetic diversity and to increase among-population differentiation, the reverse is expected under balancing selection, where gene frequencies tend to some equilibrium that maintains polymorphism. In general, Kelley et al. [20] found that the outlier approaches determine genes that have been targets of positive selection. Hoffman et al. [32] and Minder and Widmer [33] show that it is possible to find candidates for balancing selection using the Beaumont & Nichols [22] approach, but it is difficult because the lower 95% confidence limit is typically close to zero [34].
Among the 6 outlier SNPs, 3 of them had FST values lower than the 99% confidence limit in the FDIST2 test. We hypothesize that balancing or stabilizing selection may be responsible for these outliers, two of which (IL2 and ITGB2) have defence-immunity function. Indeed, genes involved in immunity are affected by balancing selection [35] and proteins that perform a defence/immunity function present low FST values [11].
In contrast, 3 loci had FST values exceeding the 99% confidence limits of FDIST2 and two of them were identified also by SAM. These genes might be important in adaptation to different environments and were probably subject to human selection, as well. SAM is able to identify both loci under directional and in stabilizing selection, but requires the comparison with the results produced by FDIST2 to differentiate the two types of selection, as is the case for IL2. Moreover, SAM can independently validate the FST outcomes and find correlations wit geographical variables, pinpointing the probable cause of the selection.
The population divergence methods implemented in FDIST2 have been widely used in several papers [7,15,17,32] and have been shown to be quite robust by simulations among various demographic scenarios [34]. Setting the same significance threshold, SAM detects more markers than FDIST2 does, but it was shown that when gradually lowering the confidence level in FDIST2, the method was also able to identify those additional loci detected by SAM [24]. SAM's sensitivity is not mastered yet, and further studies will be necessary to establish precise relationships between population genomics approaches and these statistical measures of association.
Conclusion
Data appear congruent by using the two methods for FST values exceeding the 99% confidence limits, more markedly than shown in the Joost et al. [8] paper where microsatellites were used as markers in sheep populations.
It may surprise that a high percentage of loci appear under selection, but it should be considered that the loci were purposely chosen for influencing potentially selected traits. Moreover, an active trade of livestock was historically present in the studied area [36] and adaptation to novel environment should have been crucial for the survival of imported populations.
The methods of FST and SAM can independently detect signatures of selection and therefore can reduce the probability of finding false positives if employed together.
The outlier loci identified in this study can be important because they could indicate adaptive variation in the analysed species, which is characterized by a large range of climatic conditions in the rearing areas and by a history of intense trade, that implies plasticity in adapting to new environments.
Methods
Goat breeds
A total of 16 autochthonous goat breeds, originating from Italy, Albania and Greece, were sampled for 30–32 unrelated animals per breed in farms spread over the traditional rearing area of each breed (tab. 2). No more than 3 individuals per farm were sampled to reduce the relationship among animals and to increase the breed representativeness. DNA from a total of 497 blood samples was extracted with a conventional method.
Genotyping was performed by K Biosciences (www Kbioscience.com), using Amplifluor™ (Serologicals™) and Taqman™ (Applied Biosystems™) chemistries using twenty-seven SNP markers, as described in Cappuccio et al. [25]. Generally, accuracy greater than 99% was achieved. Quality control criteria were adopted (water as negative control, inter- and intra-plate duplicate testing of a known DNA). Allelic frequency from a total of 13392 genotype assayed was compared to environmental parameters.
FDIST2 analysis
To detect the effects of selection, the approach used was that proposed by Beaumont and Nichols [22], further developed by Beaumont and Balding [34], and implemented in the FDIST2 software http://www.rubic.rdg.ac.uk/~mab/software.html. For each locus, the allele frequencies are used to compute FST values conditional on heterozygosity and to calculate P-values for each locus.
Each simulation included 32 individuals per population, 16 populations, 27 loci and an expected FST of 0.078. This method provides evidence for divergent selection by looking for outliers with FST values higher than expected, controlling for heterozygosity [22].
Population datasets were built using 100000 simulations on real data using the coalescent model. Upper and lower confidence limits of 95% quantiles were assumed for conditional joint distribution of FST versus mean heterozygosity. Loci showing atypical differentiation behaviour (i.e. FST) and lying outside the simulated neutral distribution are then detected as outliers.
SAM analysis
The Spatial Analysis Method (SAM) described in detail by Joost et al. [8] is based on an evaluation of the incidence of spatial coincidence, one of the six concepts of spatial analysis distinguished by Goodchild [37]. Spatial coincidence relates the genetic profile of the organisms studied to the environmental parameters measured at the geographic coordinates of its habitat. The data set used for analysis is in the form of a matrix. Each row of the matrix corresponds to an individual and to the geographic coordinates where it was sampled, while the columns contain a) binary information (1 or 0), relating to the status of the genetic marker (for AFLP markers, 1 or 0 respectively indicate the phenotypes « presence of band » and « absence of band »; for microsatellite and SNP markers, the numbers 1 and 0 respectively, indicate the presence or absence of a given allele at the locus in question) and b) values of environmental parameters at the location in question. Univariate logistic regression analysis are calculated by the SAM Program [38] to determine the degree of association between the frequencies of each allele and the values of the environmental parameters. By calculating the significance of the models generated by all possible pair-wise combinations (allele versus environmental parameter), the markers implicated in the models that emerge as statistically significant can identified. It can be inferred that such loci are likely to influence the process of adaptation to the environment.
Environmental data
The environmental information used in this study is comprised of altitude and climatic data. Altitude was estimated with the help of the digital elevation model SRTM30 (Shuttle Radar Topography Mission) developed by NASA, which has a resolution of 30 arc seconds. The climatic data described in Table 1 are based upon grids of 10 minutes of resolution (equivalent to approximately 12 km at the latitude of Switzerland). They are presented in the form of 9 monthly averages and an annual average. These variables characterise continental regions during the time period between 1961 and 1990 [39]. The data have been collected by the Climatic Research Unit (CRU) in Norwich, UK. For this study, monthly variables were separately analysed in order to take account of the seasonality of kidding [40-42].
Table 1.
SNPs showing simulated FST < sample FST after FDIST2 analysis.
| SNP | locus | sample He | sample FST, | test statistic | P* |
| LIPE | lipase | 0.50678 | 0.26101 | 3.95642 | 0.99995 |
| GHR | growth hormone receptor | 0.50032 | 0.01206 | -2.77215 | 0.00350 |
| CSN1-5 | casein alphaS1 | 0.50540 | 0.15638 | 2.06217 | 0.97596 |
| ITGB1 | integrin beta-1 | 0.46289 | 0.02699 | -2.01899 | 0.02662 |
| IL2_5 | interleukin 2 | 0.50477 | 0.14332 | 1.77708 | 0.95433 |
| IL2 IN2 | interleukin 2 | 0.03265 | 0.00721 | -5.00000 | 0.00000 |
*P(simulated FST < sample FST)
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LP: designed the study, performed the FST analysis, and drafted the manuscript. SJ: developed the SAM approach, carried out the relative computations, and formulated parts of the paper. PAM: leader of he Econogene project, provided DNA samples and guidance for the project. AV: participated in developing ideas, in supervision and revision of the manuscript.
Acknowledgments
Acknowledgements
This work has been partially supported by the ECONOGENE project, funded by the European Union (project QLK5-CT2001-02461). The content of the publication does not necessarily represent the views of the Commission or its services. The authors wish to thank Flora Jane Dause for help with the language.
Members of the ECONOGENE Consortium are listed here:
Abo-Shehada Mahamoud, Ajmone Marsan Paolo, Al Tarrayrah Jamil, Angiolillo Antonella, Baret Philip, Baumung Roswitha, Beja-Pereira Albano, Bertaglia Marco, Bordonaro Salvatore, Bruford Mike, Caloz Régis, Canali Gabriele, Canon Javier, Cappuccio Irene, Carta Antonello, Cicogna Mario, Crepaldi Paola, Dalamitra Stella, Daniela Krugmann, Dobi Petrit, Dominik Popielarczyk, Dunner Susana, D'Urso Giuseppe, El-Barody M. A. A, England Phillip, Erhardt Georg, Ertugrul Okan, Eva-Maria Prinzenberg, Eveline Ibeagha-Awemu, Ewa Strzelec, Fadlaoui Aziz, Fornarelli Francesca, Garcia David, Georgoudis Andreas, Gesine Lühken, Giovenzana Stefano, Gutscher Katja, Hewitt Godfrey, Hoda Anila, Horst Brandt, Istvan Anton, Juma Gabriela, Joost Stéphane, Jones Sam, Karetsou Katerina, Kliambas Georgios, Koban Evren, Kutita Olga, Lazlo Fesus, Lenstra Johannes A, Ligda Christina, Lipsky Shirin, Luikart Gordon, Marie-Louise Glowatzki, Marilli Marta, Marletta Donata, Milanesi Elisabetta, Negrini Riccardo, Nijman, Isaäc J, Obexer-Ruff Gabriela, Papachristoforou Christos, Pariset Lorraine, Pellecchia Marco, Peter Christina, Perez Trinidad, Pietrolà Emilio, Pilla Fabio, Roman Niznikowski, Roosen Jutta, Scarpa Riccardo, Sechi Tiziana, Taberlet Pierre, Taylor Martin, Togan Inci, Trommetter Michel, Valentini Alessio, Van Cann, Lisette M, Vlaic Augustin, Wiskin Louise, Zundel Stéphanie.
Contributor Information
Lorraine Pariset, Email: pariset@unitus.it.
Stephane Joost, Email: stephane.joost@epfl.ch.
Paolo Ajmone Marsan, Email: paolo.ajmone@unicatt.it.
Alessio Valentini, Email: alessio@unitus.it.
Econogene Consortium (EC), Email: paolo.ajmone@unicatt.it.
References
- Cavalli-Sforza LL. Population structure and human evolution. Proc R Soc Lond B Biol Sci. 1966;164:362–379. doi: 10.1098/rspb.1966.0038. [DOI] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nature Reviews Genetics. 2003;4:99–111. doi: 10.1038/nrg999. [DOI] [PubMed] [Google Scholar]
- Vallender EJ, Lahn BT. Positive selection on the human genome. Human Molecular Genetics. 2004;13:R245–254. doi: 10.1093/hmg/ddh253. [DOI] [PubMed] [Google Scholar]
- Luikart G, Gielly L, Excoffier L, Vigne J-D, Bouvet J, Taberlet P. Multiple maternal origins and weak phylogeographic structure in domestic goats. Proceedings of the National Academy of Sciences. 2001;98:5927–5932. doi: 10.1073/pnas.091591198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gil MR, Mikkonen M, Savolainen O. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Molecular Ecology. 2003;12:1195–1206. doi: 10.1046/j.1365-294X.2003.01826.x. [DOI] [PubMed] [Google Scholar]
- Pariset L, Cappuccio I, Joost S, D'Andrea MS, Marletta D, Ajmone Marsan P, Valentini A, ECONOGENE Consortium. Characterization of single nucleotide polymorphisms (SNPs) in sheep and their variation as an evidence of selection. Animal Genetics. 2006;37:290–292. doi: 10.1111/j.1365-2052.2006.01441.x. [DOI] [PubMed] [Google Scholar]
- Joost S, Bonin A, Bruford MW, Després L, Conord C, Erhardt G, Taberlet P. A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Molecular Ecology. 2007;16:3955–3969. doi: 10.1111/j.1365-294X.2007.03442.x. [DOI] [PubMed] [Google Scholar]
- Kane NC, Rieseberg LH. Selective Sweeps Reveal Candidate Genes for Adaptation to Drought and Salt Tolerance in Common Sunflower, Helianthus annuus. Genetics. 2007;175:1823–1834. doi: 10.1534/genetics.106.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveno E, Collada C, Guevara MA, Léger V, Soto A, Díaz L, Léger P, González-Martínez SC, Cervera MT, Plomion C, Garnier-Géré PH. Contrasting patterns of selection at Pinus pinaster Ait. Drought stress candidate genes as revealed by genetic differentiation analyses. Molecular Biology and Evolution. 2008;25:417–437. doi: 10.1093/molbev/msm272. [DOI] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Research. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payseur BA, Cutter AD, Nachman MW. Searching for evidence of positive selection in the human genome using patterns of microsatellite variability. Mol Biol Evol. 2002;19:1143–1153. doi: 10.1093/oxfordjournals.molbev.a004172. [DOI] [PubMed] [Google Scholar]
- Kayser M, Brauer S, Stoneking M. A genome scan to detect candidate regions influenced by local natural selection in human populations. Molecular Biology Evolution. 2003;20:893–900. doi: 10.1093/molbev/msg092. [DOI] [PubMed] [Google Scholar]
- Storz JF, Payseur BA, Nachman MW. Genome scans of DNA variability in humans reveal evidence for selective sweeps outside of Africa. Molecular Biology And Evolution. 2004;21:1800–1811. doi: 10.1093/molbev/msh192. [DOI] [PubMed] [Google Scholar]
- Vasemägi A, Primmer CR. Challenges for identifying functionally important genetic variation: the promise of combining complementary research strategies. Molecular Ecology. 2005;14:3623–3642. doi: 10.1111/j.1365-294X.2005.02690.x. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biology. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proceedings of the National Academy of Sciences. 2006;103:135–140. doi: 10.1073/pnas.0509691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera CM, Bazaga P. Population-genomic approach reveals adaptive floral divergence in discrete populations of a hawk moth-pollinated violet. Molecular Ecology. 2008;17:5378–5390. doi: 10.1111/j.1365-294X.2008.04004.x. [DOI] [PubMed] [Google Scholar]
- Bonin A, Taberlet P, Miaud C, Pompanon F. Explorative Genome Scan to Detect Candidate loci for Adaptation Along a Gradient of Altitude in the Common Frog (Rana temporaria) Molecular Biology and Evolution. 2006;23:773–783. doi: 10.1093/molbev/msj087. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Research. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Wong A, Aquadro CF. Approaches for identifying targets of positive selection. Trends in Genetics. 2007;23:568–577. doi: 10.1016/j.tig.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London series B: Biological Sciences. 1996;263:1619–1626. doi: 10.1098/rspb.1996.0237. [DOI] [Google Scholar]
- Jump AS, Hunt JM, Martínez-Izquierdo JA, Peñuelas J. Natural selection and climate change: temperature-linked spatial and temporal trends in gene frequency in Fagus Sylvatica. Molecular Ecology. 2006;15:3469–3480. doi: 10.1111/j.1365-294X.2006.03027.x. [DOI] [PubMed] [Google Scholar]
- Joost S, Bonin A, Taberlet P, Caloz R. Un rôle pour la science de l'information géographique en écologie moléculaire. La détection de régions du génome soumises à la sélection naturelle. Revue Internationale de Géomatique. 2008;2:215–237. doi: 10.3166/geo.18.215-237. [DOI] [Google Scholar]
- Cappuccio I, Pariset L, Ajmone-Marsan P, Dunner S, Cortes O, Erhardt G, Lühken G, Gutscher K, Joost S, Nijman IJ, Lenstra JA, England PR, Zundel S, Obexer-Ruff G, Beja-Pereira A, Valentini A, Econogene Consortium. Allele frequencies and diversity parameters of 27 single nucleotide polymorphisms within and across goat breeds. Molecular Ecology Notes. 2006;6:992–997. doi: 10.1111/j.1471-8286.2006.01425.x. [DOI] [Google Scholar]
- Taberlet P, Valentini A, Rezaei HR, Naderi S, Pompanon F, Negrini R, Ajmone-Marsan P. Are cattle, sheep, and goats endangered species? Molecular Ecology. 2007;17:275–284. doi: 10.1111/j.1365-294X.2007.03475.x. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR, LatheWC III, Ramensky VE, Bork P. SNP frequencies in human genes: An excess of rare alleles and differing modes of selection. Trends in Genetics. 2000;16:335–337. doi: 10.1016/S0168-9525(00)02058-8. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA. Adaptation and speciation: what can F(st) tell us? Trends in Ecology and Evolution. 2005;20:435–440. doi: 10.1016/j.tree.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Lao O, van Duijn K, Kersbergen P, de Knijff P, Kayse M. Proportioning Whole-Genome Single-Nucleotide-Polymorphism Diversity for the Identification of Geographic Population Structure and Genetic Ancestry. The American Journal of Human Genetics. 2006;78:680–690. doi: 10.1086/501531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MJ. Applications of selective neutrality tests to molecular ecology. Molecular Ecology. 2002;11:1245–1262. doi: 10.1046/j.1365-294X.2002.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EA, Schueler FW, Jones AG, Blouin MS. An analysis of selection on a colour polymorphism in the northern leopard frog. Molecular Ecology. 2006;15:2627–2641. doi: 10.1111/j.1365-294X.2006.02934.x. [DOI] [PubMed] [Google Scholar]
- Minder AM, Widmer A. A population genomic analysis of species boundaries: neutral processes, adaptive divergence and introgression between two hybridizing plant species. Molecular Ecology. 2008;17:1552–1563. doi: 10.1111/j.1365-294X.2008.03709.x. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Molecular Ecology. 2004;13:969–980. doi: 10.1111/j.1365-294X.2004.02125.x. [DOI] [PubMed] [Google Scholar]
- Richman A. Evolution of balanced genetic polymorphism. Molecular Ecology. 2009;9:1953–1963. doi: 10.1046/j.1365-294X.2000.01125.x. [DOI] [PubMed] [Google Scholar]
- Columella LIM. De arietibus eligendis. De re rustica, liber VII III circa 60 AD. http://www.centrumlatinitatis.org/testi_latini/Columellae/DeReRustica07.txt
- Goodchild MF. Geographic Information Systems and spatial analysis in the social sciences. In: Aldenderfer M, Maschner HDG, editor. Anthropology, space, and Geographic Information Systems. New York, Oxford University Press; 1996. pp. 214–250. [Google Scholar]
- Joost S, Kalbermatten M, Bonin A. Spatial Analysis Method (SAM): a software tool combining molecular and environmental data to identify candidate loci for selection. Molecular Ecology Resources. 2008;8:957–960. doi: 10.1111/j.1755-0998.2008.02162.x. [DOI] [PubMed] [Google Scholar]
- New M, Lister D, Hulme M, Makin I. A high-resolution dataset of surface climate over global land areas. Climate Research. 2002;21:1–25. doi: 10.3354/cr021001. [DOI] [Google Scholar]
- Mavrogenis AP, Constantinou A, Louca A. Environmental and genetic causes of variation in production traits of Damascus goats. 2. Goat productivity. Animal Production. 1984;38:99–104. [Google Scholar]
- Galina MA, Silva E, Morales R, Lopez B. Reproductive performance of Mexican dairy goats under various management systems. Small Ruminant Research. 1995;18:249–253. doi: 10.1016/0921-4488(95)00693-6. [DOI] [Google Scholar]
- Kominakis A, Rogdakis E, Vasiloudis Ch, Liaskos O. Genetic and environmental sources of variation of milk yield of Skopelos dairy goats. Small Ruminant Research. 2000;36:1–5. doi: 10.1016/S0921-4488(99)00105-4. [DOI] [Google Scholar]